
Abstract
Gas vesicle nanoparticles (GVNPs) are hollow, buoyant prokaryotic organelles used for cell flotation. GVNPs are encoded by a large gas vesicle protein (gvp) gene cluster in the haloarchaeon, Halobacterium sp. NRC-1, including one gene, gvpC, specifying a protein bound to the surface of the nanoparticles. Genetically engineered GVNPs in the Halobacterium sp. have been produced by fusion of foreign sequences to gvpC. To improve the versatility of the GVNP platform, we developed a method for displaying exogenously produced GvpC fusion proteins on the haloarchaeal nanoparticles. The streptococcal IgG-binding protein domain was fused at or near the C-terminus of GvpC, expressed and purified from E. coli, and shown to bind to wild-type GVNPs. The two fusion proteins, GvpC3GB and GvpC4GB, without or with a highly acidic GvpC C-terminal region, were found to be able to bind nanoparticles equally well. The GVNP-bound GvpC-IgG-binding fusion protein was also capable of binding to an enzyme-linked IgG-HRP complex which retained enzyme activity, demonstrating the hybrid system capability for display and delivery of protein complexes. This is the first report demonstrating functional binding of exogenously produced GvpC fusion proteins to wild-type haloarchaeal GVNPs which significantly expands the capability of the platform to produce bioengineered nanoparticles for biomedical applications.
Key points
• Haloarchaeal gas vesicle nanoparticles (GVNPs) constitute a versatile display system.
• GvpC-streptococcal IgG-binding fusion proteins expressed in E. coli bind to GVNPs.
• IgG-binding proteins displayed on floating GVNPs bind and display IgG-HRP complex.
Graphical abstract

Similar content being viewed by others


Introduction
Gas vesicles (GVs) are widely distributed among prokaryotic microorganisms (Walsby 1994; DasSarma and DasSarma 2021). GVs are advantageous for cell survival in aquatic environments, where they promote cell flotation and increase the availability of light and oxygen to cells, enhancing their photosynthetic or phototrophic capabilities (DasSarma and DasSarma 2021). GV buoyancy is dependent on exclusion of water from its interior as a consequence of the hydrophobicity of the interior surface of the vesicle membrane. The purified organelles are of interest for their monodispersed, nanoparticular size, structural stability, and bioengineerable properties, as well as their non-toxicity and self-adjuvanticity, which together make them ideal for use in biotechnological applications such as for antigen or drug delivery (DasSarma et al. 1999, 2015; Halladay et al. 1993; Stuart et al. 2001, 2004; DasSarma and DasSarma 2015; Andar et al. 2017; Hill and Salmond 2020).
In the model halophilic Archaeon (haloarchaeon) Halobacterium sp. NRC-1, a large gene cluster (gvpMLKJIHGFEDACNO) encoding more than a dozen proteins was found to be implicated in GVNP biogenesis (Jones et al. 1989; 1991; DasSarma and Arora 1997; DasSarma and DasSarma 2021). Detailed analysis of mutants constructed with interruptions in each of the gvp genes exhibited a partially or completely gas vesicle-deficient phenotype (DasSarma et al. 1994). Immunoblotting analysis using antisera directed against individual gas vesicle proteins showed the presence of seven gvp gene products in the nanoparticles, including the major GVNP protein, GvpA, which forms the striated GV membrane; the externally located GvpC protein, which strengthens and promotes growth; and five other gene products (GvpF, GvpG, GvpJ, GvpL, and GvpM) of yet unknown function (DasSarma et al. 1987; Halladay et al. 1993; Shukla and DasSarma 2004).
GvpC is remarkable in being an extremely highly acidic (pI 3.57) hydrophilic protein with a predicted molecular mass of 42,391 Da, which has been shown to be valuable for bioengineering GVNPs and displaying foreign protein sequences on their surface (DasSarma et al. 1999). In Halobacterium sp. NRC-1, GvpC is capable of accommodating insertions (up to 1,200 bp) at its C-terminal end which contains an extremely acidic region thought to stabilize the nanoparticles at the high salinity occurring in the cytoplasm of the extreme halophile (DasSarma et al. 2013). Insertion mutations further upstream in the gvpC gene resulted in formation of smaller GVNPs less than half the length and width of the wild-type, consistent with its requirement for growth and stability of larger, wild-type structures (DasSarma et al. 1994). In cyanobacteria, a distantly related GvpC has also been shown to serve a strengthening role against collapse from hydrostatic pressure (Damerval et al. 1987; Griffiths et al. 1992; Dunton et al. 2006).
A variety of genetic fusions to gvpC genes (and derivatives) have been used successfully to produce bioengineered nanoparticles with target proteins or peptides displayed in Halobacterium sp. NRC-1 (DasSarma and DasSarma 2015). This includes the use of a system in which a strain with a gvpC gene deletion was genetically complemented with an enzymatically active GvpC-luciferase fusion protein which bound in vivo to GVNPs (DasSarma et al. 2013). A series of GvpC fusion proteins with different lengths were tested for display. Those with GvpC-luciferase fusions at or near the C-terminus, but lacking the highly acidic region, performed better than shorter derivatives. This display system utilized Halobacterium strains as host for both genetic engineering and expression of the bioengineered GVNPs.
