
Abstract
The purpose of the work is to calculate accurate values of molecular properties of tetracyanoquinodimethane (TCNQ) and anions using the complete active space self-consistent field and complete active space second-order perturbation theory methods. The accuracy has been evaluated using several basis sets and active spaces. The calculated properties have, in many cases, been confirmed by experimental data (within parentheses), e.g., 9.54 eV (9.61 eV) and 3.36 eV (3.38 eV) for the ionization potential and electron affinity, respectively, of TCNQ; 3.12 eV (3.01 eV) and 3.54 eV (3.42 or 3.60 eV) for transition energies to the two lowest-lying excited singlet states of TCNQ; − 0.03, 0.46 and 1.44 eV (0, 0.5 and 1.4 eV) for electronic energies in electron attachment of TCNQ forming \(\hbox {TCNQ}^-\); and 3.88 eV (3.71 eV) for the transition energy to the second lowest-lying excited singlet state of \(\hbox {TCNQ}^{2-}\). Further, the calculations have brought insight into some experimental observations, e.g., the shape of the fluorescence spectrum of TCNQ at 3–4 eV.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
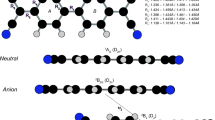
The TCNQ molecule, see Fig. 1, is a potent electron acceptor and it has gained a lot of attention in the fields of organic electronics [1] and photovoltaics [2, 3]. It was synthesized in 1962 [4] and a decade later, in 1973, it constituted, in complex with tetrathiafulvalene (TTF), the first organic metal discovered [1, 5]. Over the years experimental data concerning TCNQ and anions have accumulated and will allow the evaluation of quantum chemical methodologies.

The TCNQ molecule
The purpose of the study is twofold: calculation of accurate values of molecular properties of TCNQ and anions; and evaluation of the accuracy with respect to parameter values used in the applied methods. The evaluation also includes comparison with experimental data and shows that the results bring new insight into some experimental observations. In Sect. 2 the methods are presented and thereafter in Sect. 3 the molecular properties of TCNQ, \(\hbox {TCNQ}^-\) and \(\hbox {TCNQ}^{2-}\) are discussed. Finally, in Sect. 4, the conclusions are given.
2 Methods
In this study MOLCAS version 8.4 [6] is used throughout, and in particular the complete active space self-consistent field (CASSCF) and CAS second-order perturbation theory (CASPT2) methods. State specific calculations are performed (no state averaging) and all orbitals are optimized at CASSCF level of theory. The molecule is constrained to the point group \(\hbox {D}_{\text {2h}}\), i.e., a highly symmetric molecule with dipole moment equal to zero. For transition dipole moments (and oscillator strengths) the CASSCF state interaction (CASSI) method is used. Moreover, the default keywords are used (unless otherwise stated). The remaining parameters concern basis sets and active spaces and they are considered in Sects. 2.1 and 2.2, respectively. Finally, in Sect. 2.3, the applied geometries (resulting from ground state (GS) energy optimizations) are discussed.
2.1 Basis sets
The basis sets used are based on the atomic natural orbital (ANO) concept and they are denoted ANO-S and ANO-L, where ANO-S is designed to be small and to give good results for many molecular properties of large systems [7]. ANO-L, on the other hand, use considerably larger primitive sets and is designed to give sensible basis sets for molecular calculations [8].
The basis set sizes used in the study are given in Table 1, where basis sets \(\hbox {S}_{\text {l}}\) and \(\hbox {L}_{\text {l}}\) should give almost converged results compared to the primitive sets [7, 8]. Basis sets \(\hbox {S}_{\text {s}}\) and \(\hbox {L}_{\text {s}}\) represent minimum sizes for getting reasonable results (with the given primitives) [7, 8].
2.2 Active spaces
The first step, in deciding the active spaces, is to determine the optimum geometry of TCNQ in its GS by using second-order many-body perturbation theory (MBPT2) and the \(\hbox {S}_{\text {s}}\) basis set. SCF orbital energies and orbitals (for this geometry) are given in Figs. 2 and 3, respectively. The out-of-molecular-plane \(\pi\) and \(\pi ^*\) orbitals belong to the irreducible representations \(\hbox {b}_{\text {3u}}\), \(\hbox {b}_{\text {1g}}\), \(\hbox {b}_{\text {2g}}\) and \(\hbox {a}_{\text {u}}\) (of the \(\hbox {D}_{\text {2h}}\) point group). The highest occupied (HO) molecular orbital (MO) and the lowest unoccupied (LU) MO are two such orbitals (see first row of Fig. 3). The in-molecular-plane \(\pi _\perp\) and \(\pi _\perp ^*\) (CN) orbitals belong to the irreducible representations \(\hbox {a}_{\text {g}}\), \(\hbox {b}_{\text {2u}}\), \(\hbox {b}_{\text {1u}}\) and \(\hbox {b}_{\text {3g}}\) together with \(\sigma\) orbitals.

