
Abstract
The acetic acid bacterium (AAB) Gluconobacter oxydans incompletely oxidizes a wide variety of carbohydrates and is therefore used industrially for oxidative biotransformations. For G. oxydans, no system was available that allows regulatable plasmid-based expression. We found that the l-arabinose-inducible PBAD promoter and the transcriptional regulator AraC from Escherichia coli MC4100 performed very well in G. oxydans. The respective pBBR1-based plasmids showed very low basal expression of the reporters β-glucuronidase and mNeonGreen, up to 480-fold induction with 1% l-arabinose, and tunability from 0.1 to 1% l-arabinose. In G. oxydans 621H, l-arabinose was oxidized by the membrane-bound glucose dehydrogenase, which is absent in the multi-deletion strain BP.6. Nevertheless, AraC-PBAD performed similar in both strains in the exponential phase, indicating that a gene knockout is not required for application of AraC-PBAD in wild-type G. oxydans strains. However, the oxidation product arabinonic acid strongly contributed to the acidification of the growth medium in 621H cultures during the stationary phase, which resulted in drastically decreased reporter activities in 621H (pH 3.3) but not in BP.6 cultures (pH 4.4). These activities could be strongly increased quickly solely by incubating stationary cells in d-mannitol-free medium adjusted to pH 6, indicating that the reporters were hardly degraded yet rather became inactive. In a pH-controlled bioreactor, these reporter activities remained high in the stationary phase (pH 6). Finally, we created a multiple cloning vector with araC-PBAD based on pBBR1MCS-5. Together, we demonstrated superior functionality and good tunability of an AraC-PBAD system in G. oxydans that could possibly also be used in other AAB.
Key points
• We found the AraC-PBAD system from E. coli MC4100 was well tunable in G. oxydans.
• In the absence of AraC or l-arabinose, expression from PBAD was extremely low.
• This araC-PBAD system could also be fully functional in other acetic acid bacteria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Controlled expression of target genes to produce proteins for various purposes in a bacterial cell culture at a defined time point is often beneficial or even required in basic research and in biotechnological applications. For inducible expression, various plasmids have been developed and established in many bacteria including Escherichia coli, Pseudomonas sp., Ralstonia sp., Bacillus sp., Lactoccocus sp., Streptomyces sp., mycobacteria, corynebacteria, halophilic bacteria, and others (reviewed in, for example, Chen 2012; Connell 2001; Dilworth et al. 2018; Evans and Mizrahi 2015; Forstner et al. 2007; Gruber et al. 2015; Parachin et al. 2012; Schnappinger and Ehrt 2014; Terpe 2006; Valero 2012). Prominent classical examples are the well-known LacI-, TetR-, and AraC-dependent systems (as well as optimized or modified versions thereof) for inducible expression by addition of the respective inducer. For acetic acid bacteria (AAB), including the most frequently used and studied genera Acetobacter, Gluconobacter, Gluconacetobacter, Komagataeibacter, and Acidiphilium, a low-cost, tight, and strongly inducible expression system has not been reported yet in the literature to the best of our knowledge. Apparently, for the heterologous systems, leakiness is a major issue responsible for the relatively low induction ratios (induced/non-induced) in AAB. In Komagataeibacter rhaeticus iGEM, the TetR-dependent system from transposon Tn10 with the inducible promoter Ptet exhibited approximately only 1.5-fold induction due to high leakiness in the absence of the inducer anhydrotetracycline (Florea et al. 2016). The native l-arabinose-inducible AraC-dependent PBAD system from E. coli exhibited approximately only 5- to 12-fold induction in Gluconacetobacter xylinus ATCC 700178, Gluconacetobacter hansenii ATCC 53582, and Komagataeibacter rhaeticus iGEM due to high basal expression and required a high concentration (4%, w/v) of the inducer l-arabinose (Teh et al. 2019). The performance of IPTG-inducible LacI-dependent expression has not been reported yet for AAB according to the literature.
The AAB G. oxydans is industrially used for oxidative biotransformations of carbohydrates to produce, e.g., l-sorbose, a precursor in vitamin C production, dihydroxyacetone, a substance used for tanning lotions, or 6-amino-l-sorbose, a precursor of the antidiabetic drug miglitol (Ameyama et al. 1981; Gupta et al. 2001; Hekmat et al. 2003; Saito et al. 1997; Tkac et al. 2001; Wang et al. 2016). The beneficial ability of G. oxydans is the regio- and stereoselective incomplete oxidation of a variety of substrates (e.g., sugars and sugar alcohols) in the periplasm by membrane-bound dehydrogenases (mDHs) and release of resulting products into the cultivation medium (Mamlouk and Gullo 2013; Mientus et al. 2017; Pappenberger and Hohmann 2014). For the fully functional expression of mDHs in G. oxydans, the promoters of the alcohol dehydrogenase (PGOX1067-68) and the inositol dehydrogenase (PGOX1857) have been used in shuttle vectors (Mientus et al. 2017). While PGOX1857 is repressed in the presence of glucose (Hölscher et al. 2007), PGOX1067-68 showed constitutive activity with good expression (Mientus et al. 2017). Other G. oxydans promoters classified as strong, moderate, and weak are the constitutive promoters PGOX0264, PGOX0452, and PGOX0384 from genes encoding ribosomal proteins (Kallnik et al. 2010). Expression from moderate PGOX0452 has been used to successfully produce membrane-bound PQQ-dependent glucose dehydrogenase (GOX0265) for purification and characterization (Meyer et al. 2013). Expression from weak PGOX0384 has been used to successfully produce the succinate dehydrogenase from Acetobacter pasteurianus in G. oxydans as a first step toward a complete tricarboxylic acid cycle (Kiefler et al. 2015). In earlier work with G. oxydans, constitutive PtufB from G. oxydans and from E. coli as well as Plac from E. coli have been used (Merfort et al. 2006a, b; Schleyer et al. 2008; Tonouchi et al. 2003; Zhang et al. 2010). Since there is no Lac repressor homolog in G. oxydans, also Plac is constitutive in G. oxydans. Together, the resulting expression plasmids with all these promoters do not allow for gradually induced expression of target genes at a desired time.
Therefore, to provide for G. oxydans a tight and strongly inducible plasmid, we tested AraC-PBAD from the E. coli K12 derivative MC4100 in a pBBR1-based vector. The plasmids constructed in this study exhibited very low basal reporter gene expression and l-arabinose-dependent induction ratios up to 480-fold. GC-TOF-MS analysis confirmed oxidation of the inducer l-arabinose to l-arabinonic acid contributing to the acidification of the growth medium in shake flasks. This additional acidification turned out to be critical for the activity of intracellular reporter proteins in the stationary phase and could be eliminated by mDH deletion or pH-controlled conditions.
Materials and methods
Bacterial strains, plasmids, and culture conditions
Strains and plasmids used or created in this study are listed in Table 1. G. oxydans strains were routinely cultivated at 30 °C and 180 rpm in d-mannitol medium containing 4% (w/v) d-mannitol, 5 g L-1 yeast extract, 1 g L-1 KH2PO4, 1 g L-1 (NH4)2SO4, and 2.5 g L-1 MgSO4 × 7H2O and supplemented with 50 μg mL-1 cefoxitin. Unless stated otherwise, for shake flask cultivations, 50 mL of d-mannitol medium in 500 mL shaking flasks with three baffles was inoculated from overnight starter cultures to an initial optical density at 600 nm (OD600) of 0.3 (UV-1800, Shimadzu). If required for induction tests, l-arabinonic acid was supplemented as Li+ salt directly to the medium. The pH of the supplemented medium was adjusted to pH 6, cold sterile filtered and directly used for cultivation. Cultivations of G. oxydans harboring pBBR1MCS-2- or pBBR1MCS-5-based plasmids were supplemented with 50 μg mL-1 kanamycin or 10 μg mL-1 gentamicin, respectively (Kovach et al. 1995). Escherichia coli strains were routinely cultivated in lysogeny broth (LB) medium at 37 °C and 160 rpm. If appropriate, 50 μg mL-1 kanamycin or 10 μg mL-1 gentamicin was added to the medium. G. oxydans was transformed by conjugation using E. coli S17-1 as a donor (Kiefler et al. 2017). All E. coli strains were made competent and transformed by CaCl2 procedure (Hanahan 1983).
Enzymatic determination of l-arabinose concentrations
l-Arabinose concentrations in the medium of the G. oxydans strains 621H and BP.6 were determined using the enzymatic l-arabinose and d-galactose rapid assay kit (Megazyme). All samples were measured in microplates according to the manufacturer’s instructions by monitoring NADH formation as increase in absorption at 340 nm in a multi-well reader (infinite M1000 PRO, Tecan).
DNA microarray analysis
To analyze short-term gene expression changes in response to a pulse of 1% (w/v) l-arabinose, the transcriptomes of G. oxydans 621H cultivated in complex medium with 4% (w/v) d-mannitol were compared with the control. In the mid-exponential growth phase, 1% (w/v) l-arabinose was supplemented and the same volume of water in another 621H culture as a control. After 30 min of cultivation, each cell suspension was harvested by centrifugation (4500×g, 5 min, 4 °C). The resulting cell pellets were directly frozen in liquid nitrogen and stored at − 80 °C until RNA preparation. The preparation of RNA, cDNA synthesis, hybridization using Agilent’s 4-plex DNA microarray platform, and data analysis were carried out as described (Kranz et al. 2018).