GVNPs exhibit properties for biomedical uses including non-toxicity toward mammalian cells and animals, stability without a cold chain, light refraction and visual contrast, and delivery using microneedles (Andar et al. 2017; Yan et al. 2020). Their applications include display of Gag coat protein of simian immunodeficiency virus (SIV) (DasSarma et al. 1999; Stuart et al. 2004; Sremac and Stuart 2008; 2010), antigens of Chlamydia responsible for venereal and ocular diseases (Childs and Webley 2012), Salmonella invasion protein, SopB (DasSarma et al. 2014; 2015), and malaria parasite antigens, the circumsporozoite protein and enolase (Pecher et al. 2016; Dutta et al. 2015). GVNPs displaying a portion of bactericidal permeability-increasing protein exhibited anti-inflammatory and endotoxin-neutralizing activity as well as protective activity in a mouse model of endotoxic shock (Balakrishnan et al. 2016).
To further expand the versatility of the GVNP system, we sought to develop a hybrid approach for display utilizing Halobacterium sp. NRC-1 for production of wild-type nanoparticles and GvpC fusion protein expression and production in E. coli, a host amenable to facile gene expression with a faster growth rate. In order to develop and test such a next-generation GVNP display system (Fig. 1), we selected the classic immunoglobulin-binding streptococcal protein G (SPG) for fusion to GvpC (Sjöbring et al. 1991; Nomellini et al. 2007). SPG is a cell surface receptor protein with a multiple domain structure containing tandem repeats of serum albumin-binding and immunoglobulin-binding domains. We targeted the immunoglobulin-binding domain B (GB) of SPG, which is readily detected using commercial antibodies. This protein is also of considerable value for purification, immobilization, and detection of immunoglobulins as well as for binding of IgGs of choice for display and therapeutic applications, allowing ample uses for biomedical applications (Rigi et al. 2019).

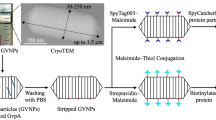
Next-generation strategy for displaying target-specific monoclonal antibodies on the surface of GVNPs. Pink and yellow spirals indicate wild-type GvpC molecules (with the yellow portion representing the acidic tail) that are predicted to lay across several GvpA ribs, holding the subunits together (DasSarma and DasSarma 2021). Upon binding of recombinant GvpC fused with IgG-binding domain (purple spiral portion), monoclonal antibodies are displayed on the surface of GVNPs (GVNP::GvpCGB::mAb). GVNP complexes associated with monoclonal antibodies (Y-shaped) specific to target cells, potentially more than one type depicted by red and yellow blocks, either alone or in combination, can be applicable for mAb-guided targeting or therapy (patent pending)
As detailed in this report, we successfully designed, constructed, expressed, and purified GvpC fusion proteins containing the functional Streptococcus GB domain employing an E. coli expression system, followed by binding and displaying the fusion proteins on the haloarchaeal GVNPs. Our findings significantly enhance the versatility of bioengineered gas vesicles for displaying foreign proteins on the nanoparticle surface.
Materials and methods
Strains and culturing
E. coli BL21(DE3) (New England Biolabs) was grown using LB media. Halobacterium sp. NRC-1 (ATCC 700,922/JCM11081) was grown in CM+ media. For GVNP preparation, NRC-1 cultures were plated on CM+ agar plates with incubation at 37 °C for 3 weeks until distinct pink turbid lawns were formed (DasSarma and Fleischmann 1995).
Gas vesicle nanoparticle preparation
GVNPs were prepared by lysing the lawns of Halobacterium sp. NRC-1 on agar plates with 5 ml of 1 × PBS [137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate dibasic, and 2 mM potassium phosphate monobasic (pH 7.2)] containing 1 mM MgSO4 and 1 mg/ml DNAse I (Sigma-Aldrich) followed by incubation for 3 h at 37 °C. The lysates were then centrifuged at 400 rpm (60 × g) overnight in a clinical centrifuge using a swinging bucket rotor. Intact buoyant GVNPs were collected and resuspended in 2–3 volumes of 1 × PBS followed by centrifugation, and processed through four additional rounds of accelerated floatation until a milky white suspension of purified GVNPs was obtained (DasSarma et al. 2013).
Recombinant DNA for bioengineering GVNPs
The streptococcal protein GB1 domain was selected for expression as an N-terminal His-tagged GvpCGB fusion protein (Nomellini et al. 2007). Four oligonucleotides corresponding to the region encoding the 54-amino acid IgG-binding domain were designed to correspond to the forward and reverse strands with ends flanked with sequences compatible for restriction endonucleases AfeI and BamHI, respectively (see below). After annealing, the synthetic oligonucleotides were cloned into AfeI/BamHI-digested pETGvpC3, which is the pET19b-derived plasmid containing the C-terminal-shortened gvpC gene (gvpC3) from the pARK-C series plasmids (DasSarma et al. 2013) for expression in E. coli BL21(DE3) to obtain the pETG19b-GvpC3GB construct. Recombinant plasmid pETGvpC4GB encoding the full-length (C4) GvpC fused with GB was constructed by replacing the 0.2 kb AatII/BamHI fragment of pET19b-GvpC3GB with the corresponding 0.5 kb AatII/BamHI fragment of the pARK-derived plasmid containing the complete GvpC4 gene fused with GB (Fig. 2).