SCF orbital energies of TCNQ. The active spaces S, M, L and \(\hbox {L}_\perp\) are indicated in the figure. Weak orbital energy lines mark diffuse Rydberg-like orbitals, which are suppressed in the calculations

SCF orbitals (\(\pi\), \(\pi _\perp\), \(\pi ^*\) and \(\pi ^*_\perp\))
The second step is to determine which occupied and virtual orbitals should form the active space. Four active spaces, denoted S, M, L and \(\hbox {L}_\perp\), are used in the study and they are defined in Figs. 2 and 3 and Table 2. The HOMO and LUMO constitute active space S, which allows the calculation of ionization potential (IP), electron affinity (EA) and low-energy states. Active space M includes the three HOMOs (HOMO{0,− 1,− 2}) and the three LUMOs (LUMO{0,+1,+2}), which allow also the calculation of higher energy states. M consists to a large extent of CC \(\pi\) and \(\pi ^*\) orbitals (see Fig. 3). The fourth out-of-molecular-plane OMO of mainly CC character is the lowest \(\pi\) orbital (\(\pi _{\text {min}}\)). Its antibonding counterpart is \(\pi ^*_{\text {max}}\) (see last row of Fig. 3). These two orbitals are not included in any of the active spaces used in the study. To investigate the impact of CN orbitals the active spaces L and \(\hbox {L}_\perp\) are introduced.
In summary, it is clear from Table 2 that \(\hbox {S} \subset \hbox {M}\), \(\hbox {M} \subset \hbox {L}\), \(\hbox {M} \subset \hbox {L}_\perp\) and \(\hbox {L} \cap \hbox {L}_\perp = {\text {M}}\). Active spaces S, M and L include only \(\pi\) and \(\pi ^*\) orbitals, while \(\hbox {L}_\perp\) includes \(\pi _\perp\) and \(\pi _\perp ^*\) orbitals as well. In Table 2\(\{\pi \}\) denotes the set of the eight occupied \(\pi\) orbitals (\(\{\pi ^*\}\) is the antibonding counterpart).
2.3 Geometries
The optimum structures of TCNQ and anions (in the GS) at CASPT2 level of theory are presented in the Supplementary Information. The optimum structures have been obtained for several combinations of basis sets and active spaces. Overall the trends and tendencies are the same for the three molecules. The smallest basis set, \(\hbox {S}_{\text {s}}\), is too limited and overestimates the bond lengths by about 0.015 Å, on average, compared to those obtained with the largest basis set, \(\hbox {L}_{\text {l}}\). The bond angles differ, on average, by about \(0.2^\circ\) between these two basis sets.
The bond lengths obtained with basis sets \(\hbox {S}_{\text {l}}\) and \(\hbox {L}_{\text {s}}\) (of equal size) differ, on average, by a few milli-Ångström with those obtained with basis set \(\hbox {L}_{\text {l}}\). \(\hbox {S}_{\text {l}}\) is somewhat better than \(\hbox {L}_{\text {s}}\), which is due to the design of the basis sets. For the dianion the bond angles are almost identical for basis sets \(\hbox {S}_{\text {l}}\), \(\hbox {L}_{\text {s}}\) and \(\hbox {L}_{\text {l}}\). For the neutral molecule and anion there is a difference, on average, by about \(0.1^\circ\) between \(\hbox {S}_{\text {l}}\) and \(\hbox {L}_{\text {s}}\), on the one hand, and the largest basis set, \(\hbox {L}_{\text {l}}\), on the other.
A third basis set of type ANO-L, \(\hbox {L}_{\text {m}}\), with size between \(\hbox {L}_{\text {s}}\) and \(\hbox {L}_{\text {l}}\) improves the geometry compared to the \(\hbox {S}_{\text {l}}\) basis set, except for the CN bond length. It can be concluded that basis sets \(\hbox {S}_{\text {l}}\) and \(\hbox {L}_{\text {m}}\) give good descriptions of the geometry, for all molecules, with overestimation of bond lengths of a few milli-Ångström and inaccuracies in bond angles of a few tenths of a degree. For time-efficient calculations basis set \(\hbox {S}_{\text {l}}\) is the better choice.
In general the variation of bond lengths and angles is a few milli-Ångström and a few tenths of a degree, respectively, between the active spaces (S, M, L and \(\hbox {L}_\perp\)) for the three molecules. However, there are some exceptions. First, for the neutral molecule the inaccuracy of several bond lengths is of the order of 0.01 Å using active space S. Second, the variation in the CN bond length is of the order of 0.01 Å for all three molecules, which may not be surprising since active spaces L and \(\hbox {L}_\perp\) contain more CN orbitals than S and M. Third, the largest variation (\(0.5^\circ\)) in bond angles occurs for CCN for the anion (between active spaces M and \(\hbox {L}_\perp\)). The conclusion is nevertheless that active space M is good enough for the geometry determination of all molecules. In Table 3 optimum structures obtained with basis set \(\hbox {S}_{\text {l}}\) and active space M are presented for TCNQ and anions in the GS. For the neutral molecule the optimum structure for two excited states are also presented. The subscripts for bond lengths and angles in Table 3 are given in Fig. 1 together with angle definitions.
