
Abstract
This is an overview of the metabolic reactions of drugs, natural products, physiological compounds, and other (general) chemicals catalyzed by flavin monooxygenase (FMO), monoamine oxidase (MAO), NAD(P)H quinone oxidoreductase (NQO), and molybdenum hydroxylase enzymes (aldehyde oxidase (AOX) and xanthine oxidoreductase (XOR)), including roles as substrates, inducers, and inhibitors of the enzymes. The metabolism and bioactivation of selected examples of each group (i.e., drugs, “general chemicals,” natural products, and physiological compounds) are discussed. We identified a higher fraction of bioactivation reactions for FMO enzymes compared to other enzymes, predominately involving drugs and general chemicals. With MAO enzymes, physiological compounds predominate as substrates, and some products lead to unwanted side effects or illness. AOX and XOR enzymes are molybdenum hydroxylases that catalyze the oxidation of various heteroaromatic rings and aldehydes and the reduction of a number of different functional groups. While neither of these two enzymes contributes substantially to the metabolism of currently marketed drugs, AOX has become a frequently encountered route of metabolism among drug discovery programs in the past 10–15 years. XOR has even less of a role in the metabolism of clinical drugs and preclinical drug candidates than AOX, likely due to narrower substrate specificity.
Similar content being viewed by others