Expression of recombinant GvpC3GB and GvpC4GB fusion proteins in E. coli. a Above: structure of gene constructs encoding the gvpC-IgG-binding domain (GB) fusion proteins containing either the full-length (382 amino acids) GvpC (GvpC4) or a shorter (280 amino acids) GvpC (GvpC3). Restriction endonuclease sites used for cloning are indicated by arrows. Below: nucleic acid and protein sequences of the 54-amino acid IgG-binding domain. b Immunoblot analysis of GvpC3GB (lane 1) and GvpC4GB (lane 2) purified from E. coli using HRP-linked goat anti-rabbit IgG antibody
Expression and purification of fusion proteins
Expression of the fusion proteins in E. coli was induced with 1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) at 20 °C for 4 h. Cells were harvested by centrifugation for 10 min at 3,000 × g. After resuspension in LEW buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0), lysozyme was added to a final concentration of 1 mg/ml, followed by the addition of 2 mM DTT and 10 mM CaCl2 and incubation for 30 min and freeze (− 80 °C)-thawing (at room temperature) three times. Inclusion bodies were collected by centrifugation at 10,000 × g for 10 min followed by resuspension in 10 ml of 8 M urea LEW buffer, and sonicated at 20% amplitude and pulse durations of 5 s on and 5 s off for 5 min using SCIENTZ-IID Ultrasonic homogenizer (Ningbo Sci Biotech, China). The supernatant was diluted with LEW buffer to reduce the urea concentration to 2 M and passed through a Protino® Ni-TED column (Macherey–Nagel™, Düren, Germany) to bind the His-tagged GvpCGB fusion proteins. After washing the column with 2 M urea LEW buffer three times, the bound fusion protein was eluted with 2 M urea LEW buffer including 250 mM imidazole. The protein concentrations were measured using BCA protein assays (Thermo Scientific) according to the manufacturer’s protocol. Expression of the GvpCGB fusion proteins was confirmed by immunoblot analysis using a 1:5000 dilution of HRP-linked goat anti-rabbit- and anti-mouse-IgG polyclonal antibodies (Thermo Scientific, Rockford, IL) (Fig. 2).
Binding and stabilization of E. coli GvpC fusion proteins to GVNPs
Binding of haloarchaeal GVNPs with exogenously produced GvpC fusion proteins was tested in the presence of 5 M NaCl or basal salt solution containing 250 g NaCl, 20 g MgSO4.7H2O, 2 g KCl, and 3 g Na3C6H5O7.2H2O (DasSarma and Fleischman 1995). To compare the binding ability of GvpC with different lengths of C-terminal region, incubation was also carried out with different amounts of GvpC3GB and GvpC4GB at 25 °C for 1.5 h. The mixture was next centrifuged at 300 × g for 30 min to separate the GVNP (top) fraction from the subnatant fraction. Levels of the recombinant GvpCGBs in each fraction were determined by SDS-PAGE and immunoblotting analysis. Briefly, each fraction was mixed with an equal volume of 2 × SDS-PAGE gel loading buffer containing 100 mM Tris–Cl (pH 6.8), 4% (w/v) SDS, 20% glycerol, and 200 mM β-mercaptoethanol and boiled for 4 min. The proteins were then analyzed by SDS-PAGE and immunoblotting and quantified using ImageJ (Sambrook and Russell 2001; Schneider et al. 2012).
Immunoblotting analysis of GvpC3GB and GvpC4GB fusion proteins
Immunoblotting analysis of purified gas vesicles was determined in a manner similar to that described previously (Shukla and DasSarma 2004). Briefly, GVNP (top) or subnatant fractions were mixed in an equal volume of 2 × sample loading buffer as above, and boiled for 4 min. After cooling to room temperature, the samples were briefly vortexed and electrophoresed on a 10% SDS–polyacrylamide gel for 90 min at 100 V at room temperature on a Bio-Rad (San Diego, CA) Mini-PROTEAN Electrophoresis system. The electrophoretically fractionated GVNPs were electroblotted onto a 0.45 μm Immobilon-PVDF (Millipore Corp., Boston, MA) membrane for 2 h at 100 V using a Bio-Rad mini gel blotter. The membranes were washed twice for 5 min with TBS-T buffer, blocked for 1 h with 3% milk proteins in TBS-T, and followed by washing three times for 5 min each. The washed membranes were incubated for 30 min at room temperature by gentle shaking with HRP-linked anti-rabbit IgG antibody diluted 1:3,000. The membranes were then washed 3 times each for 5 min with TBS-T and then subjected to chemiluminescence reaction using ECL reagent (GE Healthcare) according to the manufacturer’s specifications. Band intensity was measured by ImageQuant LAS 500 (GE Healthcare) followed by quantification using ImageJ (Schneider et al. 2012).