3 Results and discussion
In Sects. 3.1–3.3 the calculated molecular properties of TCNQ, \(\hbox {TCNQ}^-\) and \(\hbox {TCNQ}^{2-}\), respectively, are discussed and compared to experimental data. Since the calculations involve unbound anionic states, wave functions have to be carefully analysed in order to detect non-reliable solutions. An analysis of wave functions are given in Sect. S4 in Supplementary Information. That CASSCF calculations still may provide reliable well-localized solutions has been discussed by Rubio et al. [9].
3.1 TCNQ
In order to find out how able the applied methodology is in matching experimental data, the IP and EA of TCNQ are determined. In Table 4, the two properties are given for various combinations of the five basis sets and the four active spaces. For correspondence with experiment IP is the vertical IP (VIP) (using the geometry of optimized GS of TCNQ at CASPT2 level of theory) and EA is the vertical detachment energy (VDE) (using the geometry of optimized GS of \(\hbox {TCNQ}^-\)).
The calculated IP and EA both increase with increasing size of basis set and active space. The results differ by only about \(0.05~\hbox {eV}\) between the two largest basis sets, \(\hbox {L}_{\text {m}}\) and \(\hbox {L}_{\text {l}}\), and is most likely converged for the largest basis set. The inclusion of CN orbitals in the active space matters somewhat and has a larger effect on the EA than the IP, and CN \(\pi\) (and \(\pi ^*\)) orbitals affect the results more than CN \(\pi _\perp\) (and \(\pi _\perp ^*\)) orbitals. For the largest basis set, \(\hbox {L}_{\text {l}}\), and active space M, the calculated IP of 9.54 eV agrees well with the experimental value of 9.61 eV obtained from photoelectron spectrum of TCNQ in the gaseous state [10]. For the same basis set and active space, the calculated EA of 3.36 eV agrees well with the experimental value of \(3.383\pm 0.001~\hbox {eV}\) obtained from photoelectron spectrum of \(\hbox {TCNQ}^-\) produced using electrospray ionization [11].
From the photoelectron spectrum of \(\hbox {TCNQ}^-\), Zhu and Wang conclude that the geometry difference between the TCNQ anion and neutral molecule is small due to the short vibrational progression [11]. In Table 3, the optimized geometries of the GS of TCNQ (\(1^1\hbox {A}_{\text {g}}\)) and \(\hbox {TCNQ}^-\) (\(1^2\hbox {B}_{\text {2g}}\)) are given. The structures differ somewhat, particularly the \(\hbox {CC}_3\) bond (0.03 Å) and the ring (0.02 Å and 1–\(2^\circ\)).
The excited states of TCNQ, below 5 eV, at CASPT2 level of theory are summarized in Table 5 for various combinations of the five basis sets and the four active spaces. The excitation energies obtained with basis set \(\hbox {S}_{\text {s}}\) differ, on average, by 0.06 eV with those obtained with basis set \(\hbox {L}_{\text {l}}\). The excitation energies obtained with basis sets \(\hbox {S}_{\text {l}}\), \(\hbox {L}_{\text {s}}\) and \(\hbox {L}_{\text {m}}\) are similar and they differ, on average, by 0.02 eV with those obtained with basis set \(\hbox {L}_{\text {l}}\). The largest differences are 0.11 eV for \(\hbox {S}_{\text {s}}\) and 0.05, 0.03 and 0.02 eV for \(\hbox {S}_{\text {l}}\), \(\hbox {L}_{\text {s}}\) and \(\hbox {L}_{\text {m}}\), respectively. The inclusion of CN orbitals in the active space affects some states substantially. By including the CN \(\pi\) (and \(\pi ^*\)) orbitals in the active space the excitation energy to \(2^3\hbox {B}_{\text {1u}}\) is reduced by almost 0.3 eV (for the seven excited states the difference is 0.1 eV on average). The inclusion of CN \(\pi _\perp\) (and \(\pi _\perp ^*\)) orbitals has a less pronounced effect on the excitation energies (for the seven excited states the difference is 0.05 eV on average).
The \(1^1\hbox {B}_{\text {1u}}\) state of TCNQ is strongly absorbing from the GS with an oscillator strength of 1.5 at CASSCF level of theory. The calculated excitation energy, 3.49 eV (obtained with the largest basis set, \(\hbox {L}_{\text {l}}\), and active space M), does not agree well with (non gas-phase) experimental values: 3.08\(-\)3.09 eV (polymer matrices) [12]; 3.10 eV (mixture of polar and non-polar solvents) [13]; 3.09\(-\)3.17 eV (various polarities of solvents) [14]; and 3.17 eV (non-polar solvents) [15, 16]. A further discussion of the reasons for this difference is outside the scope of the present article.