Introduction
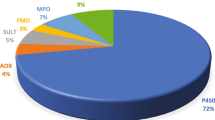
In our previous reports, we analyzed the properties and participation of human enzymes in the metabolism of physiological and xenobiotic compounds, including natural products (Rendić and Guengerich 2012, 2015, 2021). The analysis showed an overwhelming participation of the cytochrome P450 (P450, CYP) enzymes (~ 95%) in the metabolism of the compounds. P450 enzymes catalyze a great number of metabolic reactions and have important effects on the biological activities (physiologic, therapeutic, and/or toxic) of xenobiotics such as drugs, natural products, “general chemicals” (e.g., pesticides, pro-carcinogens, various environmental chemicals), and physiological compounds. In addition to P450s, other enzymes such as microsomal flavin-containing monooxygenase (FMO), monoamine oxidase (MAO), and aldehyde oxidase (AOX) enzymes participate in the metabolism of these compounds, although to a lower extent (~ 2%, 1%, and 2%, respectively). Other oxidoreductase enzymes participate to an extent of < 1% (Rendić and Guengerich 2012, 2015). The mechanism, kinetics, and metabolic properties of P450 enzymes (Guengerich 2022) and oxidative metabolism, and the gene regulation of non-cytochrome P450 enzymes have been discussed recently (Pang et al. 2022). In the present paper, we discuss mechanisms and metabolic properties of human FMO, MAO, NAD(P)H quinone oxidoreductase (NQO), molybdenum-containing hydroxylases (AOX and xanthine oxidoreductase (XOR) enzymes) in the oxidation of drugs, physiological and natural products, and other (general) chemicals as substrates and inhibitors of these enzymes, in the context of their participation in the metabolism of the compounds and also possible toxic effects that might result from oxidation and reduction reactions.
The review is divided into four parts, addressing these four sets of enzymes. Only the human enzymes are discussed and data are only presented for these. The experimental data are presented in tables, and the published kinetic values were categorized according to the values and effects presented in Table 1.
Results and discussion
Flavin-containing monooxygenase (FMO)
As reported previously, FMOs participate in ~ 2% of reactions involved in the metabolism of xenobiotics, natural products, and physiological compounds (Rendić and Guengerich 2015). There is a higher fraction of FMO enzymes involved in the metabolism of general chemicals when compared to the metabolism of drugs, natural products, or physiological compounds. The reactions catalyzed by FMO enzymes are predominately detoxication reactions and include N-, S-, P-, and Se-atom oxygenations, depending on the substrate structure (Table 2).
In some cases, FMOs are involved in the activation of substrates to toxic products. When calculating the participation of FMO enzymes in activation reactions, we found that FMOs participate in ~ 1% in the reactions, catalyzed predominately by FMO1 and FMO3 and related to the formation of N- or S-oxides. These results show equal participation of FMO enzymes in detoxication reactions and the formation of potentially toxic products. For comparison, P450 enzymes participate in ~ 66% of reactions involving the formation of toxic products and in 95% of the overall oxidations and reductions of xenobiotics and natural products (Rendić and Guengerich 2012, 2015).
Enzymes
In the literature, different terminology has been used for these enzymes: FMO(s), FAD-containing amine oxidases, microsomal oxygenases containing flavin, and mixed-function microsomal amine oxidases. The enzyme was discovered by the late Prof. Daniel Ziegler, who worked with the enzyme from swine, and frequently scientists simply referred to this as “Ziegler’s Enzyme” for many years (Pettit et al. 1964; Ziegler 1988, 2002; Ziegler and Pettit 1966). The enzymes are found in the endoplasmic reticulum of most organs and tissues, predominately in the liver and in the lungs, kidneys, digestive tract, brain, and others (Dannan and Guengerich 1982).
The human FMO enzymes are characterized by the following features: FMO enzymes contain 1 mol FAD/mol enzyme, Mr ~ 65 kDa, and about 535 amino acids. In humans, 11 FMO genes have been identified, encoding five active FMOs (FMO1–5) and six pseudogenes. FMOs are differentially distributed in organs, and the amino acid sequences of the orthologous forms of the enzymes in different animal species are 80–90% similar (Cashman 2004; Henderson et al. 2014; Hines 2006; Huang et al. 2021; Koukouritaki et al. 2002; Krueger et al. 2009; Nagashima et al. 2009; Phillips and Shephard 2017, 2020; Shimizu et al. 2011, 2015; Ziegler 1988).
The most frequently represented reactions catalyzed by FMO enzymes are N- and S-oxygenations (Elfarra 1995; Furnes and Schlenk 2004; Krause et al. 2003), although some oxygenations are known for phosphorus and selenium atoms (Hodgson and Levi 1992; Jones et al. 2017; Rooseboom et al. 2001) (Table 2).
FMO1 is the major form expressed in the neonatal liver and kidneys and small intestine of adults. FMO2 is the most abundant in human lungs and is expressed in the liver and kidneys at a minor level. The non-functional variant FMO2*2 is predominant in humans, but in some ethnic groups that have been studied (Afro- and Hispanic-Americans) the variant FMO2*1 is present (Krueger et al. 2005). The developmental expression pattern for human hepatic FMO1 and FMO3 shows that relatively high levels of FMO1 expression are observed throughout prenatal development, in particular during the embryonic period, but FMO3 is essentially absent in the fetal liver. In the human liver, FMO3 is the most abundant enzyme and predominantly oxidizes tertiary amines, including a large number of clinically important drugs and amines ingested in food. FMO3 is a highly polymorphic enzyme, and polymorphism is related to a rare hereditary disorder of the inability to metabolize trimethylamine (a disorder called trimethylaminuria) (Phillips et al. 1995). FMO3 enzymes have been associated with some clinically relevant drug–drug or drug–chemical interactions because a large number of clinically important drugs (as well as natural products, e.g., indoles, tyramine, trimethylamine) possess amine structures. FMO4 is present at a low level in multiple tissues (e.g., liver, kidneys, brain). FMO5 is highly expressed in the adult human liver.
There are significant differences between individuals and ethnic groups in both expression and functional activity. Genetic polymorphism in the human FMO genes (in major part associated with the FMO3 gene) may lead to changes in N- and/or S-oxygenations of drugs, xenobiotics, and endogenous substances.
Following the P450s (Rendić 2002; Rendić and Di Carlo 1997), FMOs are the most important enzymes involved in the monooxygenation of amine-containing xenobiotics or amines that are formed during the biotransformation of drugs, general chemicals, natural products, and physiological compounds (Rendić and Guengerich 2015). Reactions catalyzed by FMO enzymes have been generally considered as detoxications but there are exceptions to this rule. All FMO enzymes possess the structural features by which FAD and NADPH are bound. Important endogenous roles for the FMO family have been suggested, including the regulation of cellular stress resistance and major cellular metabolic activities that involve central carbon metabolism (Huang et al. 2021; Krueger and Williams 2005).
Typical substrates include aliphatic, basic amines and some aromatic primary amines, secondary amines, tertiary amines, N-arylamides, heteroaromatic amines, hydroxylamines, and hydramines (e.g., metamizole, N,N-dimethylaniline). Substrates of FMOs (e.g., N-alkyl arylamines including N-methylaniline and N,N-dimethylaniline) can be substrates for both FMO and P450 enzymes, depending on the structural and electronic properties of substituents and basicity of the amines. FMO enzymes predominantly catalyze N-oxidation of most of the cyclic and acyclic secondary amines (Hanson et al. 2010) (Tables 3, 4, 5), while P450s tend to catalyze N-dealkylation reactions because of the chemical mechanisms involved (Seto and Guengerich 1993).