Nucleic acids
Four oligonucleotides corresponding to the Streptococcus gene region encoding the 54-amino acid IgG-binding domain (GenBank: OL890683) were designed to correspond to the forward and reverse strands with each end flanked with sequence compatible for AfeI and BamHI (underlined) restriction endonuclease sites, respectively:
-
GB1F 5′- GCT TAC AAG CTC ATC CTG AAC GGC AAG ACC CTC AAA GGG GAG ACC ACG ACC GAA GCC GTC GAC GCC GCG ACC GCC GAG
-
GB2F 5′- AAG GTC TTC AAG CAG TAC GCG AAC GAC AAC GGC GTG GAC GGC GAA TGG ACG TAC GAC GAC GCC ACC AAG ACG TTC ACC GTC ACC GAA GCT TGA G
-
GB1R 5′- GTA CTG CTT GAA GAC CTT CTC GGC GGT CGC GGC GTC GAC GGC TTC GGT CGT GGT CTC CCC TTT GAG GGT CTT GCC GTT CAG GAT GAG CTT GTA AGC
-
GB2R 5′- GATCC TCA AGC TTC GGT GAC GGT GAA CGT CTT GGT GGC GTC GTC GTA CGT CCA TTC GCC GTC CAC GCC GTT GTC GTT CGC
Results
Cloning, expression, and purification of GvpC-IgG-binding domain (GB) fusion proteins
Two versions of GvpC-Streptococcal IgG-binding (GB) domain fusion proteins with a 54-amino acid region of the GB domain and two different GvpC lengths were designed. GvpC4GB contains the full-length GvpC, while GvpC3GB contains most of GvpC but lacks the acidic tail in the C-terminal region (Fig. 2). The two fusion genes were inserted into the E. coli expression plasmid pET19b, resulting in the constructs pET19b-GvpC3GB and pET19b-GvpC4GB. The plasmids were transformed into E. coli BL21(DE3), induced, and fusion proteins purified by affinity column chromatography (as described in the “Materials and methods” section). The fusion proteins were detected as polypeptide bands of 60 kDa (GvpC3GB) and 70 kDa (GvpC4GB) by immunoblotting analysis (Fig. 2b). The measured electrophoretic mobility anomaly of GvpC protein and its derivatives on SDS–polyacrylamide gels was expected (48 kDa for GvpC4GB and 37 kDa for GvpC3GB), as has been consistently observed for highly acidic Halobacterium proteins (Halladay et al. 1993; Shukla and DasSarma 2004).
GVNP binding and competition analysis of GvpC3GB and GvpC4GB fusion proteins
To test the binding properties of GvpCGBs and effects of salts and salt concentration on binding with GVNPs, the two fusion proteins, GvpC3GB and GvpC4GB, were incubated with GVNPs from Halobacterium sp. NRC-1. The purified fusion proteins were mixed with GVNPs in the presence of 0.7 or 3.5 M NaCl and 0.07 or 0.7 × basal salt (BS) solutions. The mixtures were incubated for 1.5 h at 25 °C and subjected to floatation facilitated by low-speed centrifugation. The capacity of the recombinant GvpCGB association with GVNP was determined by immunoblot analysis of the relative amounts of GvpCGBs present in the GVNP-containing top (floating) phase and the subnatant (Fig. 3). In contrast to a clear single band observed in GvpC3GB (Fig. 3a), GvpC4GB (Fig. 3b) often appeared as doublet bands possibly due to a proteolytic cleavage of the purified protein or by an aberrant mobility stemming from incomplete denaturation associated with extremely acidic stretches. The significant increase in the level of GvpCGBs in the top phase as compared to that of the subnatant demonstrates that both GvpC3GB and GvpC4GB can form a complex with purified wild-type GVNPs. A more dramatic increase in the level of both GvpC3GB and GvpC4GB association with GVNP complex was observed at the higher salt concentrations (Fig. 3c). The results indicate a more facile binding of the recombinant GvpCGBs to the GVNP platform in the presence of high salt concentrations.

Binding of a GvpC3GB and b GvpC4GB to wild-type GVNPs. GvpC3GB or GvpC4GB purified from E. coli were incubated with GVNPs for 1.5 h in the indicated solutions followed by centrifugation (300 × g for 30 min) and immunoblot analysis of the separated top and subnatant fractions: 0.7 M NaCl (lanes 1–2), 3.5 M NaCl (lanes 3–5), 0.07 × basal salts (lanes 6–7), or 0.7 × basal salts (lanes 8–10). Controls contain the GvpCGB and GVNP mixtures collected prior to centrifugation (lanes 3 and 8) together with top fraction (lanes: 2, 5, 7, and 10) and subnatant fraction (lanes: 1, 4, 6, and 9). c Densitometric quantification of GvpC3GB signals detected (blue: subnatant fraction, orange: top fraction)
To test whether the acidic GvpC C-terminal region in GvpC4GB conferred an advantage in binding or stabilization of GVNP-fusion-protein complexes, competitive binding experiments were carried out with different ratios (5:1 to 1:1, and 1:5) of GvpC3GB to GvpC4GB in 1 × PBS solution containing 3 M NaCl. In the first set of binding, carried out with an excess of the recombinant GvpCGBs as compared to GVNPs, the levels of the GvpC3GB and GvpC4GB detected by immunoblotting of centrifugation separated subnatant (Fig. 4a, lanes 1, 3, and 5) and top phases (lanes 2, 4, and 6) were similar and found to be consistent with the predetermined ratios (Fig. 4a).