In a recent study, Chaki et al. investigated the \(1^1\hbox {B}_{\text {1u}}\) state of TCNQ, in the gas phase, using laser induced fluorescence [16]. They observed a weak (0,0) band at 3.01 eV followed by additional vibronic bands at higher energies, and strong (broad) bands at 3.42 and 3.60 eV [16]. The observed strong bands fit well with the calculated value of 3.49 eV. What remains to be explained is the vibrational spectrum at lower energies. In the interpretation of the spectrum two issues are addressed. Firstly, the intensity pattern of the observed vibrational spectrum suggests a considerable geometric change in the excited state due to a weak (0,0) band followed by stronger vibrational bands [16]. Secondly, the fluorescence lifetimes for some vibronic bands, including the (0,0) band, are much longer [16] than measured in non gas-phase experiments (non-polar hexane solution) [14].
In resolving the two issues the geometries of the two lowest singlet excited states of TCNQ (\(2^1\hbox {A}_{\text {g}}\) and \(1^1\hbox {B}_{\text {1u}}\)) are optimized. The results are given in Table 3. The structures of the \(1^1\hbox {A}_{\text {g}}\) and \(1^1\hbox {B}_{\text {1u}}\) states differ somewhat, particularly the \(\hbox {CC}_3\) bond (0.03 Å), but they are not considerably different. The optimized structure of \(1^1\hbox {B}_{\text {1u}}\) is similar to the optimized GS of \(\hbox {TCNQ}^-\) and in both states the LUMO is singly occupied.
The structures of the \(1^1\hbox {A}_{\text {g}}\) and \(2^1\hbox {A}_{\text {g}}\) states, on the other hand, differ to a larger extent, particularly the \(\hbox {CC}_3\) bond (0.08 Å). The optimized structure of \(2^1\hbox {A}_{\text {g}}\) is similar to the optimized GS of \(\hbox {TCNQ}^{2-}\) and in both states the LUMO is doubly occupied (in the dominant configuration).

The lowest electronic states of TCNQ at CASPT2 level of theory (using basis set \(\hbox {S}_{\text {l}}\), active space M, and shift = 0.20 H). The lines are given for visual purposes. The bold line represents the strongly absorbing \(1^1\hbox {B}_{\text {1u}}\) state. From left to right on the x-axis the CC single bonds are decreasing and the CC double bonds increasing (especially \(\hbox {CC}_3\))
The electronic spectrum of TCNQ (below 5 eV) at the optimized geometries of the three lowest singlet states is plotted in Fig. 4. Between 3 and 4 eV there are three singlet and two triplet states, which may interfere with each other. The strongly absorbing \(1^1\hbox {B}_{\text {1u}}\) state seems to intersect with the \(2^1\hbox {A}_{\text {g}}\) state, which implies that the coupling between the electronic and nuclear motion has to be considered. The adiabatic excitation energy between the \(1^1\hbox {A}_{\text {g}}\) and \(2^1\hbox {A}_{\text {g}}\) states is 3.12 eV, which is close to the measured (0,0) band at 3.01 eV. If the \(2^1\hbox {A}_{\text {g}}\) state is responsible for the observed (0,0) band then its irreducible representation (forbidden transition to the GS) and geometry (different from the GS geometry) could explain the long lifetime and the intensity pattern of the observed vibrational spectrum.
The lowest triplet state of TCNQ has been measured at 1.96 and 1.85 eV by Khvostenko et al. using UV–vis absorption spectroscopy with Br-containing solvents [15]. The calculated value of 1.38 eV (using \(\hbox {L}_{\text {l}}\) and M) is different from the measured values, possibly due to solvent effects. A further discussion of the reasons for this difference is outside the scope of the present article.
3.2 TCNQ anion
The excited states of \(\hbox {TCNQ}^-\), below 5 eV, at CASPT2 level of theory are summarized in Table 6 for various combinations of the five basis sets and the four active spaces. The excitation energies are somewhat more stable for the anion than the neutral molecule regarding the choice of basis set and active space. But the trends are similar. The similar results for basis sets \(\hbox {S}_{\text {l}}\) and \(\hbox {L}_{\text {l}}\) using active space M are noteworthy.
Using glass measurements Haller and Kaufman identified the two lowest \(^2\hbox {B}_{\text {3u}}\) states at 1.45 and 2.84 eV [17]. From absorption spectra (in solution) Jonkman and Kommandeur identified three \(^2\hbox {B}_{\text {3u}}\) states at 1.46, 2.95 and 4.40 eV [13]. The (non gas-phase) experimental transition energies to the three lowest \(^2\hbox {B}_{\text {3u}}\) states thus are redshifted 0.1\(-\)0.5 eV compared to calculated values. Further, Jonkman and Kommandeur identified a \(^2\hbox {A}_{\text {u}}\) state at 2.95 eV [13], which is similar to the calculated values of 2.92\(-\)3.04 eV.