Recombinant DNA work
All DNA oligonucleotides used in this study were synthesized by Eurofins MWG and are listed in Table S1. All enzymes for recombinant DNA work were purchased from Thermo Scientific. DNA manipulations by polymerase chain reaction (PCR), restriction, and ligation reactions followed standard protocols (Sambrook et al. 1989). Reporter plasmids were constructed from amplified DNA fragments and the restricted broad-host vectors pBBR1MCS-2 or pBBR1MCS-5 in a one-step isothermal Gibson assembly (Gibson et al. 2009). The terminator sequence BBa_B1002 from the iGEM parts library was placed downstream of the reporter genes. For DNA amplification by PCR, Q5 polymerase was used according to the conditions recommended by the manufacturer (New England Biolabs). All DNA modifications used to obtain designed plasmids were performed with E. coli DH5α. Plasmids were isolated from E. coli using a QIAprep spin miniprep kit (Qiagen). The plasmid inserts constructed in this work were checked by DNA sequencing (Eurofins MWG).
Construction of plasmids
Plasmid pBBR1MCS-2-araC-PBAD-uidA was constructed using the primer pairs PF1/PF2 and PF3/PF4 to generate a 1253-bp DNA fragment with araC-PBAD from the commercially available plasmid pBAD/Myc-His A (Invitrogen/Thermo Fischer) and a 1886-bp DNA fragment with uidA from E. coli K12 genomic DNA and the terminator BBa_B1002 inserted by elongating the 5′-end of PF4. Plasmid pBBR1MCS-2-araC-PBAD-mNG was constructed using the primer pairs PF1/PF5 and PF6/PF7 to generate a 1,238 bp DNA fragment with araC-PBAD and a 790 bp DNA fragment with mNG and terminator BBa_B1002, respectively. For insertion of the two overlapping DNA fragments into pBBR1MCS-2 by Gibson assembly (50 °C; 1 h), pBBR1MCS-2 was restricted by the endonucleases SacI and KpnI. Elongated 5′-ends in primers PF2/PF3 and PF5/PF6 were used to introduce downstream of PBAD and 6 bp upstream of the start codon of mNG or uidA, the artificial Shine-Dalgarno sequence AGGAGA (Hentschel et al. 2013). To change the plasmid backbone from pBBR1MCS-2 to pBBR1MCS-5, araC-PBAD-uidA and araC-PBAD-mNG were excised and ligated into pBBR1MCS-5 using SacI and Eco81I.
The plasmid pBBR1MCS-5-PBAD-mNG was constructed using the primer pair PF8/PF9 to obtain a 1164-bp DNA fragment PBAD-mNG from pBBR1MCS-5-araC-PBAD-mNG. The resulting fragment was cloned into pBBR1MCS-5 using BamHI and XhoI. To prevent undesired transcripts initiating from PBAD, the G. oxydans terminator of gdhM (GOX0265) was inserted upstream of PBAD.
Plasmid pBBR1MCS-5-araE-araC-PBAD-mNG was constructed by insertion of araE into pBBR1MCS-5-araC-PBAD-mNG. For that, pBBR1MCS-5-araC-PBAD-mNG was restricted by the enzymes AscI and Eco81I and a 1576-bp DNA fragment comprising araE without own promoter, amplified with the primer pair PF10/PF11 from E. coli K12 genomic DNA, was integrated downstream of araC by Gibson assembly. PF10 and PF11 contained elongated 5′-ends to insert the T7 terminator sequence downstream of araE and a Shine-Dalgarno sequence upstream of araE between araE and araC. Thus, araE and araC are expected to be expressed as a polycistronic transcript from the araC promoter.
Plasmid pBBR1MCS-5-araC-PBAD-MCS as an empty vector with a ribosome-binding site, a new NdeI site upstream of the multiple cloning site (MCS) and the iGEM terminator sequence BBa_B1002 was constructed in two steps. In the first step, the DNA fragment araC-PBAD generated with the primer pair PF12/PF13 was inserted into BshTI/SacI-restricted pBBR1MCS-5 by Gibson assembly keeping the original MCS from pBBR1MCS-5. In the second step, the terminator sequence BBa_B1002 was integrated downstream of the MCS by integration of a DNA fragment amplified with the primer pair PF14/PF15. For the integration of the terminator sequence, the plasmid obtained in step one was restricted with XhoI and SphI.
Plasmid pBBR1MCS-5-araC-PBAD-MCS-mNG was constructed from pBBR1MCS-5 by integration of the DNA fragments araC-PBAD and mNG-BBa_B1002 in a Gibson assembly. In the amplification of the DNA fragments, the restriction sites NdeI and XhoI were integrated resulting in the same sequence as it would have been obtained via classical restriction cloning of mNG into pBBR1MCS-5-araC-PBAD-MCS using the restriction enzymes NdeI and XhoI. The DNA fragments araC-PBAD and mNG-BBa_B1002 were amplified with the primer pairs PF1/PF16 and PF17/PF18, respectively. For integration, pBBR1MCS-5 was restricted using the restriction enzymes BshTI and KpnI.
Measurements of fluorescence protein and enzyme activity
For online monitoring of expression induction and relative promoter strengths of promoter–reporter constructs in G. oxydans, the fluorescence protein mNeonGreen (mNG) was used (Shaner et al. 2013). In shake flask experiments, mNG expression in G. oxydans was induced by addition of 1% (w/v) l-arabinose from a 50% (w/v) stock solution. Reference cultures were supplemented with an equal volume of water. In intervals, growth (OD600) and fluorescence emission was monitored by a spectrophotometer (UV-1800, Shimadzu) and a Tecan Reader (λex 504 nm/λem 517 nm; gain 60; ex/em bandwidth 5 nm; infinite M1000 PRO, Tecan), respectively. In BioLector cultivations using 48-well Flowerplates® (m2p-labs), batches of 800 μL of d-mannitol medium were inoculated from overnight precultures and incubated (1200 rpm; 85% humidity, 30 °C). Over a period of up to 30 h, cell growth and fluorescence was monitored in each well simultaneously by measuring the backscatter (A620 nm; gain 20) and fluorescence emission (λex 510 nm/λem 532 nm; gain 60). Typically, mean values and standard deviations of all experiments were derived from at least three biological replicates.
For enzymatic reporter assays, β-d-glucuronidase (UidA) was used (Kallnik et al. 2010). UidA activity was determined in a Miller assay essentially as described (Miller 1992). Immediately after inoculation of G. oxydans shake flask cultures, uidA expression was induced by the addition of 1% (w/v) l-arabinose. Non-induced cultures supplemented with an equal volume of water were used as controls. In intervals, 500 μL samples were taken to determine OD600 and UidA activity. For permeabilization of the cells, 30 μL of the culture broth in appropriate dilutions in assay buffer was incubated in a 96-well plate (20 min; 30 °C) with 100 μL of prewarmed Z-mix (54.6 mM Na2HPO4 × 7H2O, 36.4 mM NaH2PO4 × H2O, 9.1 mM KCl, 0.9 mM MgSO4, 45.5 mM DTT, 4.5% (v/v) chloroform, 0.5% (w/v) SDS, pH 7). After the addition of 100 μL of prewarmed 4-nitrophenyl-β-d-glucopyranoside (4 g L-1) using a multichannel pipette, β-d-glucuronidase activity was monitored measuring the absorption of p-nitrophenol in intervals of 1 min at 420 nm (infinite M1000 PRO, Tecan). From all constructs, at least three biological replicates were measured in triplicates to determine mean values and standard deviations.
Cell flow cytometer analysis
For single cell analysis of reporter gene expression, the G. oxydans strains 621H and BP.6 carrying the plasmid pBBR1MCS-5-araC-PBAD-mNG were analyzed using a BD FACSAria II cell sorter (BD Biosciences) equipped with a 70-μm nozzle run with a sheath pressure of 70 psi. Using the 488 nm laser beam, the front scatter (FSC) and side scatter (SSC) were recorded as small-angle (axial) and perpendicular scatter, respectively. Combining a 502-nm-long-pass and 530/30 nm band-pass filter, the emitted mNG fluorescence from the SSC signal was detected. Fluorescence data were acquired using a two-step gating strategy: at first, signals from cell debris and electronic noise were excluded by gating a population in a FSC-H vs. an SSC-H plot. Secondly, to perform singlet discrimination, from the resulting population the FSC-H signal was plotted against FSC-W. The gated singlet population was used for fluorescence acquisition in all experiments. While the total event rate during measurements never exceeded 15,000 events/s, for each sample, the signals of 100,000 events were recorded. For FACS device control and data analysis, FACSDiva 7.0.1 software (BD Biosciences) was used. Gated events (n = 100,000) were used for data analysis in FlowJo for Windows 10.4.2 (FlowJo, LLC) and Prism 7.04 (GraphPad Software) to visualize FACS data.