Detection of GvpC3GB and GvpC4GB binding with GVNPs. a Binding was carried out by incubation of GvpC3GB and GvpC4GB with a ratio of 1:5 (lanes: 1 and 2), 1:1 (lanes: 3 and 4), and 5:1 (lanes: 5 and 6) with GVNPs in 1 × PBS containing 3 M NaCl at room temperature for 1 h and then separated by centrifugation into the top (lanes: 2, 4, and 6), and subnatant (lanes: 1, 3, and 5) fractions for immunoblotting. Top fractions containing the GVNP complex were collected, mixed with 5 volumes of 1 × PBS containing 3 M NaCl, followed by b a second round of centrifugation. Immunoblotting was performed on the top (lanes: 2, 4, and 6) and subnatant (lanes: 1, 3, and 5) fractions
The GVNP-enriched top phase (Fig. 4a, lanes 2, 4, and 6) was collected, diluted with five volumes of 3 M NaCl binding solution, followed by fractionation by a second low-speed centrifugation. Immunoblotting analysis (Fig. 4b) of the top (lanes 2, 4, and 6) and subnatant (lanes 1, 3, and 5) fractions detected more GvpC fusion protein in the GVNP::GvpCGB-containing top phase compared to the subnatant fraction, indicating that a substantial fraction remained bound to the GVNPs. The ratios of GvpC3GB and GvpC4GB fusion proteins detected from both fractions were similar to each other when the ratio was 1:1. Additionally, in samples where the ratios of fusion proteins differed greatly (5:1 or 1:5), the top phase contained the dominant fusion protein proportional to the ratio originally added to the mixture. These results confirmed that there was no preferential binding by either the shorter or longer fusion protein in vitro. The results are consistent with previous findings that the acidic amino acid-rich region present in the C-terminal region does not substantially impact GvpC binding to the GVNPs in vivo (DasSarma et al. 2013).
Demonstration of functionality of IgG-binding (GB) domain displayed on GVNPs
To determine whether the IgG-binding (GB) domain displayed on GVNPs was functional, we used HRP-linked goat anti-rabbit IgG antibodies to test binding by via measuring the enzymatic activity. The binding assay was carried out with the smaller GvpC3GB fusion protein and an excess amount of GVNPs in either 1 × PBS or 3 M NaCl solution. Upon centrifugal separation, the GVNP::GvpC3GB-containing top phase was collected and further incubated in the same salt concentration with the addition of HRP-linked antibody followed by centrifugation. Levels of GvpC3GB-bound IgG in each fraction were determined by chemiluminescence reactions catalyzed by HRP conjugated to the antibodies (Fig. 5a). The results demonstrated a greater chemiluminescence in the upper GVNP-containing fractions (spots 2 and 4), compared to that of the subnatant (spots 1 and 3), confirming antibody binding to the GVNP complex. In addition, a higher level of GVNP::GvpC3GB bound IgG is in agreement with the results in Fig. 3a in which a higher level of GvpC3GB remained associated with the GVNPs in higher salt concentrations.

Immunoplate assays of GvpC3GB fusion protein functionality on GVNP surface. a GVNPs and GvpC3GB were first incubated in 1 × PBS (spots 1 and 2) or 3 M NaCl (spots 3 and 4) at room temperature for 1 h followed by centrifugation at 300 × g for 30 min. The top phase containing GVNP::GvpC3GB was collected and further incubated with HRP-linked goat anti-rabbit IgG antibody followed by the centrifugal separation as above. b GVNPs were also incubated with HRP-linked goat anti-rabbit IgG antibody (spots 1 and 2) or with the GvpC3GB plus antibodies (lanes 3 and 4) in 3 M NaCl followed by centrifugal separation as above. An equal volume collected from GVNP-containing top (spots 2 and 4) and subnatant (spots 1 and 3) fractions were subject to chemiluminescence followed by densitometric quantification as shown in the graphs (blue: subnatant fraction, orange: top fraction)
In order to rule out potential background from non-specific association of HRP-linked antibodies to the components of wild-type GVNPs, a further binding assay was carried out in the presence or absence of the fusion protein (Fig. 5b). The mixture containing GVNP and HRP-linked antibody, with or without GvpC3GB (cf. spots 1 and 2 to 3 and 4), showed that exogenously produced GvpC3GB fusion protein binding to GVNPs significantly enhances the binding of HRP-linked antibodies on the exterior surface of the GVNP complex.