In 1977 Compton and Cooper presented properties of \(\hbox {TCNQ}^-\) in the gaseous phase [18]. They concluded that TCNQ attaches electrons with energies of \(\sim 0\), 0.7 and 1.3 eV forming long-lived negative ions [18]. In Fig. 5, the lowest electronic states of TCNQ and \(\hbox {TCNQ}^-\) are plotted (with 0 eV representing the GS of TCNQ). It is clear from the figure that there are \(\hbox {TCNQ}^-\) states around 0 eV (\(-0.23\), \(-0.14\), \(-0.03\)); 0.7 eV (0.46, 0.76, 0.86); and 1.3 eV (1.31, 1.31, 1.44).

The lowest electronic states of TCNQ, \(\hbox {TCNQ}^-\) and \(\hbox {TCNQ}^{2-}\) at CASPT2 level of theory (using basis set \(\hbox {S}_{\text {l}}\), active space L and shift = 0.20 H). The geometries are the optimized structures of the GS for each molecule
In 2018 Khvostenko et al. [19] found that TCNQ (in the gaseous phase) attaches electrons with energies of 0, 0.5 and 1.4 eV, i.e., similar energies as those obtained earlier by Compton and Cooper. Khvostenko et al. [19] concluded that the ion doublet formed at 0 eV is due to the second lowest core-excited Feshbach resonance, where an electron from the second highest OMO enters the LUMO together with the incident electron. The resulting doublet state is \(1^2\hbox {B}_{\text {1g}}\), which is calculated at \(-0.03~\hbox {eV}\) (using basis set \(\hbox {S}_{\text {l}}\) and active space L). Another possibility for the electron attachment at 0 eV (also suggested by Khvostenko et al.) is vibrational Feshbach resonances [19], where the molecule is vibrationally excited by the incident electron, which enters an UMO of the molecule. Close to 0 eV there are two such states, \(1^2\hbox {A}_{\text {u}}\) and \(2^2\hbox {B}_{\text {3u}}\), calculated at \(-0.23\) and \(-0.14~\hbox {eV}\), respectively.
The electron attachment at 0.5 eV is, according to Khvostenko et al., due to the third lowest core-excited Feshbach resonance [19], where an electron from the third highest OMO enters the LUMO together with the incident electron. The resulting doublet state is \(2^2\hbox {B}_{\text {2g}}\), which is calculated at 0.46 eV (using basis set \(\hbox {S}_{\text {l}}\) and active space L). The anomalously long-lived anion created at this energy is, according to Khvostenko et al., due to the formation of a quartet state via intersystem crossing [19]. Indeed, in this energy region there are two quartet states, \(1^4\hbox {B}_{\text {1g}}\) and \(1^4\hbox {B}_{\text {2g}}\), calculated at 0.76 and 0.86 eV, respectively. The energy gap between the doublet and the lowest quartet state is large (0.30 eV) using active space L and smaller using active space M (0.11 eV). However, by changing the geometry to the one optimal for the \(1^4\hbox {B}_{\text {1g}}\) state, the \(1^4\hbox {B}_{\text {1g}}\) state falls below \(2^2\hbox {B}_{\text {2g}}\) by 0.33 eV (see Fig. 6) and the two states seem to cross each other. In Fig. 6, the results of using the optimized geometry of the \(2^2\hbox {B}_{\text {2g}}\) state are included as well. These results indicate possible crossings of the \(2^2\hbox {B}_{\text {2g}}\) state with the three states at 0 eV (\(1^2\hbox {A}_{\text {u}}\), \(2^2\hbox {B}_{\text {3u}}\) and \(1^2\hbox {B}_{\text {1g}}\)) and therefore other explanations of the long-livedness of the anion.

The lowest electronic states of \(\hbox {TCNQ}^-\) at CASPT2 level of theory (using basis set \(\hbox {S}_{\text {l}}\), active space L and shift = 0.20 H). The lines are given for visual purposes. The bold lines represent the strongly absorbing \(1^2\hbox {B}_{\text {3u}}\) and \(2^2\hbox {B}_{\text {3u}}\) states. To the left on the x-axis \(\hbox {CC}_3\) and \(\hbox {CCC}_2\) are large and to the right \(\hbox {CC}_1\) and \(\hbox {CCC}_1\) are large
Finally, the electron attachment at 1.4 eV is, according to Khvostenko et al., due to the fourth and fifth lowest core-excited Feshbach resonances [19]. However, the fourth and fifth highest OMOs belong to \(\hbox {L}_\perp \setminus\)M (see Fig. 2). The core-excited Feshbach resonances from \(\hbox {L}_\perp \setminus\)M to LUMO are calculated at 2.26–2.49 eV, i.e., around 1 eV above the measured value. Instead the electron attachment at 1.4 eV could be due to core-excited Feshbach resonances from L\(\setminus\)M to LUMO. Three of these four states are given in Table 6 and the states are 3\(^2\hbox {B}_{\text {3u}}\), 3\(^2\hbox {B}_{\text {1g}}\) and \(2^2\hbox {A}_{\text {u}}\) calculated at 1.44, 1.65 and 1.74 eV (above the GS of TCNQ). The 3\(^2\hbox {B}_{\text {3u}}\) state is the most probable candidate and just below this doublet state there is a quartet state (\(1^4\hbox {B}_{\text {3u}}\)) at 1.31 eV, which could explain the long-livedness of the anion via intersystem crossing.