Human FMO enzymes participating in the metabolism of drugs (data calculated for major and minor enzymes from Table 3; a total of 114 drugs used in calculations)
`
FAD, NADPH, and O2 are required for the FMO catalyzed reactions, but the FAD is tightly bound to the enzyme and does not need to be added (i.e., acts as a prosthetic group instead of a cofactor (Dixon and Webb 1964)). Of the human FMO enzymes, FMO3 is the prominent enzyme that converts nucleophilic heteroatom-containing chemicals, drugs, and xenobiotics to more polar materials, which are generally more efficiently excreted in the urine. The substrate specificity for FMO3 is distinct from that of FMO1. Of the five FMO families, FMO1 and FMO3 are the most prevalent in drug metabolism in humans (Fig. 1, Table 3). A similar participation pattern of the enzymes was found for general chemicals (Table 4). For natural products and physiological compounds, the most prominent enzymes were FMO3 and FMO1, followed by FMO2 and FMO4, with low participation of FMO5 (Table 5).
In general, FMO enzymes have not been reported to be very inducible. However, induction of FMO4 and FMO5 cDNA has been reported in human hepatocytes by the drug rifampicin (Rae et al. 2001), and the tricyclic antidepressants imipramine and chlorpromazine were reported to upregulate recombinant FMO3 catalyzed methimazole S-oxidation in a concentration-dependent manner (Adali et al. 1998, 1999; Cherrington et al. 1998) (Table 3). In addition, FMO5 mRNA was upregulated in HepG2 cells by the natural product (herbal medicine) St. John’s wort and its active component hyperforin, as well as by the synthetic progestin R5020 in a breast cancer cell line that stably expresses B-receptors (YB cells) (Miller et al. 1997).
Inhibition of FMO3 was reported by dietary indoles such as indole-3-carbinol (contained in Brussels sprouts (Cashman et al. 1999a)) and decreased expression and activity of FMO3 was observed for endogenously formed nitric oxide (Ryu et al. 2004) (Table 6).
The potential for adverse reactions due to drug–drug interactions is less likely for drugs predominately metabolized by FMO than for P450 enzymes. However, physiological factors can influence FMO function, and this may have clinical implications (Cashman and Zhang 2006; Ryu et al. 2004). For instance, in the case of mammalian FMO3, which does not appear to be very inducible (vide supra), inter-individual variations in FMO3-dependent metabolism of drugs, other chemicals, and endogenous compounds are more likely to be caused by genetic and ethnic polymorphisms (Cashman 2002b; Cashman et al. 2000; Cashman and Zhang 2002; Hisamuddin and Yang 2007). However, human FMO enzymes can activate drugs (e.g., antibiotics, antibacterial, antitubercular, CNS stimulants), natural products, and general chemicals to toxic products, resulting in adverse reactions (Table 7).
Reactions
Human FMO3 N-oxygenates primary, secondary, and tertiary amines but only human FMO1 is highly efficient at N-oxygenating tertiary amines. Both human FMO1 and FMO3 S-oxygenate many nucleophilic sulfur-containing substrates, and in some cases, reactions proceed with high stereoselectivity (Cashman 2000).
N-oxygenations
The N-oxygenation reactions of primary amines catalyzed by FMO enzymes, which occur without splitting the C–N bond, can result in the formation of toxic nitroso compounds. The reaction usually creates potentially toxic hydroxylamines in the first step, which can be further oxidized into oxime and nitroso compounds (Fig. 2) (e.g., sulfamethoxazole and amphetamine N-oxidation). N-Oxygenations of secondary amines, e.g., cyclic and acyclic secondary amines, are catalyzed by FMO enzymes, and those of N-alkyl- and N-aryl amines are generally catalyzed by both FMO and P450 enzymes (e.g., N-methylamphetamine, Tables 3, 7).