Discussion
The genetic system of the haloarchaeal model organism, Halobacterium sp. NRC-1, has been employed to produce a variety of bioengineered GVNPs which can be scaled and purified by hypotonic lysis and flotation (DasSarma et al. 1999; DasSarma et al. 2012; 2013; DasSarma and DasSarma 2017; 2021). All previous bioengineered GVNPs were produced by fusion of foreign protein sequences to GvpC entirely in the haloarchaeon using its genetic system (DasSarma and Fleischman 1995; Berquist et al. 2006). Examples of successful GVNP-displayed sequences constructed in Halobacterium sp. NRC-1 include those for vaccine development, e.g., Salmonella and Plasmodium antigens, and others for the delivery of immunological therapeutics, such as bactericidal permeability-increasing (BPI) protein for protection against sepsis (DasSarma and DasSarma 2015; DasSarma et al. 2015; Balakrishnan et al. 2016). For such biomedical applications, additional valuable features of GVNPs reported include their successful delivery via syringe needles through several routes of administration, as well as by microneedles, and their stability, safety, and non-toxicity (Andar et al. 2017; DasSarma et al. 2015; Balakrishnan et al. 2016).
While all past display work using GVNPs employed genetic engineering of Halobacterium sp. NRC-1 in vivo, with the current investigation, we determined that it is possible to express the GvpC fusion proteins in E. coli, followed by binding in vitro to wild-type GVNPs purified from the haloarchaeon and successfully displaying the fusion proteins on the nanoparticle surface. This advanced hybrid system should allow for the design and production of GvpC fusion proteins more expeditiously, given the ease of genetically engineering E. coli, and significantly enhance the capability of the GVNP display system to meet rapidly changing demands (Burki 2021). Success of this hybrid method enables more rapid production of GvpC fusion proteins using the bacterial expression system combined with their binding and display employing large quantities of easily purified GVNPs after hypotonic lysis and centrifugally accelerated flotation from the haloarchaeon (DasSarma et al. 1999).
No information was available previously on whether overexpressed hybrid Halobacterium sp. NRC-1 GvpC bound to wild-type GVNPs in vitro, or if a GVNP complex with GvpC fusion protein components from heterologous origins would bind and be functionally displayed on the nanoparticles. Consequently, our approach utilized wild-type GVNPs from Halobacterium sp. NRC-1 released by hypotonic lysis and tested binding of GvpC fusion proteins produced from E. coli, capitalizing on the facility of each host organism. Both the complete GvpC fusion and a slightly shortened form at the C-terminal region showed binding, with slightly higher binding levels achieved with higher salt concentration in either case. These results provide clear evidence for successful in vitro binding to the nanoparticles using components obtained from heterologous origins, Halobacterium and E. coli. Moreover, the results indicate that wild-type GVNPs are not saturated for GvpC binding sites. Our findings have opened the way to successfully developing the next-generation GVNP display system which can be loaded with one or more target protein(s) or peptide(s) obtained as easily purified GvpC fusion proteins produced exogenously (Fig. 1) (DasSarma et al. 1999).
For display as a GvpC fusion, we selected a broadly useful protein with valuable immunological characteristics, the immunoglobulin-binding domain B1 of streptococcal protein G (GB1) (Nomellini et al. 2007). The display of GvpCGB fusion proteins could be readily detected using commercial enzyme-linked antibodies via chemiluminescence. The display system developed with the addition of GB antibody-binding domain at the GvpC C-terminal region generated bioengineered GVNPs with the potential to directly bind a target molecule or cells by using a specific antibody. This new IgG (GB) display system expands the potential biomedical value of GVNPs due their ease of production and purification, and facility for immobilization and detection of immunoglobulins. In addition, the display system permits delivery of, in principle, any IgG of choice for therapeutic applications (Fig. 1). Therefore, the study significantly enhances the versatility of the GVNP display platform (patent pending).
The streptococcal IgG-binding protein display was also chosen to allow reversible binding to antibodies for antibody purification and enzyme-linked immunosorbent assays (ELISA). In addition, IgG-binding fusion proteins may also be useful for therapeutics, via attachment of molecules directed for the treatment of cell proliferation diseases. Moreover, their characteristics of non-toxicity, self-adjuvanticity, and ready deliverability will further expand the uses of a potentially self-guided drug or therapy system (Balakrishnan et al. 2016; Andar et al. 2017). Rapid, targeted, and increased specificity for therapy should be made possible employing this new hybrid production system.