In 1991 Brinkman et al. [20] reported results from electron photodetachment spectroscopy studies on \(\hbox {TCNQ}^-\) in the gaseous phase. They found that photodetachment occurs in two energy bands. The high energy band at \(> 2.5~\hbox {eV}\) (with the highest peak around 3.1 eV) [20] is most likely due to the strongly absorbing transition \(1^2\hbox {B}_{\text {2g}}\rightarrow\) \(2^2\hbox {B}_{\text {3u}}\) calculated at 3.13\(-\)3.20 eV and where the \(2^2\hbox {B}_{\text {3u}}\) state is close in energy to the GS of TCNQ. The low energy band at 1.2–2.1 eV (with the highest peaks around 1.5 and 1.6 eV) [20] requires a deeper analysis. Brinkman et al. concluded that here more than one photon is involved in the detachment process [20]. The first excitation is due to the strongly absorbing transition \(1^2\hbox {B}_{\text {2g}}\rightarrow\) \(1^2\hbox {B}_{\text {3u}}\) calculated at 1.53\(-\)1.66 eV. Brinkman et al. [20] suggest a fast internal conversion to a vibrationally hot GS ion from where the ion excites to the \(2^2\hbox {B}_{\text {3u}}\) state.
In another experiment Brinkman et al. [20] used two different frequencies, one in the low energy band (1.80 eV) and one in the region between the two bands (2.41 eV). When increasing the intensity of the latter light the electron detachment increased dramatically also indicating a two-photon process [20]. Considering the energy of the latter light, another possibility for the second photon is the strongly absorbing transition \(1^2\hbox {B}_{\text {3u}}\rightarrow\) \(2^2\hbox {B}_{\text {2g}}\) calculated at 2.07\(-\)2.24 eV.
3.3 TCNQ dianion
The TCNQ anion has several bound states (maybe as many as five according to Fig. 5). For the TCNQ dianion there is experimental evidence of a long-lived state (millisecond timescale) [21, 22] and computational evidence of boundness [21]. According to the present calculations the TCNQ dianion has no bound states (see Fig. 5 and Table 7), but the Coulomb barrier may either way prevent the dianion from immediate electron detachment. The adiabatic EA of \(\hbox {TCNQ}^-\) is increasing from \(-0.56\) to \(-0.42~\hbox {eV}\) when improving the basis set from \(\hbox {S}_{\text {s}}\) to \(\hbox {L}_{\text {l}}\) by including more diffuse functions (see \(\hbox {EA}^-\) in Table 7). Since active space M forces the two extra electrons to reside in the LUMO of TCNQ, it seems unlikely that the EA should turn positive by improving the basis set further. For a further discussion, see the end of Sect. S4 in Supplementary Information. Table 7 also includes the energy (EA\(-\hbox {EA}^-\)) for the disproportionation reaction
which is estimated at 3.5 eV by Jonkman and Kommandeur [13]. \(\hbox {EA}-\hbox {EA}^-\) is stable with respect to basis set and is increasing moderately from 3.64 to 3.70 eV when improving the basis set from \(\hbox {S}_{\text {s}}\) to \(\hbox {L}_{\text {l}}\).
The excited states of \(\hbox {TCNQ}^{2-}\), below 5 eV, at CASPT2 level of theory are summarized in Table 8 for various combinations of the five basis sets and the three largest active spaces. It is clear from the table that large basis sets with a large content of diffuse functions make it possible for the dianion to “remove” one electron, which is energetically more favourable. Another observation is the necessity to use a large active space (L) in order to capture all possible low-lying states.
From absorption spectra (in solution) Jonkman and Kommandeur identified a strong transition at 2.56 eV [13], which is considerably less than calculated values of 3.04 eV (a weak transition to \(1^1\hbox {B}_{\text {2u}}\)) and 3.88 eV (a strong transition to \(1^1\hbox {B}_{\text {1u}}\)). By using UV–visible spectroelectrochemistry Bellec et al. obtained an absorption maximum at 3.71 eV [23] (the numerical value calculated from \(\lambda _{\text {max}} = 334\) nm from Panja et al. [22]), which agrees better with the calculated value of 3.88 eV.
4 Conclusions
In this study various properties of TCNQ and anions have been calculated with the CASPT2 method using several basis sets and active spaces. One conclusion is that basis set \(\hbox {S}_{\text {l}}\) of type ANO-S and size 3s2p(H)/4s3p2d(C,N) results in property values in agreement with those obtained with the larger basis set \(\hbox {L}_{\text {l}}\) of type ANO-L and size 3s2p1d(H)/5s4p3d2f(C,N). The accuracy of geometries is of the order of a few milliÅngström for bond lengths and a few tenths of a degree for bond angles. The accuracy of EA and IP is of the order of a tenth of an electronvolt. For excitation energies the accuracy is better by about a factor of ten and is of the order of a few hundredths of an electronvolt.