Bioreactor cultivation of G. oxydans
Bioreactor cultivations were conducted in DASbox® mini-bioreactors controlled by DASware software (Eppendorf). In 385-mL glass vessels equipped with two 6-bladed Rushton-type impellers, O2 (InPro® 6800 series, Mettler-Toledo), pH (EasyFerm Plus K8 120, Hamilton), and temperature sensors, 150-mL 4% (w/v) d-mannitol medium was inoculated to an initial OD600 of 0.3 (30 °C). The initial gas flow rate was set to 6 sL h−1. Starting agitation frequency was 500 rpm. The pH was maintained at 6 by automatic titration using H2SO4 or KOH stocks (1.5 M). Dissolved oxygen tension in the medium was maintained ≥ 30% by a cascade of first adjusting the agitation speed to a maximum of 1200 rpm, then by rising the O2 concentration in the supplied gas up to 80% (v/v) and eventually by increasing the gas flow rate. In intervals, samples were taken from the culture broth and OD600 as well as fluorescence were determined as described above.
l-Arabinose biotransformation for the analysis of oxidation products
To identify the reaction products of l-arabinose oxidation by G. oxydans strains, cells were grown in 25 mL of d-mannitol medium in 500 mL shake flasks to an OD600 of 1.6, centrifuged (4000×g, 5 min) and washed twice with 1 mM HEPES (pH 7). The pellet was resuspended in 25 mL biotransformation buffer (33.9 g L-1 Na2HPO4, 15 g L-1 KH2PO4, 5 g L-1 NH4Cl, 2.5 g L-1 NaCl, 0.49 g L-1 MgSO4, 0.02 g L-1 CaCl2) supplemented with 1% (w/v) l-arabinose. The cell suspensions were incubated for 24 h at 30 °C on a shaker (180 rpm). Afterwards, cells were centrifuged (4000×g, 5 min) and the cell-free supernatant was analyzed using a gas chromatograph (Agilent 6890N, Agilent Technologies) coupled to a Waters Micromass GCT Premier high-resolution time-of-flight mass spectrometer (Waters). Sample derivatization, GC-TOF-MS operation, and peak identification were essentially conducted as described (Paczia et al. 2012). Biotransformation medium without cells was used as reference.
Results
Growth of G. oxydans 621H in the presence of l-arabinose
In the present study, we wanted to test l-arabinose-dependent gene expression in G. oxydans 621H to provide a regulatable expression plasmid for this AAB, similar to AraC-dependent vectors constructed for E. coli (Guzman et al. 1995). In such a system, the inducer l-arabinose needs to enter the cell where it binds to the transcriptional regulator AraC (Schleif 2010). In the case of G. oxydans 621H, l-arabinose was reported to be oxidized already in the periplasm by the membrane-bound glucose DH GdhM (GOX0265) (Mientus et al. 2017; Peters et al. 2013). Thus, the presence of l-arabinose could affect the growth of G. oxydans 621H by using it as an energy source through oxidation, thereby inactivating the inducer. The G. oxydans multi-deletion strain BP.6 lacks GdhM and exhibited no l-arabinose-oxidizing activity anymore (Peters et al. 2013). Therefore, we first tested the impact of l-arabinose on growth of the G. oxydans strains 621H and BP.6 in shake flasks. In complex medium supplemented with 1% (w/v) l-arabinose instead of d-mannitol, growth of G. oxydans 621H was very poor and OD600 merely doubled within 8 h, while strain BP.6 did not grow at all (Fig. 1a). In medium supplemented with 4% (w/v) d-mannitol plus 1% (w/v) l-arabinose, both strains grew very similar (μ = 0.18 ± 0.01 h−1 for 621H and 0.17 ± 0.01 h−1 for BP.6) compared with the control condition with 4% (w/v) d-mannitol, yet without arabinose (μ = 0.18 ± 0.01 h-1 for 621H and 0.17 ± 0.01 h−1 for BP.6). In the stationary phase, both strains reached similar final OD600 values independent of the l-arabinose supplementation (621H OD600 with d-mannitol 3.84 ± 0.18 and with d-mannitol + l-arabinose 3.65 ± 0.28; BP.6 OD600 with d-mannitol 3.48 ± 0.26 and with d-mannitol + l-arabinose 3.66 ± 0.08). The concentration of l-arabinose supplemented to the medium did not decrease without cells or with BP.6 cells, while in the 621H cultures l-arabinose was decreased by approximately 80% within 24 h (Fig. 1b). This suggested that l-arabinose or its oxidation product did not impair growth of G. oxydans.

Comparisons of the G. oxydans strains 621H and BP.6. a Growth (OD600) in shake flasks in complex medium with 4% (w/v) d-mannitol or 1% (w/v) l-arabinose as well as both 4% (w/v) d-mannitol plus 1% (w/v) l-arabinose. b Arabinose concentrations in complex medium with 4% (w/v) d-mannitol plus 1% (w/v) l-arabinose in shake flasks. Data represent mean ± SD from three biological replicates
l-Arabinose is oxidized to l-arabinonic acid and hardly affected global gene expression
Based on the measurement of whole cell l-arabinose-oxidation activities of G. oxydans mDH deletion strains and of specifically complemented strains using DCPIP as electron acceptor, the membrane-bound glucose DH is expected to be responsible for l-arabinose oxidation (Mientus et al. 2017; Peters et al. 2013). Therefore, we tested the G. oxydans strains 621H and BP.6 for L-arabinose oxidation and analyzed the product(s) by GC-TOF-MS analysis. The multi-deletion strain BP.6 lacks six mDHs including the membrane-bound glucose DH GdhM (GOX0265) and still possesses the cytosolic glucose DH (GOX2015) GdhS (Peters et al. 2013). Cell suspensions of each strain with an OD600 of 1.6 were incubated for 24 h at 30 °C and 180 rpm in biotransformation buffer supplemented with 1% (w/v) l-arabinose. Afterwards, cell-free supernatants were obtained for GC-TOF-MS measurements. Strain 621H clearly oxidized l-arabinose, and l-arabinonic acid was identified as the product, while strain BP.6 consumed only minor amounts of l-arabinose and formed minor amounts of l-arabinonic acid (Fig. S1). The 621H sample showed two new peaks at retention times (Rt) 13.49 and 14.74 min. The peak at Rt 13.49 min had an m/z value of 364, corresponding to arabinonic acid γ-1,4-lactone with 3 TMS groups. The peak at Rt 14.74 min had an m/z value of 511, corresponding to arabinonic acid with 5 TMS groups and a methyl group split off. According to the comparable arbitrary peak areas, data indicated that GdhM did significantly contribute to l-arabinose oxidation. For strain BP.6, only a very low amount of l-arabinonic acid was found (~ 3%) compared with 621H. Together, this indicated that l-arabinose was indeed mainly oxidized by the membrane-bound glucose DH, while the contribution of the cytosolic glucose DH GdhS or another enzyme remaining in strain BP.6 was negligible.
To check whether the addition of l-arabinose as an inducer possibly affects short-term global gene expression in G. oxydans 621H, transcriptomes were compared using DNA microarrays. Addition of 1% (w/v) l-arabinose in the mid-exponential growth phase to 621H cells cultured in complex medium with d-mannitol had only very little effects on the relative mRNA levels within 30 min (Table S2). The highest mRNA level increase (2-fold) was found for GOX0707 encoding the DNA protection during starvation protein Dps followed by six hypothetical proteins (1.6- to 1.9-fold). The strongest mRNA level decrease (0.46-fold) was observed for GOX0536 encoding a hydroxamate-type ferrisiderophore receptor followed by its neighboring genes GOX0532 (0.57-fold) and GOX0531 (0.59-fold) encoding the ExbB and ExbD proteins of the TonB-ExbB-ExbD system as well as GOX0758 (0.56-fold) encoding a porin protein. There is no obvious functional link between these genes and l-arabinose metabolism.
Construction of AraC-dependent l-arabinose-inducible expression plasmids
Based on the tight high-level expression vectors containing the PBAD promoter developed for E. coli (Guzman et al. 1995), Invitrogen commercialized the AraC-PBAD system for dose-dependent expression of genes with plasmid pBAD/Myc-His A useful for expression of potentially toxic or essential genes in E. coli. The araC sequence used in pBAD/Myc-His A corresponds to the araC sequence of the widely used MC4100 lineage of E. coli K12 (Casadaban 1976). It differs in nine codons from araC of the E. coli K12 reference strain MG1655, which could be of advantage for expression in G. oxydans. Six of the nine different codons cluster in the region of the helix-turn-helix motif responsible for DNA binding at the C terminus of AraC (Brunelle and Schleif 1989). Five of these six codons exhibit much higher usage frequency in G. oxydans when using araC from MC4100 instead of MG1655 (Table S3). Consequently, the AraC-dependent promoter PBAD flanked by araC from MC4100 and the reporter gene uidA or mNeonGreen was integrated into the multiple cloning site (MCS) as described in “Materials and methods.” To enable efficient translation of the reporter, 16 nt containing a proven Shine-Dalgarno sequence functional in G. oxydans was placed between PBAD and the reporter gene. Furthermore, as a terminator sequence, BBa_B1002 was inserted downstream of the reporter genes. We also constructed a reporter plasmid without araC to test the AraC dependence of PBAD in G. oxydans. Another reporter plasmid additionally carried the l-arabinose transporter gene araE to test the sensitivity of induction towards l-arabinose (Fig. S2).