General reaction of N-oxygenation of primary amines by FMO
S-oxygenations
Compounds containing a sulfur atom as a part of the structure are present in physiological compounds such as amino acids and derivatives (e.g., cysteine, methionine, glutathione), lipids, and enzyme cofactors (e.g., biotin, thioredoxin, lipoic acid, coenzyme A) and in natural products (e.g., the toxin amanitin and various compounds isolated from onions, radishes, and watercress). The characteristic odor and healing properties of plants of the genus Allium are attributed to sulfur-containing compounds. A number of drugs and general chemicals (e.g., solvents, insecticides) are substrates for S-oxygenation.
S-Oxygenation reactions (Fig. 3) occur by mechanisms similar to N-oxygenation (vide infra), catalyzed by FMO enzymes (also called sulfoxidases). In addition, P450s may be involved (Rendić 2002). Substrates in these reactions include thiocarbamides, thiones, thioamides, sulfides (aromatic and aliphatic), thiols, and mercaptopurines (Table 2). Some intermediates formed in S-oxidations (e.g., sulfenes, sulfines) are reactive and potentially toxic because they can react with proteins and lipids in cells (Table 7). The final products (S-oxides) of the S-oxygenation reactions may also exert toxic effects (Furnes and Schlenk 2004; Shimizu et al. 2007; Siddens et al. 2014).

Typical oxygenation reaction catalyzed by FMO enzymes, where R denotes part of the molecule and X is a heteroatom, usually N or S
Mechanism of oxygenation of heteroatoms (N- and S-oxygenation)
Compounds possessing a soft nucleophilic heteroatom are substrates of FMO enzymes. Structure–activity studies suggest that in addition to nucleophilicity, the size and charge of potential substrates are important parameters limiting access to the enzyme-bound hydroxylating intermediate form of the enzyme (4a-hydroperoxide) (Ziegler 2002).
The mechanism of oxygenation of nucleophilic groups catalyzed by FMO enzymes is presented in the context of the following three steps (Phillips and Shephard 2019; Siddens et al. 2014; Ziegler 1988) (Fig. 4): (1) NADPH binds to the enzyme and reduces FAD to FADH2 (a rapid reaction). The result is the formation of a ternary complex (Enzyme-FADH2-NADP+). (2) FADH2 binds molecular oxygen, as a co-substrate, and produces a relatively stable C4a-hydroperoxyflavin (also a rapid reaction). The cofactor NADP+ remains attached to the enzyme during the reaction, stabilizing the complex. (3) The C4a-hydroperoxyflavin is a strong electrophile and can oxygenate a nucleophilic group, with an attack of activated oxygen (electrophile) atom from the C4a-hydroperoxyflavin molecule on the nucleophilic atom (nitrogen, sulfur, phosphorus) in the substrate molecule, without prior binding of the substrate to the enzyme. The transfer of the oxygen atom to a substrate (reaction of monooxygenation of the substrate) results in the formation of 4a-hydroxyflavin. (3a) If there is no substrate that can be oxygenated near the enzyme, the C4a-hydroperoxyflavin releases H2O2, the oxidized form of the enzyme, and NADP+. (4) Removal of the water molecule (dehydration) (and release of NADP+ from the complex) regenerates the oxidized form of the enzyme (slow reaction).

Oxygenation of substrates with FMO enzymes
Access to the active form of oxygen on the prosthetic group (flavin) is observed for non-ionizable lipophilic amines and amines that are found in the form of mono-cations at physiological pH (step 3). Amines that possess two cationic groups at physiological pH (and amines with one or more anionic groups) cannot approach the active site and are not preferred substrates for FMO enzymes. These structural requirements prevent many endogenous substances from being substrates of the enzymes.
The catalytic cycle and mechanism of monooxygenation catalyzed by FMO enzymes differ significantly from the mechanism that P450s generally use in catalysis. The latter mechanism takes place via an intermediate reactive form of oxygen (FeO3+) that involves radical species (Ziegler 2002). An interesting kinetic feature of the FMO mechanism is that (in general, with a given FMO) the kcat does not vary much and the Km varies among substrates, and the Km is not a measure of inherent affinity for the enzyme (Kd).
Oxidations of ketones by FMOs in Baeyer–Villiger oxidations
FMOs, like other flavin-based monooxygenases in general, utilize flavin 4a-hydroperoxides in their mechanisms (Walsh 1979), with the hydroperoxide acting as an electrophile to oxygenate nitrogen or sulfur (Fig. 4). Flavin 4a-hydroperoxides can also act as nucleophiles, when deprotonated, catalyzing Baeyer–Villiger reactions with carbonyls (Fig. 5) (Walsh and Chen 1988). This is an important reaction in some bacteria, allowing the breaking of a (ketone) ring structure to generate acidic products that can be degraded (e.g., by fatty acid oxidation enzymes) for use as a carbon source. An example of a mammalian enzyme that does this is human FMO5 (Fiorentini et al. 2016; Walsh 1979).