Further, it can be concluded that the active orbital space, denoted M, consisting of the three HOMOs and the three LUMOs (\(\pi\) and \(\pi ^*\) orbitals of mainly CC character) is sufficient for obtaining most of the studied properties. The exceptions concern higher excited states (4–5 eV) of \(\hbox {TCNQ}^-\) and \(\hbox {TCNQ}^{2-}\), where larger active spaces including also \(\pi\) (and \(\pi ^*\)) orbitals of CN character are required. The influence of \(\pi _\perp\) (and \(\pi _\perp ^*\)) orbitals (CN) is minor and they can be excluded from the active space.
Finally, it can be concluded that with the presented methodology it is possible to suggest explanations to several experimental observations on TCNQ and anions and to calculate fairly accurate values of properties of these molecules. For example, the calculated values of the IP and EA of TCNQ are 9.54 and 3.36 eV, respectively, using basis set \(\hbox {L}_{\text {l}}\). The experimental values are 9.61 [10] and \(3.383\pm 0.001\) [11] eV, respectively. Another example concerns the two lowest-lying excited singlet states, \(2^1\hbox {A}_{\text {g}}\) and \(1^1\hbox {B}_{\text {1u}}\), of TCNQ with calculated excitation energies of 3.12 (adiabatic) and 3.54 eV, respectively, using basis set \(\hbox {S}_{\text {l}}\). The two states may explain the shape of the fluorescence spectrum of TCNQ at 3–4 eV obtained by Chaki et al. [16], and data from the experiment suggest the excitation energies 3.01 eV for \(2^1\hbox {A}_{\text {g}}\) and 3.42 or 3.60 eV for \(1^1\hbox {B}_{\text {1u}}\) [16]. A third example has to do with electron attachment of TCNQ. According to experiment electrons with energies 0, 0.5 and 1.4 eV are attached [19]. The calculations suggest the values \(-0.03\), 0.46 and 1.44 eV (using basis set \(\hbox {S}_{\text {l}}\)) and the corresponding states \(1^2\hbox {B}_{\text {1g}}\), \(2^2\hbox {B}_{\text {2g}}\) and \(3^2\hbox {B}_{\text {3u}}\). The fourth and final example involves the TCNQ dianion and its second lowest-lying excited singlet state (\(1^1\hbox {B}_{\text {1u}}\)). The calculated transition energy from the GS is 3.88 eV (with basis set \(\hbox {S}_{\text {l}}\)), which agrees well with the experimental value of 3.71 eV [23].
References
Goetz KP, Vermeulen D, Payne ME, Kloc C, McNeil LE, Jurchescu OD (2014) Charge-transfer complexes: new perspectives on an old class of compounds. J Mater Chem C 2:3065–3076. https://doi.org/10.1039/c3tc32062f
Harvey PD, Fortin D (1998) Photoproperties of the polymeric \(\{\)[M(dmb)\(_2\)]Y\(\}_n\) materials: photoinduced intrachain energy and intermolecular electron transfers, and design of photovoltaic cells. Coord Chem Rev 171:351–354
Fujisawa J (2020) Interfacial charge-transfer transitions for direct charge-separation photovoltaics. Energies 13:2521–2534. https://doi.org/10.3390/en13102521
Acker DS, Hertler WR (1962) Substituted quinodimethans. I. Preparation and chemistry of 7,7,8,8-tetracyanoquinodimethan. J Am Chem Soc 84(17):3370–3374. https://doi.org/10.1021/ja00876a028
Ferraris J, Cowan DO, Walatka V Jr, Perlstein JH (1973) Electron transfer in a new highly conducting donor-acceptor complex. J Am Chem Soc 95(3):948–949
Aquilante F, Autschbach J, Carlson RK, Chibotaru LF, Delcey MG, De Vico L, Fdez Galván I, Ferré N, Frutos LM, Gagliardi L, Garavelli M, Giussani A, Hoyer CE, Li Manni Giovanni, Lischka H, Ma D, Malmqvist PÅ, Müller T, Nenov A, Olivucci M, Pedersen TB, Peng D, Plasser F, Pritchard B, Reiher M, Rivalta I, Schapiro I, Segarra-Martí J, Stenrup M, Truhlar DG, Ungur L, Valentini A, Vancoillie S, Veryazov V, Vysotskiy VP, Weingart O, Zapata F, Lindh R (2016) MOLCAS 8: new capabilities for multiconfigurational quantum chemical calculations across the periodic table. J Comput Chem 37:506–541
Pierloot K, Dumez B, Widmark P-O, Roos BO (1995) Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. IV. Medium size basis sets for the atoms H–Kr. Theor Chim Acta 90:87–114
Widmark P-O, Malmqvist P-Å, Roos BO (1990) Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. I. First row atoms. Theor Chim Acta 77:291–306