Performance of the AraC-PBAD system with enzyme reporter UidA in shake flasks
With the pBBR1MCS-5-araC-PBAD-uidA plasmid, we tested the basal expression and the induction by l-arabinose using the reporter enzyme β-d-glucuronidase. Besides G. oxydans 621H, which oxidizes l-arabinose by mDHs, we also used the multi-deletion strain BP.6 that is almost unable to oxidize l-arabinose to check whether AraC-PBAD performs better in BP.6 than in 621H. Both strains were grown in shake flasks using d-mannitol medium with and without 1% (w/v) l-arabinose. For strain 621H, the highest UidA activity was observed at the end of the exponential phase after approximately 9 h with 12,772 ± 1604 MU compared with 146 ± 47 MU in the control cultures without l-arabinose, the latter indicating very low basal expression (Fig. 2a). Strain BP.6 exhibited 11,780 ± 813 MU after 9 h in induced cultures compared with 225 ± 29 MU in the non-induced controls. Surprisingly, UidA activity in l-arabinose-supplemented G. oxydans 621H was highly reduced after 24 h (2,007 ± 583 MU), while UidA activity in BP.6 remained high after 24 h (13,209 ± 715 MU). When compared with the non-induced cultures, maximal UidA induction ratios of 87 and 59 were calculated for 621H and BP.6, respectively (Table 2). The lower ratio calculated for BP.6 was primarily due to the somewhat higher basal expression under non-induced condition, while the absolute UidA activity in BP.6 under induced conditions was similar as that in 621H. Generally, the UidA activities in the non-induced cultures did barely surpass the background activity values determined in cell-free control samples. This indicated that the expression plasmid pBBR1MCS-5-araC-PBAD-uidA showed very low basal expression of PBAD in the absence of l-arabinose.

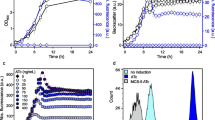
Comparisons of the G. oxydans strains 621H and BP.6. a Growth and UidA activity in Miller units (MU) in strains 621H and BP.6 carrying plasmid pBBR1MCS-5-araC-PBAD-uidA in l-arabinose-induced and non-induced condition in shake flasks. b Growth and specific mNeonGreen (mNG) fluorescence in strains 621H and BP.6 carrying plasmid pBBR1MCS-5-araC-PBAD-mNG in l-arabinose-induced and non-induced condition in shake flasks. The mNG fluorescence was measured in a Tecan reader. The specific fluorescence was calculated from absolute fluorescence per OD600. c Growth according to backscatter and specific mNG fluorescence in strains 621H and BP.6 carrying plasmid pBBR1MCS-5-araC-PBAD-mNG in l-arabinose-induced and non-induced condition in microscale BioLector cultivations. The specific fluorescence was calculated from absolute fluorescence per backscatter. For induction, always 1% (w/v) l-arabinose was added to the d-mannitol medium. For all experiments, data represent mean ± SD from three biological replicates
Performance of the AraC-PBAD system with the fluorescence reporter protein mNeonGreen
For continuous determination of reporter gene expression and its dynamics as well as to verify the strong induction of PBAD and AraC dependence in G. oxydans, we also tested the fluorescence reporter protein mNeonGreen (mNG). In all tests with plasmid pBBR1MCS-5-araC-PBAD-mNG, the mNG fluorescence signals in non-induced cultures did barely surpass the background signals of cell-free control samples, suggesting also a very low basal expression of mNG. In shake flask cultivations with 1% (w/v) l-arabinose, the mNG fluorescence in G. oxydans 621H peaked approximately after 8 h (Fig. 2b). At the end of the cultivation (24 h), the mNG fluorescence was much lower in strain 621H, while in strain BP.6, the mNG fluorescence was further increased. The maximal induction ratios based on the absolute or the biomass-specific mNG fluorescence were calculated to be 289 ± 75 or 327 ± 71 for strain 621H and 431 ± 14 or 481 ± 38 for BP.6, respectively (Table 3). In microscale BioLector cultivations, a similar induction profile was observed (Fig. 2c). The maximal induction ratios based on specific fluorescence were calculated to be 222 ± 8 for 621H and 192 ± 8 for BP.6. Again, after entering the stationary phase, the specific mNG fluorescence steadily decreased and reached zero after about 16 h in strain 621H, while for strain BP.6, the decrease was much weaker reduced over time and remained high. This was in contrast with the shake flask cultivations, where mNG fluorescence increased in the stationary phase in strain BP.6 (Fig. 2b). In the late stationary phase (16–24 h), the backscatter values of the 621H cultures supplemented with 1% (w/v) l-arabinose steadily increased further. Measurements of the OD600 in a photometer showed no differences between l-arabinose-supplemented strain 621H and the non-supplemented 621H cultures. Additional control measurements with cell-free complex medium adjusted to different pH values (pH 6, 4.5, 4, and 3.3) revealed differences in backscatters already in cell-free medium due to the differences in pH that could not be observed in OD600 measurements in a photometer. This suggested that the steady increase of backscatter values in l-arabinose-induced 621H cultures in the stationary phase resulted from a stronger acidification of the growth medium that cannot be detected by OD600 measurements in a photometer. Taken together, the newly constructed expression plasmids for G. oxydans based on the AraC-PBAD system exhibited very low basal expression of the reporter proteins UidA and mNG and very strong induction by 1% (w/v) l-arabinose both in shake flask and in microscale BioLector cultivations (up to 480-fold).
The PBAD promoter is AraC-dependent in G. oxydans and tunable by varying the l-arabinose concentrations
In E. coli, the regulator AraC acts as a repressor of PBAD in the absence of l-arabinose by bending the promoter DNA and stimulates transcription from PBAD when l-arabinose is present (Schleif 2010; Soisson et al. 1997). To rule out the possibility that in G. oxydans PBAD is either repressed or activated by a G. oxydans protein, the plasmid pBBR1MCS-5-PBAD-mNG missing the araC gene was constructed and tested in G. oxydans 621H. In this experiment, no differences in fluorescence were observed between induced and non-induced cells, thus no induction was observed with 1% (w/v) l-arabinose when AraC was absent (Fig. S3). This result showed that the inducibility of PBAD in G. oxydans is independent of endogenous proteins and indeed specifically dependent on heterologous AraC. Furthermore, in the absence of AraC the reporter is almost not expressed from PBAD, thus repression of PBAD by AraC seemed not to be required in G. oxydans.
The tunability of the AraC-PBAD system in G. oxydans was tested with a range of l-arabinose concentrations. The mNG fluorescence gradually increased similarly in strains 621H and BP.6 with increasing concentrations of l-arabinose (Fig. 3). Compared with 1% (w/v) l-arabinose, supplementation with 2% (w/v) l-arabinose did not lead to a significant increase in mNG fluorescence, indicating saturation of induction at close to 1% (w/v) inducer. Whereas only a small decrease of the mNG fluorescence was observed in the stationary phase for the BP.6 cultures induced with the highest l-arabinose concentrations, mNG fluorescence for strain 621H decreased to zero in the stationary phase for the cultures induced with more than 0.5% (w/v) l-arabinose. In contrast, no decrease was observed for the 621H cultures with up to 0.25% l-arabinose, clearly suggesting a correlation between l-arabinonic acid formation and loss in mNG fluorescence signals.

l-Arabinose-dependent modulation of expression in the G. oxydans strains 621H (a) and BP.6 (b) carrying plasmid pBBR1MCS-5-araC-PBAD-mNG in microscale BioLector cultivations. Reporter gene mNG expression was induced with increasing concentrations of l-arabinose from 0.03 to 2% (w/v) as indicated. Data represent mean ± SD from three biological replicates
Overall, in the exponential growth phase, the performance of the AraC-PBAD system was very similar in the l-arabinose-oxidizing strain 621H compared with the multi-deletion strain BP.6. Therefore, we asked whether the oxidation product l-arabinonic acid could possibly also act as an inducer on AraC, thereby compensating for the decrease in l-arabinose by its oxidation in 621H cultures. To test this, we added 1% (w/v) of l-arabinonic acid to mannitol medium and monitored the mNG fluorescence of strain 621H with pBBR1MCS-5-araC-PBAD-mNG in BioLector cultivations. l-Arabinonic acid did not induce the AraC-PBAD system, since the cultures exhibited a similar background fluorescence as non-induced 621H cultures without l-arabinose or l-arabinonic acid (Fig. S4).