FMO5 appears to be adapted for the nucleophilic Baeyer–Villiger chemistry. Examples of reactions attributed to FMO5 are presented in Fig. 5, including four drugs (Fiorentini et al. 2016, 2017; Lai et al. 2011; Meng et al. 2015). This is an interesting reaction, in that the lactones can be readily cleaved to open-chain products by the action of esterases or by non-enzymatic base-catalyzed hydrolysis (Fig. 6).

Some Baeyer–Villiger C–C oxidations of drugs catalyzed by FMO (Guengerich and Yoshimoto 2018)
Thus, a C–C oxygen insertion reaction can be utilized to cleave a C–C bond (Guengerich and Yoshimoto 2018). Recently an alternate flavin mechanism involved in some oxygenations has been shown to involve a flavin N5-oxide (Teufel et al. 2015), but it is unknown whether this intermediate could also be involved in Baeyer–Villiger oxidations.
Substrates and reactions catalyzed by human FMO enzymes
Substrates contain nucleophilic heteroatoms nitrogen, sulfur, phosphorus, or selenium. As already pointed out, the best substrates are cyclic and acyclic amines that are not ionized at physiological pH (Kim and Ziegler 2000; Rettie et al. 1994; Rooseboom et al. 2001; Ziegler 1988). Many drugs possessing nucleophilic heteroatoms in their structure are substrates of these enzymes (Phillips and Shephard 2017; Sawada and Yokosawa 1991; Yamazaki et al. 2014; Cashman, 2000) (Table 3), as well as general chemicals (Table 4) and natural products and physiological compounds (Table 5). Additional substrates are iodides and boron-containing compounds (Jones and Ballou 1986). Drug oxidations are the most studied group of reactions with human FMOs (Tables 3, 4, 5), followed by general chemicals and physiological compounds. In addition, FMO-catalyzed reactions are predominately detoxication reactions, with some examples of contributions of the reactions to bioactivation and formation of toxic products or intermediates (Table 7) (Cashman 2002a).
In many of the cases, the results presented were obtained using purified and recombinant human enzymes expressed in different systems. Although information obtained by studies in such systems is of great value for further research, the results obtained may not be representative of the most important processes occurring in cells or tissues. In addition, some FMO-catalyzed reactions can also be catalyzed by other enzymes in cells, e.g., P450 (Tables 3, 4, 5) and AOX enzymes (Table 3). The participation of P450 enzymes in the metabolism of the FMO substrates by N-oxidation may be a minor contribution to overall metabolic reactions of the compound in some cases (e.g., N-oxygenations of cediranib, C-1311, benzydamine, selegiline, dapsone (Table 3)) or might predominate in the overall metabolic pathway of a compound, e.g., disulfoton, methiocarb, phorate, sulprofos (Table 4), M-04579 (Table 3). Dapsone N-oxygenation is, for instance, catalyzed by several P450 enzymes (P450s 1A2, 2C, 2D6, 2E1, 3A4) with high or intermediate Km values, contributing to its activation to toxic N-hydroxylamine formation (Li et al. 2003; Winter et al. 2000). Dapsone was, in addition, reported to be a substrate-dependent activator of P450 2C9 enzyme activity and thus activating its own oxidation (Hummel et al. 2004). However, P450-catalyzed N-oxidation of dapsone appears to be of minor importance to its overall metabolism (Rendić and Guengerich 2021). In addition to being substrates of P450 enzymes, FMO substrates can also be either strong P450 inhibitors with the potential for drug–drug interactions (e.g., cimetidine Rendić et al. 1983, 1979) (Fig. 9), or weak inhibitors of P450 enzymes with minor potential for inducing drug–drug interactions (e.g., ranitidine) (Fig. 10) (Rendić et al. 1982, 1983).
An additional characteristic of the reactions catalyzed by FMO enzymes is stereoselectivity which, depending on the substrate, can occur with high or low selectivity for a substrate or product formed. Stereoselectivity can occur regarding both N- and S-oxygenations (Tables 3, 4, 5). For instance, no selectivity is observed for product formation by N-oxygenation of two geometric isomers of clomiphene, but high regioselectivity in the conversion of only one of the two isomers of GSK5182 has been reported (the Z-isomer) (Table 3). For sulindac sulfide (a sulindac metabolite), a high degree of stereoselectivity towards the R-isomer was observed (Table 3), and stereoselectivity for N-oxidation is reported for deprenyl (Table 3) and trans-(S)-(-)-N-1′-nicotine oxide (Table 5). Stereoselectivity was also reported for S-oxidation of the L-isomer (FMO4) and the D-isomer of methionine (FMO3) (Table 5), N-oxygenation of (S)-N-methylamphetamine, and S-oxygenation of (R)-sulindac sulfide (Table 3).
In addition to their interaction with FMO and/or P450 enzymes, the drugs/chemicals that interact with FMOs can also induce or inhibit the activity of drug transporters. Clozapine, for instance, is a substrate for FMO3-catalyzed N-oxygenation (Table 3) and also a substrate and/or inhibitor of P450 enzymes (Rendić 2002). The drug is a substrate in P450 1A2, 2D6, and 3A4 catalyzed N-demethylations, and P450 1A2 and 3A4 catalyzed N-oxygenation (Fig. 7) (Buur-Rasmussen and Brøsen 1999; Murray et al. 2018; Tugnait et al. 1999). Furthermore, clozapine N-oxide is reported to be an inhibitor of P450 2B6 and 2C19 enzymes (Giri et al. 2017). In addition, clozapine was reported to be an inhibitor of the drug transporter P-glycoprotein, with the potential to affect the pharmacokinetic properties of co-administered drugs (Liu et al. 2021b; Wang et al. 2006). This example illustrates the complexity of predicting possible drug–drug interactions when a drug is a substrate and/or inhibitor of multiple drug-metabolizing enzymes and/or drug transporters, the properties which are also affected by the properties of the co-administered drug(s).