Rubio M, Merchán M, Ortí E, Roos BO (1995) Theoretical study of the electronic spectra of the biphenyl cation and anion. J Phys Chem 99:14980–14987
Ikemoto I, Samizo K, Fujikawa T, Ishii K, Ohta T, Kuroda H (1974) Photoelectron spectra of tetracyanoethylene (TCNE) and tetracyanoquinodimethane (TCNQ). Chem Lett 3:785–790
Zhu G-Z, Wang L-S (2015) Communication: vibrationally resolved photoelectron spectroscopy of the tetracyanoquinodimethane (TCNQ) anion and accurate determination of the electron affinity of TCNQ. J Chem Phys 143:221102. https://doi.org/10.1063/1.4937761
Iimori T, Ishikawa T, Torii Y, Tamaya H, Nakano H, Kanno M (2020) Effect of rigidity of microenvironment on fluorescence of 7,7,8,8-tetracyanoquinodimethane (TCNQ). Chem Phys Lett 738:136912. https://doi.org/10.1016/j.cplett.2019.136912
Jonkman HT, Kommandeur J (1972) The UV spectra and their calculation of TCNQ and its mono- and di-valent anion. Chem Phys Lett 15(4):496–499
Tamaya H, Nakano H, Iimori T (2017) 7,7,8,8-Tetracyanoquinoidimethane (TCNQ) emits visible photoluminescence in solution. J Lumin 192:203–207. https://doi.org/10.1016/j.jlumin.2017.06.051
Khvostenko OG, Kinzyabulatov RR, Khatymova LZ, Tseplin EE (2017) The lowest triplet of tetracyanoquinodimethane via UV-vis absorption spectroscopy with Br-containing solvents. J Phys Chem A 121:7349–7355. https://doi.org/10.1021/acs.jpca.7b05623
Chaki N, Muramatsu S, Iida Y, Kenjo S, Inokuchi Y, Iimori T, Ebata T (2019) Laser spectroscopy and lifetime measurements of the S\(_{1}\) state of tetracyanoquinodimethane (TCNQ) in a cold gas-phase free-jet. ChemPhysChem 20:996–1000. https://doi.org/10.1002/cphc.201900214
Haller I, Kaufman FB (1976) Spectra of tetracyanoquinodimethane monovalent anion: vibrational structure and polarization of electronic transitions. J Am Chem Soc 98(6):1464–1468
Compton RN, Cooper CD (1977) Negative ion properties of tetracyanoquinodimethan: electron affinity and compound states. J Chem Phys 66(10):4325–4329. https://doi.org/10.1063/1.433743
Khvostenko OG, Khatymova LZ, Lukin VG, Kinzyabulatov RR, Tuimedov GM, Tseplin EE, Tseplina SN (2018) Long-lived negative molecular ions of TCNQ formed by the resonant capture of electrons with above zero energies. Chem Phys Lett 711:81–86. https://doi.org/10.1016/j.cplett.2018.09.032
Brinkman EA, Günther E, Brauman JI (1991) Bound excited electronic states of anions studied by electron photodetachment spectroscopy. J Chem Phys 95:6185–6187. https://doi.org/10.1063/1.461589
Nielsen SB, Nielsen MB (2003) Experimental evidence for the 7,7,8,8-tetracyano-p-quinodimethane dianion in vacuo. J Chem Phys 119:10069–10072. https://doi.org/10.1063/1.1618216
Panja S, Kadhane U, Andersen JU, Holm AIS, Hvelplund P, Kirketerp M-BS, Nielsen SB, Støchkel K, Compton RN, Forster JS, Kilså K, Nielsen MB (2007) Dianions of 7,7,8,8-tetracyano-p-quinodimethane and perfluorinated tetracyanoquinodimethane: information on excited states from lifetime measurements in an electrostatic storage ring and optical absorption spectroscopy. J Chem Phys 127:124301. https://doi.org/10.1063/1.2771177
Bellec V, De Backer MG, Levillain E, Sauvage FX, Sombret B, Wartelle C (2001) In situ time-resolved FTIR spectroelectrochemistry: study of the reduction of TCNQ. Electrochem Commun 3:483–488
Acknowledgements
The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC), partially funded by the Swedish Research Council through grant agreement no. 2018-05973. The author also gratefully acknowledges the comments and suggestions of reviewers.
Funding
Open access funding provided by Karlstad University. No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
All work was performed by KA (single author).
Corresponding author
Ethics declarations
Conflict of interest
The author has no competing interests to declare that are relevant to the content of this article.
Consent for publication
The author has obtained consent from the department where the work has been carried out.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Andersson, K. Molecular properties of TCNQ and anions. Theor Chem Acc 142, 58 (2023). https://doi.org/10.1007/s00214-023-02998-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-023-02998-7