The experiments described above showed that l-arabinose is able to activate AraC in G. oxydans, yet it is unknown how l-arabinose enters the G. oxydans cytoplasm. Knowledge on sugar transport systems in G. oxydans is scarce in general and no data are available for l-arabinose to our knowledge. The E. coli protein AraE is a low-affinity high-capacity l-arabinose transporter (Khlebnikov et al. 2001). To facilitate l-arabinose uptake by G. oxydans and thereby possibly improving the sensitivity of the l-arabinose-inducible system, plasmid pBBR1MCS-5-araE-araC-PBAD-mNG was constructed containing araE including a Shine-Dalgarno sequence directly downstream of araC. This should enable co-transcription of araC and araE from ParaC. When analyzing strain 621H carrying plasmid pBBR1MCS-5-araE-araC-PBAD-mNG with 0.1 and 0.5% (w/v) l-arabinose, however, the specific fluorescence was 40 to 60% lower compared with strain 621H carrying the plasmid without araE (Fig. S5). Thus, no increase in sensitivity towards the inducer was observed with pBBR1MCS-5-araE-araC-PBAD-mNG under the conditions tested.
Acidification of the growth medium is responsible for the decrease in reporter activities
In the experiments described above, both UidA activity and mNG fluorescence strongly decreased in the stationary phase of the 621H cultures induced with 1% (w/v) l-arabinose but not in the BP.6 cultures. The decrease correlated with the l-arabinose concentration and was absent at concentrations up to 0.25% (w/v). This clearly indicated that oxidation of l-arabinose affected the dynamics of the reporter activities. According to previous data and our GC-TOF-MS results, strain 621H oxidizes l-arabinose to l-arabinonic acid by the membrane-bound glucose DH, which is absent in strain BP.6 (Mientus et al. 2017). With a pKa of 3.39 (https://hmdb.ca/metabolites/HMDB0000539), formation of l-arabinonic acid caused an additional acidification of the growth medium. The pH values of the d-mannitol media of strains 621H and BP.6 supplemented with 1% (w/v) l-arabinose decreased from initially pH 6.0 to 3.3 ± 0.1 and 4.4 ± 0.1, respectively, after 24 h. In contrast, the pH values of the non-induced cultures were 4.7 ± 0.1 and 4.6 ± 0.1 for 621H and BP.6, respectively, after 24 h. These data confirm that l-arabinose oxidation to l-arabinonic acid by strain 621H leads to a stronger acidification of the medium.
Bacteria typically exhibit energy-dependent mechanisms for cytoplasmic pH homeostasis in order to survive during exposure to acidic or alkaline conditions. The observed decreases in mNG fluorescence suggested that due to the formation of l-arabinonic acid, the cytoplasmic pH in strain 621H increasingly acidified in the stationary phase when cells starved for energy. Recently, it was shown for the mNG protein that a shift from pH 6 to 4 is sufficient to reduce the mNG fluorescence by approximately 75% (Steiert et al. 2018). As described above, the pH values of induced 621H cultures after 24 h were even lower than pH 4. Consequently, we tested whether fresh d-mannitol-free and l-arabinose-free medium adjusted to different pH values could restore the mNG fluorescence of stationary phase 621H cells cultivated with 1% (w/v) l-arabinose. For this test, we used plasmid pBBR1MCS-2-PGOX0264-mNG in order to constitutively express mNG from the strong promoter of GOX0264. As expected, when cultivated with l-arabinose the mNG fluorescence was clearly decreased in the stationary phase, while without l-arabinose the mNG fluorescence remained high (Fig. S6a). Incubation of the cells grown with l-arabinose for 1 h in fresh medium adjusted to pH 3.5, 4.9, and 6.2 gradually recovered the mNG fluorescence (Fig. S6b). This result indicated that the decrease in mNG fluorescence in 621H cells was hardly due to degradation of the mNG protein in the stationary phase. Rather, the strong acidification of the growth medium by l-arabinonic acid formation in 621H cultures affected the cytoplasmic pH during the stationary phase which in turn was responsible for the decrease in mNG fluorescence. Furthermore, for G. oxydans 621H carrying pBBR1MCS-5-araC-PBAD-uidA grown with 1% (w/v) l-arabinose, the loss of UidA activity in the stationary phase (24 h) was also restored partly when cells were transferred for 1 h into fresh medium adjusted to pH 6 (Fig. S6c, d).
To analyze the induction of the AraC-PBAD system in G. oxydans and the pH-dependent decrease and recovery in mNG fluorescence on the single cell level, flow cytometer analysis was applied. For strain 621H, 8 h after induction, 91% of all cells exhibited high fluorescence signals (approximately 40,000 a.u.) within the chosen gate, indicating a high population homogeneity close to the end of the exponential growth phase (Fig. 4a). 26 h after induction, when the pH of the medium was 3.3 ± 0.1, the overall fluorescence was diminished and two distinct populations at approximately 10,000 a.u. and approximately 600 a.u. emerged. Transferring the 621H cells into d-mannitol-free and l-arabinose-free medium adjusted to pH 6 resulted in a recovery of the mNG fluorescence in a single population with 83% exhibiting a fluorescence signal of approximately 30,000 a.u.. For G. oxydans BP.6, also a strong induction with a high homogeneity and a fluorescence of about 40,000 a.u. was observed after 8 h. These signals were hardly decreased in the stationary phase after 26 h (pH 4.4 ± 0.1) and were not affected by the transfer into fresh medium adjusted to pH 6 (Fig. 4b). These single cell results are in line with the assumption that additional acidification of the growth medium causes a stronger intracellular acidification in the stationary phase. Furthermore, the single cell data showed that the AraC-PBAD system enabled a highly uniform induction response in the cells.

FACS analysis of the G. oxydans strains 621H (a) and BP.6 (b) carrying plasmid pBBR1MCS-5-araC-PBAD-mNG. Cells were grown in shake flasks with d-mannitol medium and induced with 1% (w/v) l-arabinose. FACS analysis was performed 8 and 26 h after induction. After 26 h, cells were transferred into fresh l-arabinose- and d-mannitol-free medium adjusted to pH 6 followed by incubation on a rotary shaker for 2 h. As a control, 621H or BP.6 cells without plasmids also grown in d-mannitol medium with 1% (w/v) l-arabinose were used. Total counts per sample represent 100,000 events and only appears to be different due to the logarithmic scale on the x-axis
UidA activity and mNG fluorescence are stable in pH-controlled bioreactor cultivations
In the following, pH-controlled bioreactor cultivations were carried out to circumvent effects due to acidification of the medium when assaying the performance of the l-arabinose-inducible AraC-PBAD-system in G. oxydans. Therefore, G. oxydans strains 621H and BP.6 carrying pBBR1MCS-5-araC-PBAD-uidA and pBBR1MCS-5-araC-PBAD-mNG were grown in DASbox® mini bioreactors under controlled conditions at pH 6 and dissolved oxygen concentrations above 30%. Sufficient oxygen levels are crucial for maturation of the chromophore (Shaner et al. 2013). After the 24-h fermentation in d-mannitol medium with 1% (w/v) l-arabinose, the total volume of potassium hydroxide solution consumed to maintain pH 6 in the 621H cultures (11.2 ± 0.7 mL) was much higher compared with the BP.6 cultures (6.0 ± 1.0 mL), which is in line with the l-arabinose oxidation by strain 621H yielding l-arabinonic acid. As expected, when maintaining a constant pH of 6 throughout the cultivation, neither the mNG fluorescence nor the UidA activity decreased in the stationary phase in 621H cells oxidizing l-arabinose (Fig. 5).

l-Arabinose-inducible reporter gene expression in DASbox fermentations in pH-controlled conditions (pH 6). Both, mNG and UidA activity remained high in G. oxydans 621H carrying plasmid pBBR1MCS-5-araC-PBAD-mNG (a) or pBBR1MCS-5-araC-PBAD-uidA (b) 24 h after induction with 1% (w/v) l-arabinose when pH 6 was maintained
Providing an AraC-PBAD-dependent plasmid with a multiple cloning site as empty vector
Albeit an AraC-PBAD-dependent expression plasmid with genes of interest could be easily obtained by Gibson assembly in an all-in-one fragment mixture cloning, we wanted to provide an AraC-PBAD-dependent plasmid for G. oxydans with a multiple cloning site as an empty vector for classical restriction enzyme-based cloning attempts to insert genes of interest. Therefore, we assembled araC-PBAD with the multiple cloning site from pBBR1MCS-5 and the terminator sequence BBa_B1002 from the iGEM parts library in plasmid pBBR1MCS-5 to obtain plasmid pBBR1MCS-5-araC-PBAD-MCS as an empty vector (Fig. 6). In this plasmid, the ribosome binding site AGGAGA was included upstream of the NdeI site newly created upstream of the original MCS by exchanging a single G to a C. To check the functionality and inducibility of AraC-PBAD in this empty vector as a multiple cloning reference plasmid, we created the reporter plasmid pBBR1MCS-5-araC-PBAD-MCS-mNG by Gibson assembly resulting in the same sequence as it would have been obtained via classical restriction cloning of mNG using the restriction enzymes NdeI and XhoI. When the resulting plasmid pBBR1MCS-5-araC-PBAD-MCS-mNG was tested in strain 621H, again strong induction in the presence of l-arabinose was observed and the basal expression in the absence of l-arabinose was again very low (Fig. S7). However, for unknown reasons the specific mNG fluorescence reached only approximately 40% of the maximum observed with the test plasmid always used before and not having the multiple cloning site or remaining parts thereof. Nevertheless, this demonstrated that plasmid pBBR1MCS-5-araC-PBAD-MCS was also functional and could serve as a multiple cloning vector to insert fragments containing genes of interest and if required own ribosome binding sites.