In the reactions of drug substrates of FMO enzymes, the oxygenated products produced are usually more polar (Table 3) and may be more rapidly eliminated from the body or maybe substrates in conjugation reactions. As shown in Table 3, drugs belonging to several important therapeutical categories are substrates of FMO enzymes, e.g., anticancer (cediranib), antiulcer (cimetidine, ranitidine), antidepressants, CNS stimulants (amphetamine and derivatives), and antibacterial drugs (sulfamethoxazole). In some cases, substrates of the FMO enzymes are metabolites produced by the catalytic activity of other enzymes, e.g., S-methyl esonarimod, sulindac sulfide, 3-hydroxynabumetone, tazarotenic acid, and S-methyl-N,N-diethyldithiocarbamate (a disulfiram metabolite). The data also show that in humans FMO3 and FMO1 are the most frequently represented among the FMO enzymes catalyzing the metabolism of drugs (Fig. 1), as well as with the general chemicals (possessing a tertiary amine group, thiols, thiolates, sulfides, thiourea derivatives, and organothiophosphate insecticides) (Table 4), and natural products (e.g., (S)-nicotine, phenethylamine, cysteamine, and methionine-containing compounds) (Table 5). In the case of natural compounds as substrates the enzymes often exert stereoselectivity for a particular isomer (e.g., l-methionine as substrate) or for the formation of a particular isomer (e.g., formation of trans-(S)-(-)-N-1′-nicotine oxide). Also, the products of the reactions are, in some cases, more toxic than the parent compounds (Table 7). Prominent among the reactions producing reactive metabolites are those involving thiourea and derivatives (e.g., thiourea, thioacetazone, ethionamide) as substrates. The metabolite(s) of the compounds are potentially carcinogenic compounds formed by the oxygenation of a sulfur atom. Exposure to thiourea, for instance, can damage bone marrow, causing reductions in the number of red blood cells, white blood cells, and/or blood platelets. Thiourea and derivatives are oxidized by FMO1, FMO2, and FMO3 enzymes with the formation of sulfinic and sulfenic acids (Tables 4, 7); however, the toxicity of thiourea and its derivatives was assigned to the activity of the FMO3 enzyme (Smith and Crespi 2002). In some cases, the same activation reaction (i.e., S-oxidation) might also be catalyzed by P450 enzymes (e.g., activation of the insecticides methiocarb and aldicarb) (Costa et al. 2003; Fujino et al. 2016) (Tables 4, 7).
Examples of substrates and reactions resulting in the formation of non-toxic metabolites
Nicotine (stimulant, agonist at nicotinic acetylcholine receptors)
Nicotine N′-oxygenation is one of the direct detoxication pathways for nicotine, accounting for 4–7% of total urinary nicotine metabolites (Fig. 8). Several FMO enzymes catalyze the reaction, and the role of this reaction increases in subjects with deficient P450 2A6 activity. While all of the recombinant FMO enzymes can mediate nicotine N-oxide formation, FMO1, FMO2, and FMO3 exhibit the highest activity. It was reported that oxidation of nicotine in humans occurs with a certain degree of stereoselectivity, and the formation of trans-nicotine N-1′-oxide catalyzed by FMO3 has been reported as a highly stereoselective probe of human FMO3 (Cashman et al. 1995) (Table 5). In other animal species (rat, swine, rabbit) the oxidation is catalyzed by FMO1, and approximately the same amounts of nicotine isomers are formed (Cashman 2000; Cashman et al. 1992; Park et al. 1993; Perez-Paramo et al. 2019) (Fig. 8).