This investigation has significantly expanded the range of applications of GVNPs for the development of drug therapy and vaccine delivery. Moreover, the GVNP display system is likely to be further enhanced through deeper understanding of the nanoparticle architecture and fine-tuning the hybrid genetic system. While the genetic requirements of essential gvp genes for wild-type GVNP production have been established, the detailed mechanistic functions would still gain from further analysis of gvp gene products (Shukla and DasSarma 2004; DasSarma and DasSarma 2015, 2021). Thus, additional studies of the properties of the hybrid display system will improve the capabilities and performance of the in vitro binding and display. By using a hybrid expression and production system for presenting an IgG-binding protein on a self-adjuvanting delivery platform, we have greatly expanded its potential capability for antibody-guided GVNP targeting and multiple target display.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Change history
18 April 2022
A Correction to this paper has been published: https://doi.org/10.1007/s00253-022-11924-z
References
Andar AU, Karan R, Pecher WT, DasSarma P, Hedrich WD, Stinchcomb AL, DasSarma S (2017) Microneedle-assisted skin permeation by nontoxic bioengineerable gas vesicle nanoparticles. Mol Pharm 14:953–958. https://doi.org/10.1021/acs.molpharmaceut.6b00859
Balakrishnan AP, DasSarma P, Bhattacharjee O, Kim JM, DasSarma S, Chakravortty D (2016) Halobacterial nano vesicles displaying murine bactericidal permeability-increasing protein rescue mice from lethal endotoxic shock. Sci Rep 6:33679. https://doi.org/10.1038/srep33679
Berquist BR, Müller JA, DasSarma S (2006) Genetic systems for halophilic archaea. Methods Microbiol 35:649–680. https://doi.org/10.1016/S0580-9517(08)70030-8
Burki TK (2021) The race between vaccination and evolution of COVID-19 variants. Lancet Respir Med 9(11):e109. https://doi.org/10.1016/S2213-2600(21)00443-4
Childs TS, Webley WC (2012) In vitro assessment of halobacterial gas vesicles as a Chlamydia vaccine display and delivery system. Vaccine 30:5942–5948. https://doi.org/10.1016/j.vaccine.2012.07.038
Damerval T, Houmard J, Guglielmi G, Csiszàr K, Tandeau de Marsac N (1987) A developmentally regulated gvpABC operon is involved in the formation of gas vesicles in the cyanobacterium Calothrix 7601. Gene 54:83–92. https://doi.org/10.1016/0378-1119(87)90350-7
DasSarma S, Fleischmann EM (eds) (1995) Archaea, a laboratory manual: halophiles. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
DasSarma S, Arora P (1997) Genetic analysis of gas vesicle gene cluster in haloarchaea. FEMS Microbiol Lett 153:1–10. https://doi.org/10.1111/j.1574-6968.1997.tb10456.x
DasSarma S, DasSarma P (2015) Gas vesicle nanoparticles for antigen display. Vaccines 3:686–702. https://doi.org/10.3390/vaccines3030686
DasSarma P, DasSarma S (2021) Gas vesicle nanoparticles. In: Encyclopedia of Life Sciences, Vol 2:1–14. John Wiley & Sons, Ltd. https://doi.org/10.1002/9780470015902.a0029044
DasSarma S, Damerval T, Jones JG, Tandeau de Marsac N (1987) A plasmid-encoded gas vesicle protein gene in a halophilic archaebacterium. Mol Microbiol 1:365–370. https://doi.org/10.1111/j.1365-2958.1987.tb01943.x
DasSarma S, Arora P, Lin F, Molinari E, Yin LR-S (1994) Wild type gas vesicle formation requires at least ten genes in the gvp gene cluster of Halobacterium halobium plasmid pNRC100. J Bacteriol 176:7646–7652. https://doi.org/10.1128/jb.176.24.7646-7652
DasSarma S, Halladay J, Ng W-L (1999) Recombinant vector and process for cell flotation. U.S. patent, no. 6,008,051
DasSarma P, Zamora RC, Müller JA, DasSarma S (2012) Genome-wide responses of the model archaeon Halobacterium NRC-1 to oxygen limitation. J Bacteriol 194:5520–5537. https://doi.org/10.1128/JB.01153-12
DasSarma S, Karan R, DasSarma P, Barnes S, Ekulona F, Smith B (2013) An improved genetic system for bioengineering buoyant gas vesicle nanoparticles from haloarchaea. BMC Biotechnol 13:112. https://doi.org/10.1186/1472-6750-13-112
DasSarma P, Negi VD, Balakrishnan A, Karan R, Barnes S, Ekulona F, Chakravortty D, DasSarma S (2014) Haloarchaeal gas vesicle nanoparticles displaying Salmonella SopB antigen reduce bacterial burden when administered with live attenuated bacteria. Vaccine 32:4543–4549. https://doi.org/10.1016/j.vaccine.2014.06.021
DasSarma P, Negi VD, Balakrishnan A, Kim JM, Karan R, Chakravortty D, DasSarma S (2015) Haloarchaeal gas vesicle nanoparticles displaying Salmonella antigens as a novel approach to vaccine development. Proced Vaccinol 9:16–23. https://doi.org/10.1016/j.provac.2015.05.003
Dunton PG, Mawby WJ, Shaw VA, Walsby AE (2006) Analysis of tryptic digests indicates regions of GvpC that bind to gas vesicles of Anabaena flos-aquae. Microbiol (reading) 152:1661–1669. https://doi.org/10.1099/mic.0.28755-0
Dutta S, DasSarma P, DasSarma S, Jarori GK (2015) Immunogenicity and protective potential of a Plasmodium spp. enolase peptide displayed on archaeal gas vesicle nanoparticles. Malar J 14:406. https://doi.org/10.1186/s12936-015-0914-x