Scheme of the pBBR1MCS-5-based araC-PBAD plasmid with a multiple cloning site (a) and sequence information details (b). The ribosome-binding site AGGAGA (GOX_RBS) is included and usable when the insert cloning is carried out on the 5′-end by NdeI; otherwise, another RBS needs to be included in the insert upstream of the gene of interest. The iGEM terminator sequence of BBa_B1002 is located downstream of the multiple cloning site (MCS)
Discussion
For the AAB G. oxydans, no expression plasmid was available that shows very low background expression and allows strong and tunable target gene expression. In this study, we found that the AraC-PBAD system derived from E. coli K12 MC4100 performed very well in G. oxydans with the pBBR1 plasmid backbone. It exhibited very low basal reporter gene expression, induction ratios up to 480-fold, and gradually increased expression in an l-arabinose concentration-dependent manner. Typically, the regulatable expression plasmids tested in AAB species and reported so far suffered from very leaky expression in the absence of the respective inducer, resulting in very low induction ratios for the promoters PBAD, PTet or PLux, while the overall expression was strong according to the reporter activities (Florea et al. 2016; Teh et al. 2019). Among these three promoters, the best induction ratios were only 5- to 12-fold for the l-arabinose-inducible AraC-PBAD system from E. coli when used in G. xylinus 700178, G. hansenii 53582, or K. rhaeticus iGEM (Teh et al. 2019). The reason for the leakiness in AAB reported for the repressor-regulated promoters is not clear. It seems the binding of the repressors to their target DNA sequences is not strong enough in the cytoplasm of AAB or there is not enough repressor protein. However, in the case of AraC the mechanism of transcriptional gene regulation is different compared with those of the pure repressor proteins TetR, LuxR, and LacI, which just dissociate from their operators when their respective inducer is bound to the protein. In E. coli, AraC does not only repress PBAD by looping the DNA when bound to specific target sequences in the absence of l-arabinose but also is essential for activation of PBAD in the presence of l-arabinose via a modified binding to target sequences, causing unlooping of the promoter DNA and stimulation of both the binding of RNA polymerase and the transition from the closed to the open promoter complex (Schleif 2010; Soisson et al. 1997). Additionally, in E. coli, the cAMP receptor protein (CRP) also plays an important role in the induction of PBAD by stimulating the opening of the DNA loop and either the binding of the RNA polymerase or the transition to the open complex (Schleif 2010). In the genome of G. oxydans, there is only a single protein (GOX0974/GOX_RS06010) predicted to belong to the CRP/FNR superfamily of transcriptional regulators (Korner et al. 2003; Kranz et al. 2017; Prust et al. 2005). According to our data, this protein is a member of the FNR family and not a cAMP-binding protein of the CRP family (unpublished). Therefore, CRP appears to be absent in G. oxydans. Furthermore, with plasmid pBBR1MCS-5-PBAD-mNG missing the araC gene, we confirmed that the induction by l-arabinose specifically depended on AraC and not on an endogenous G. oxydans protein. The absence of AraC and therefore the missing repression of PBAD was not sufficient to allow transcription from PBAD in G. oxydans. A CRP likely absent in G. oxydans could be the reason. This is important in view of leakiness, yet we cannot rule out that in the absence of AraC PBAD is somehow repressed by a G. oxydans protein that is displaced from PBAD when AraC is present. Nevertheless, the activation indicated that in G. oxydans, AraC was functional and required as a transcriptional activator of PBAD, thus heterologous AraC bound to the respective promoter DNA in the cytoplasm and interacted with the RNA polymerase of G. oxydans, yet AraC was not required to repress PBAD. Therefore, using transcriptional activation instead of derepression and considering CRP may circumvent the high leakiness of the classical repressor-based expression plasmids in AAB. However, the reason why AraC-PBAD from E. coli did not perform well in G. xylinus 700178, G. hansenii 53582, or K. rhaeticus iGEM remains unclear (Teh et al. 2019). We used the araC-PBAD sequence from E. coli K12 MC4100 that exhibits much better codon usage frequencies in G. oxydans for five rare E. coli K12 MG1655 codons located in the C-terminal helix-turn-helix region of AraC responsible for DNA binding. The effects of the different araC codon usage frequencies from MC4100 vs. MG1655 in AAB are unknown. Furthermore, differences in the constructed plasmids such as terminators used and the orientations of plasmid-based reporter and resistance genes to each other, as well as further little sequence-specific differences may affect the formation or stability of their transcripts and the resulting translation, thereby affecting the overall performance of the AraC-PBAD systems in AAB. This can already be seen in the result that even in G. oxydans with plasmid pBBR1MCS-5-araC-PBAD-MCS-mNG, which was very similar to the extremely well-performing plasmid pBBR1MCS-5-araC-PBAD-mNG, only approximately 40% of the maximal specific mNG fluorescence was found. This indicated that already the single G to C exchange between the RBS and the ATG start codon to create an NdeI site or the short remaining MCS sequence from the XhoI site (CTCGAG) after the stop codon directly upstream of the terminator sequence and overlapping with it by the last G, or even both, somehow negatively affected the resulting mNG transcript or its translation more drastically than one would expect. Further studies are required to analyze these sequence effects in AAB in more detail.
For induction of the AraC-PBAD system in E. coli, typically a concentration in the range from 0.001 to 0.2% of l-arabinose is sufficient (Guzman et al. 1995; Narayanan et al. 2006). In this study with G. oxydans and plasmid pBBR1MCS-5-araC-PBAD-mNG, a range of approximately 0.1 to 1% of l-arabinose was required for minimal and maximal induction. This range is much higher compared with E. coli and reflects an altered inducer responsiveness of the AraC-PBAD system in G. oxydans. The GC-TOF-MS analysis demonstrated that l-arabinose was oxidized by the membrane-bound glucose DH in strain 621H. Nevertheless, in the multi-deletion strain BP.6 almost completely unable to oxidize l-arabinose, the l-arabinose responsiveness of the AraC-PBAD system was similar as that of strain 621H. Thus, oxidation and the resulting decrease of l-arabinose did not reduce the responsiveness of the AraC-PBAD system, although the oxidation product l-arabinonic acid did not act as an inducer on AraC. Therefore, usage of the AraC-PBAD system in G. oxydans is not limited to strains that are unable to oxidize l-arabinose.
The lowered responsiveness of the AraC-PBAD system in G. oxydans to l-arabinose might be caused by a very low uptake of the sugar into the cell. Attempts to improve uptake by expression of the E. coli araE gene encoding a secondary transporter for l-arabinose did not increase the responsiveness and even had a negative effect on the expression of the reporter gene mNG in our case. The reason for this result is unclear. It might be related to the co-expression of araE with araC resulting in a longer transcript than araE or araC mRNA alone. The longer transcript could be less stable or could form secondary structures that negatively affect the translation process, both yielding less AraC activator protein. Additional experiments with separate expression of araE from a constitutive promotor independent of araC-PBAD or alternative arabinose transporter genes are necessary to elucidate the potential to improve the responsiveness of the araC-PBAD system in G. oxydans (Khlebnikov et al. 2001). An endogenous l-arabinose transporter is not known in G. oxydans (Prust et al. 2005). A BLAST search with the E. coli AraE protein sequence in the G. oxydans proteome revealed three proteins with 46 to 56% sequence identity. GOX0808 (56% identity) is annotated as a galactose-proton symporter, GOX0649 (51% identity) as a sugar-proton symporter, and GOX1971 (46% identity) also as a galactose-proton symporter. One or several of these transporters might be promiscuous and poorly take up l-arabinose as a side reaction. No data are available on the substrates, properties, and physiological functions of these transporters.
With respect to the question if l-arabinose can be degraded within G. oxydans cells, our results in which this sugar was used in complex medium as sole supplement and as co-substrate with d-mannitol argue against this possibility. With l-arabinose alone strain 621H showed maximally one doubling while strain BP.6 showed no growth at all. This suggested that oxidation of l-arabinose somewhat contributed to generate energy in 621H for growth, yet not in BP.6 unable to oxidize l-arabinose. In E. coli and other bacteria, l-arabinose is catabolized via an initial isomerization to l-ribulose catalyzed by AraA, followed by phosphorylation to l-ribulose 5-phosphate catalyzed by AraB, and epimerization to d-xylulose 5-phosphate catalyzed by AraD (Englesberg 1961; Englesberg et al. 1962). In G. oxydans, homologs of AraA and AraD are absent and only a protein annotated as ribulokinase (GOX2186) showing 26% sequence identity to E. coli AraB was found. Therefore, l-arabinose taken up by G. oxydans is probably not converted to an intermediate like d-xylulose 5-phosphate that could be catabolized in the pentose phosphate pathway.