Griffiths AE, Walsby AE, Hayes PK (1992) The homologies of gas vesicle proteins. J Gen Microbiol 138:1243–1250. https://doi.org/10.1099/00221287-138-6-1243
Halladay JT, Jones JG, Lin F, MacDonald AB, DasSarma S (1993) The rightward gas vesicle operon in Halobacterium plasmid pNRC100: identification of the gvpA and gvpC gene products by use of antibody probes and genetic analysis of the region downstream of gvpC. J Bacteriol 175:684–692. https://doi.org/10.1128/jb.175.3.684-692.1993
Hill AM, Salmond GPC (2020) Microbial gas vesicles as nanotechnology tools: exploiting intracellular organelles for translational utility in biotechnology, medicine and the environment. Microbiology 166:501–509. https://doi.org/10.1099/mic.0.000912
Jones JG, Hackett NR, Halladay JT, Scothorn DJ, Yang C-F, Ng W-L, DasSarma S (1989) Analysis of insertion mutants reveals two new genes in the pNRC100 gas vesicle gene cluster of Halobacterium halobium. Nucleic Acids Res 17:7785–7793. https://doi.org/10.1093/nar/17.19.7785
Jones JG, Young DC, DasSarma S (1991) Structure and organization of the gas vesicle gene cluster on the Halobacterium halobium plasmid pNRC100. Gene 102:1017–1022. https://doi.org/10.1016/0378-1119(91)90549-q
Nomellini JF, Duncan G, Dorocicz IR, Smit J (2007) S-layer-mediated display of the immunoglobulin G-binding domain of streptococcal protein G on the surface of Caulobacter crescentus: development of an immunoactive reagent. Appl Environ Microbiol 73:3245–3253. https://doi.org/10.1128/AEM.02900-06
Pecher, WT, Kim JM, DasSarma P, Karan R, Sinnis P, DasSarma S (2016) Halobacterium expression system for production of full-length Plasmodium falciparum circumsporozoite protein. In: Rampelotto PH (ed) Biotechnology of Extremophiles: Advances and Challenges, Series: Grand Challenges in Biology and Biotechnology, Vol 1. Springer International Publishing, Switzerland, pp 699–709. https://doi.org/10.1007/978-3-319-13521-2_25
Rigi G, Ghaedmohammadi S, Ahmadian G (2019) A comprehensive review on staphylococcal protein A (SpA): its production and applications. Biotechnol Appl Biochem 66:454–464. https://doi.org/10.1002/bab.1742
Sambrook J, Russell J (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schneider C, Rasband W, Eliceiri K (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. https://doi.org/10.1038/nmeth.2089
Shukla HD, DasSarma S (2004) Complexity of gas vesicle biogenesis in Halobacterium sp. strain NRC-1: identification of five new proteins. J Bacteriol 186:3182–3186. https://doi.org/10.1128/JB.186.10.3182-3186.2004
Sjöbring U, Björck L, Kastern W (1991) Streptococcal protein G. Gene structure and protein binding properties. J Biol Chem 266(1):399–405. https://doi.org/10.1016/S0021-9258(18)52448-0
Sremac M, Stuart ES (2008) Recombinant gas vesicles from Halobacterium sp displaying SIV peptides demonstrate biotechnology potential as a pathogen peptide delivery vehicle. BMC Biotechnol 8:9. https://doi.org/10.1186/1472-6750-8-9
Sremac M, Stuart ES (2010) SIVsm Tat, Rev, and Nef1: functional characteristics of r-GV internalization on isotypes, cytokines, and intracellular degradation. BMC Biotechnol 10:54. https://doi.org/10.1186/1472-6750-10-54
Stuart ES, Sremac M, Morshed F, DasSarma S (2001) Antigen presentation using novel particulate organelles from halophilic archaea. J Biotechnol 88:119–128. https://doi.org/10.1016/s0168-1656(01)00267-x
Stuart ES, Morshed F, Sremac M, DasSarma S (2004) Cassette-based presentation of SIV epitopes with recombinant gas vesicles from halophilic archaea. J Biotechnol 114:225–237. https://doi.org/10.1016/j.jbiotec.2004.01.005
Walsby AE (1994) Gas vesicles. Microbiol Rev 58:94–144. https://doi.org/10.1128/mr.58.1.94-144.1994
Yan J, Yin M, Foster FS, Démoré CEM (2020) Tumor contrast imaging with gas vesicles by circumventing the reticuloendothelial system. Ultrasound Med Biol 46:359–368. https://doi.org/10.1016/j.ultrasmedbio.2019.09.009
Acknowledgements
We thank Yoo-Jeong Jeon for her assistance with immunoblot analysis.
Funding
The research leading to these results received funding from the National Research Foundation of Korea (KNRF) funded by the Korean government under grant no. NRF-2017R1D1A1A02019244 and the National Institutes of Health, USA, grant no. AI139808.
Author information
Authors and Affiliations
Contributions
YSK, YRK, MJC, PD, and JMK conducted the experimental analysis subunit localization experiment, and partially drafted the manuscript. SD, PD, and JMK designed the study and partially drafted and finalized the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
This article does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised. Conflict of interest text is now corrected.
Rights and permissions
About this article

Cite this article
Kim, JM., Kim, YS., Kim, YR. et al. Bioengineering of Halobacterium sp. NRC-1 gas vesicle nanoparticles with GvpC fusion protein produced in E. coli. Appl Microbiol Biotechnol 106, 2043–2052 (2022). https://doi.org/10.1007/s00253-022-11841-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-022-11841-1