The oxidation of l-arabinose by G. oxydans 621H did not affect the inducibility of PBAD, yet as a side effect the growth medium was much more acidified due to the formation of l-arabinonic acid. Therefore, when expressing target genes encoding pH-sensitive proteins in batch cultures without pH control, the use of a G. oxydans strain like BP.6 that lacks the ability to oxidize l-arabinose is beneficial to preserve the activity of the target protein in the stationary phase. In the case of strain 621H, additional acidification of the medium due to l-arabinonic acid formation resulted in a severe loss of UidA activity and mNG fluorescence in the stationary phase. Both, UidA activity and mNG fluorescence could be partly or almost fully recovered by solely transferring the cells into fresh medium adjusted to pH 6, indicating that at least the mNG protein was hardly degraded. The activity of the β-d-glucuronidase UidA is reduced by half when the pH is lowered to 4.3 compared with the activity in the optimal range of pH 5.0 to 7.5 (Jefferson et al. 1986). For mNG, it was shown that between pH 5 and 6, three different protonated forms of the chromophore are present. When lowering the pH from 6 to 4, the acidic form becomes predominant, which decreases the fluorescence intensity by half and at pH 3 the mNG fluorescence is completely lost (Steiert et al. 2018). Our flow cytometry analysis revealed the occurrence of two subpopulations of 621H cells with reduced mNG fluorescence 26 h after induction in medium with a pH of 3.3. These two subpopulations might reflect a different progress in cytoplasmic acidification of 621H cells coping with pH 3.3 in the stationary phase. After resuspension in fresh medium with pH 6, mNG fluorescence was recovered and the two subpopulations merged into a single population. In strain BP.6, which is unable to oxidize l-arabinose, flow cytometry revealed that the acidification of the medium from pH 6 to 4.4 had only a marginal effect on mNG fluorescence. As the studies with isolated mNG demonstrated a strong decrease in fluorescence at pH 4.4 (Steiert et al. 2018), this result indicates that the intracellular pH in BP.6 cells must be higher than that in 621H cells. Bacteria have evolved various mechanisms for pH homeostasis in order to maintain the cytoplasmic pH in a range that enables survival and growth (reviewed in, for example, Baker-Austin and Dopson 2007; Follmann et al. 2009; Krulwich et al. 2011). For G. oxydans, the mechanisms of pH homeostasis have not yet been studied. We previously compared global gene expression of G. oxydans 621H cells grown at pH 4 and at pH 6 and observed 35 genes which were upregulated at pH 4 more than twofold and 37 genes that were downregulated more than twofold (Hanke et al. 2012). Obvious mechanisms for pH homeostasis could not be deduced from these data. In summary, the comparison of mNG fluorescence in strains 621H and BP.6 suggested that G. oxydans is well able to perform reasonable pH homeostasis at pH 4.4 and less able at pH 3.3. However, when maintaining the medium at pH 6, e.g., by cultivation of the cells with pH control, the acidification due to l-arabinose oxidation can be avoided also in G. oxydans 621H, preserving the activity of proteins synthesized under the control of PBAD. Together, our results demonstrate the functionality and potential of l-arabinose-inducible gene expression as a tool for G. oxydans and possibly also for other AAB.
References
Ameyama M, Shinagawa E, Matsushita K, Adachi O (1981) d-fructose dehydrogenase of Gluconobacter industrius: purification, characterization, and application to enzymatic microdetermination of d-fructose. J Bacteriol 145(2):814–823
Baker-Austin C, Dopson M (2007) Life in acid: pH homeostasis in acidophiles. Trends Microbiol 15(4):165–171. https://doi.org/10.1016/j.tim.2007.02.005
Brunelle A, Schleif R (1989) Determining residue-base interactions between AraC-protein and araI DNA. J Mol Biol 209(4):607–622. https://doi.org/10.1016/0022-2836(89)90598-6
Casadaban MJ (1976) Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol 104(3):541–555. https://doi.org/10.1016/0022-2836(76)90119-4
Chen R (2012) Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol Adv 30(5):1102–1107. https://doi.org/10.1016/j.biotechadv.2011.09.013
Connell ND (2001) Expression systems for use in actinomycetes and related organisms. Curr Opin Biotechnol 12(5):446–449. https://doi.org/10.1016/S0958-1669(00)00243-3
Dilworth MV, Piel MS, Bettaney KE, Ma P, Luo J, Sharples D, Poyner DR, Gross SR, Moncoq K, Henderson PJF, Miroux B, Bill RM (2018) Microbial expression systems for membrane proteins. Methods 147:3–39. https://doi.org/10.1016/j.ymeth.2018.04.009
Englesberg E (1961) Enzymatic characterization of 17 l-arabinose negative mutants of Escherichia coli. J Bacteriol 81:996–1006
Englesberg E, Anderson RL, Weinberg R, Lee N, Hoffee P, Huttenhauer G, Boyer H (1962) l-Arabinose-sensitive, l-ribulose 5-phosphate 4-epimerase-deficient mutants of Escherichia coli. J Bacteriol 84:137–146
Evans JC, Mizrahi V (2015) The application of tetracyclineregulated gene expression systems in the validation of novel drug targets in Mycobacterium tuberculosis. Front Microbiol 6:812. https://doi.org/10.3389/fmicb.2015.00812
Florea M, Hagemann H, Santosa G, Abbott J, Micklem CN, Spencer-Milnes X, Garcia LD, Paschou D, Lazenbatt C, Kong DZ, Chughtai H, Jensen K, Freemont PS, Kitney R, Reeve B, Ellis T (2016) Engineering control of bacterial cellulose production using a genetic toolkit and a new cellulose-producing strain. Proc Natl Acad Sci 113(24):E3431–E3440. https://doi.org/10.1073/pnas.1522985113
Follmann M, Ochrombel I, Kramer R, Trotschel C, Poetsch A, Ruckert C, Huser A, Persicke M, Seiferling D, Kalinowski J, Marin K (2009) Functional genomics of pH homeostasis in Corynebacterium glutamicum revealed novel links between pH response, oxidative stress, iron homeostasis and methionine synthesis. BMC Genomics 10. https://doi.org/10.1186/1471-2164-10-621
Forstner M, Leder L, Mayr LM (2007) Optimization of protein expression systems for modern drug discovery. Expert Rev Proteomics 4(1):67–78. https://doi.org/10.1586/14789450.4.1.67
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6(5):343–U41. https://doi.org/10.1038/Nmeth.1318
Gruber S, Schwab H, Koefinger P (2015) Versatile plasmid-based expression systems for Gram-negative bacteria-General essentials exemplified with the bacterium Ralstonia eutropha H16. New Biotechnol 32(6):552–558. https://doi.org/10.1016/j.nbt.2015.03.015
Gupta A, Singh VK, Qazi GN, Kumar A (2001) Gluconobacter oxydans: its biotechnological applications. J Mol Microbiol Biotechnol 3(3):445–456
Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177(14):4121–4130. https://doi.org/10.1128/jb.177.14.4121-4130.1995
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166(4):557–580
Hanke T, Richhardt J, Polen T, Sahm H, Bringer S, Bott M (2012) Influence of oxygen limitation, absence of the cytochrome bc(1) complex and low pH on global gene expression in Gluconobacter oxydans 621H using DNA microarray technology. J Biotechnol 157(3):359–372. https://doi.org/10.1016/j.jbiotec.2011.12.020
Hekmat D, Bauer R, Fricke J (2003) Optimization of the microbial synthesis of dihydroxyacetone from glycerol with Gluconobacter oxydans. Bioprocess Biosyst Eng 26(2):109–116. https://doi.org/10.1007/s00449-003-0338-9
Hentschel E, Will C, Mustafi N, Burkovski A, Rehm N, Frunzke J (2013) Destabilized eYFP variants for dynamic gene expression studies in Corynebacterium glutamicum. Microb Biotechnol 6(2):196–201. https://doi.org/10.1111/j.1751-7915.2012.00360.x
Hölscher T, Weinert-Sepalage D, Görisch H (2007) Identification of membrane-bound quinoprotein inositol dehydrogenase in Gluconobacter oxydans ATCC 621H. Microbiology 153(Pt 2):499–506. https://doi.org/10.1099/mic.0.2006/002196-0
Jefferson RA, Burgess SM, Hirsh D (1986) Beta-glucuronidase from Escherichia coli as a gene-fusion marker. Proc Natl Acad Sci 83(22):8447–8451. https://doi.org/10.1073/pnas.83.22.8447
Kallnik V, Meyer M, Deppenmeier U, Schweiger P (2010) Construction of expression vectors for protein production in Gluconobacter oxydans. J Biotechnol 150(4):460–465. https://doi.org/10.1016/j.jbiotec.2010.10.069
Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD (2001) Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology 147(Pt 12):3241–3247. https://doi.org/10.1099/00221287-147-12-3241
Kiefler I, Bringer S, Bott M (2015) SdhE-dependent formation of a functional Acetobacter pasteurianus succinate dehydrogenase in Gluconobacter oxydans--a first step toward a complete tricarboxylic acid cycle. Appl Microbiol Biotechnol 99(21):9147–9160. https://doi.org/10.1007/s00253-015-6972-8
Kiefler I, Bringer S, Bott M (2017) Metabolic engineering of Gluconobacter oxydans 621H for increased biomass yield. Appl Microbiol Biotechnol 101(13):5453–5467. https://doi.org/10.1007/s00253-017-8308-3
Korner H, Sofia HJ, Zumft WG (2003) Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol Rev 27(5):559–592. https://doi.org/10.1016/S0168-6445(03)00066-4
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM 2nd, Peterson KM (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166(1):175–176. https://doi.org/10.1016/0378-1119(95)00584-1
Kranz A, Vogel A, Degner U, Kiefler I, Bott M, Usadel B, Polen T (2017) High precision genome sequencing of engineered Gluconobacter oxydans 621H by combining long nanopore and short accurate Illumina reads. J Biotechnol 258:197–205. https://doi.org/10.1016/j.jbiotec.2017.04.016
Kranz A, Steinmann A, Degner U, Mengus-Kaya A, Matamouros S, Bott M, Polen T (2018) Global mRNA decay and 23S rRNA fragmentation in Gluconobacter oxydans 621H. BMC Genomics 19(1):753. https://doi.org/10.1186/s12864-018-5111-1
Krulwich TA, Sachs G, Padan E (2011) Molecular aspects of bacterial pH sensing and homeostasis. Nat Rev Microbiol 9(5):330–343. https://doi.org/10.1038/nrmicro2549
Mamlouk D, Gullo M (2013) Acetic acid bacteria: physiology and carbon sources oxidation. Indian J Microbiol 53(4):377–384. https://doi.org/10.1007/s12088-013-0414-z
Merfort M, Herrmann U, Bringer-Meyer S, Sahm H (2006a) High-yield 5-keto-d-gluconic acid formation is mediated by soluble and membrane-bound gluconate-5-dehydrogenases of Gluconobacter oxydans. Appl Microbiol Biotechnol 73(2):443–451. https://doi.org/10.1007/s00253-006-0467-6
Merfort M, Herrmann U, Ha SW, Elfari M, Bringer-Meyer S, Gorisch H, Sahm H (2006b) Modification of the membrane-bound glucose oxidation system in Gluconobacter oxydans significantly increases gluconate and 5-keto-d-gluconic acid accumulation. Biotechnol J 1(5):556–563. https://doi.org/10.1002/biot.200600032
Meyer M, Schweiger P, Deppenmeier U (2013) Effects of membrane-bound glucose dehydrogenase overproduction on the respiratory chain of Gluconobacter oxydans. Appl Microbiol Biotechnol 97(8):3457–3466. https://doi.org/10.1007/s00253-012-4265-z
Mientus M, Kostner D, Peters B, Liebl W, Ehrenreich A (2017) Characterization of membrane-bound dehydrogenases of Gluconobacter oxydans 621H using a new system for their functional expression. Appl Microbiol Biotechnol 101(8):3189–3200. https://doi.org/10.1007/s00253-016-8069-4
Miller JH (1992) A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Narayanan N, Xu Y, Chou CP (2006) High-level gene expression for recombinant penicillin acylase production using the araB promoter system in Escherichia coli. Biotechnol Prog 22(6):1518–1523. https://doi.org/10.1021/bp060135u
Paczia N, Nilgen A, Lehmann T, Gatgens J, Wiechert W, Noack S (2012) Extensive exometabolome analysis reveals extended overflow metabolism in various microorganisms. Microb Cell Factories 11:122. https://doi.org/10.1186/1475-2859-11-122
Pappenberger G, Hohmann HP (2014) Industrial production of l-ascorbic acid (vitamin C) and d-isoascorbic acid. Adv Biochem Eng Biotechnol 143:143–188. https://doi.org/10.1007/10_2013_243
Parachin NS, Mulder KC, Viana AAB, Dias SC, Franco OL (2012) Expression systems for heterologous production of antimicrobial peptides. Peptides 38(2):446–456. https://doi.org/10.1016/j.peptides.2012.09.020
Peters B, Mientus M, Kostner D, Junker A, Liebl W, Ehrenreich A (2013) Characterization of membrane-bound dehydrogenases from Gluconobacter oxydans 621H via whole-cell activity assays using multideletion strains. Appl Microbiol Biotechnol 97(14):6397–6412. https://doi.org/10.1007/s00253-013-4824-y
Prust C, Hoffmeister M, Liesegang H, Wiezer A, Fricke WF, Ehrenreich A, Gottschalk G, Deppenmeier U (2005) Complete genome sequence of the acetic acid bacterium Gluconobacter oxydans. Nat Biotechnol 23(2):195–200. https://doi.org/10.1038/nbt1062
Saito Y, Ishii Y, Hayashi H, Imao Y, Akashi T, Yoshikawa K, Noguchi Y, Soeda S, Yoshida M, Niwa M, Hosoda J, Shimomura K (1997) Cloning of genes coding for l-sorbose and l-sorbosone dehydrogenases from Gluconobacter oxydans and microbial production of 2-keto-l-gulonate, a precursor of l-ascorbic acid, in a recombinant G. oxydans strain. Appl Environ Microbiol 63(2):454–460
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schleif R (2010) AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol Rev 34(5):779–796. https://doi.org/10.1111/j.1574-6976.2010.00226.x
Schleyer U, Bringer-Meyer S, Sahm H (2008) An easy cloning and expression vector system for Gluconobacter oxydans. Int J Food Microbiol 125(1):91–95. https://doi.org/10.1016/j.ijfoodmicro.2007.04.016
Schnappinger D, Ehrt S (2014) Regulated expression systems for mycobacteria and their applications. Microbiol Spectr 2(1). https://doi.org/10.1128/microbiolspec.MGM2-0018-2013
Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang J (2013) A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 10(5):407–409. https://doi.org/10.1038/nmeth.2413
Simon R, Priefer U, Pühler A (1983) A broad host range mobilization system for in vivo genetic-engineering - Transposon mutagenesis in Gram-negative bacteria. Bioresour Technol 1(9):784–791. https://doi.org/10.1038/nbt1183-784
Soisson SM, MacDougall-Shackleton B, Schleif R, Wolberger C (1997) Structural basis for ligand-regulated oligomerization of AraC. Science 276(5311):421–425. https://doi.org/10.1126/science.276.5311.421
Steiert F, Petrov EP, Schultz P, Schwille P, Weidemann T (2018) Photophysical behavior of mNeonGreen, an evolutionarily distant green fluorescent protein. Biophys J 114(10):2419–2431. https://doi.org/10.1016/j.bpj.2018.04.013
Teh MY, Ooi KH, Teo SXD, Bin Mansoor ME, Lim WZS, Tan MH (2019) An expanded synthetic biology toolkit for gene expression control in Acetobacteraceae. ACS Synth Biol 8(4):708–723. https://doi.org/10.1021/j.acssynbio.8b00168
Terpe K (2006) Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 72(2):211–222. https://doi.org/10.1007/s00253-006-0465-8
Tkac J, Navratil M, Sturdik E, Gemeiner P (2001) Monitoring of dihydroxyacetone production during oxidation of glycerol by immobilized Gluconobacter oxydans cells with an enzyme biosensor. Enzym Microb Technol 28(4-5):383–388
Tonouchi N, Sugiyama M, Yokozeki K (2003) Construction of a vector plasmid for use in Gluconobacter oxydans. Biosci Biotechnol Biochem 67(1):211–213. https://doi.org/10.1271/bbb.67.211
Valero F (2012) Heterologous expression systems for lipases: A review. Methods Mol Biol 861:161–178. https://doi.org/10.1007/978-1-61779-600-5_11
Wang EX, Ding MZ, Ma Q, Dong XT, Yuan YJ (2016) Reorganization of a synthetic microbial consortium for one-step vitamin C fermentation. Microb Cell Factories 15:21. https://doi.org/10.1186/s12934-016-0418-6
Zhang L, Lin JP, Ma YS, Wei DZ, Sun M (2010) Construction of a novel shuttle vector for use in Gluconobacter oxydans. Mol Biotechnol 46(3):227–233. https://doi.org/10.1007/s12033-010-9293-2
Acknowledgments
We are grateful to Lion Flachbart for providing the reporter mNeonGreen and for helpful discussions. We also thank Nick Wierckx or helpful discussion and Armin Ehrenreich for kindly providing G. oxydans strain BP.6 carrying multiple deletions of mDHs. For technical support, we thank Sabrina Derksen.
Availability of data and material
The microarray data are accessible in NCBI’s Gene Expression Omnibus through accession number GSE151596.
Funding
Open Access funding enabled and organized by Projekt DEAL. We are grateful to the Federal Ministry of Education and Research (BMBF) for generous financial support of the project IMPRES (031B0370B). The funding organization did not influence the design of the study or collection, analysis, and interpretation of data, or writing the manuscript.
Author information
Authors and Affiliations
Contributions
PMF and TL selected the reporters and constructed the plasmids. PMF carried out all growth experiments to test strains with and without constructs and performed the data analysis. JG performed the GC-TOF-MS and data analysis. PMF and CS performed the FACS analysis and analyzed the data. PMF and MO performed the DASbox cultivations. TP designed and supervised the study. PMF, MB, and TP wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1210 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fricke, P.M., Link, T., Gätgens, J. et al. A tunable l-arabinose-inducible expression plasmid for the acetic acid bacterium Gluconobacter oxydans. Appl Microbiol Biotechnol 104, 9267–9282 (2020). https://doi.org/10.1007/s00253-020-10905-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-10905-4