Oxygenation of nicotine by FMO enzymes
Cimetidine (histamine H2 receptor antagonist)
Cimetidine S-oxygenation has been suggested as a stereoselective functional probe of human FMO3 activity (Cashman 2000; Cashman et al. 1995; Lu et al. 1998). FMO1 produces more of the S-oxide-(−)-enantiomer and FMO3 generates mainly the S-oxide-( +)-enantiomer (with no activity for FMO5) (Hai et al. 2009) (Table 3) (Fig. 9).

Oxygenation of cimetidine by FMO enzymes
Ranitidine (histamine H2 receptor antagonist)
The FMO enzymes in human liver microsomes formed the S- (13–18%) and N-oxides (66–76%) as products. Recombinant human FMO1, FMO2, FMO3, and FMO5 all formed the N-oxide, with FMO3 as the major enzyme. S-Oxide formation catalyzed by FMO3 was reported to be very low, as well as N-oxide formation by FMO5. Based on these results, it has been suggested that ranitidine N-oxide formation can be used as an in vivo probe to determine hepatic FMO3 activity (Cashman 2000; Chung et al. 2000a, b; Overby et al. 1997) (Table 3, Fig. 10).

Oxygenation of ranitidine by FMO enzymes
Chlorpromazine (antipsychotic, phenothiazine)
The N-oxide derivative of chlorpromazine is a stable and pharmacologically active chlorpromazine metabolite. Chlorpromazine is a substrate for both FMO and P450 enzymes (Table 3, Fig. 11). In humans, it is metabolized to 7-hydroxy-N-desmethylchlorpromazine in reactions catalyzed by multiple P450 enzymes (Rendić 2002). Chlorpromazine N-oxide, formed by FMO1 as a major enzyme, is oxidized to a sulfoxide by P450 enzymes (chlorpromazine N,S-dioxide formation) and generates additional metabolites (7-hydroxy, N-desmethyl, 7-hydroxy-N-desmethyl, and N-desmethyl sulfoxide derivatives). The in vivo metabolites are formed in the order: chlorpromazine N-oxide > chlorpromazine sulfoxide > 7-hydroxychlorpromazine > norchlorpromazine sulfoxide > norchlorpromazine. Chlorazepine N-oxide was also reduced back to chlorpromazine (Beckett et al. 1988; Cashman et al. 1993b; Chetty et al. 1994; Jaworski et al. 1990; Ohmiya and Mehendale 1984). This example illustrates the complexity of drug metabolism and activity when metabolic reactions are components of multiple metabolic pathways and effects (Adali et al. 1998, 1999).

Chlorpromazine oxygenations by FMO and P450 enzymes
Dimethylamphetamine (CNS stimulant and anorectic)
N,N-Dimethylamphetamine is an N-methylamphetamine analog with weaker central nervous system stimulant activity. One of the metabolites of dimethylamphetamine in humans is the stable N-oxide (Fig. 12), possessing much lower neurotoxic potential compared to amphetamine and N-methylamphetamine (Lee et al. 2009a, b; Ricaurte et al. 1989). The reaction is catalyzed by FMO1 (as the major enzyme) and FMO3. The reaction catalyzed by FMO1 was reported to be enantioselective for L-N-oxide formation (Table 3).

N-Oxygenation of dimethylamphetamine
Sulfides
Sulfide drugs and general chemicals, or their metabolites, are oxidized to S-oxides by human FMO enzymes (Tables 3, 4). The reaction of sulfide oxidation showed differential structurally dependent stereoselectivity. For instance, sulfoxidation of methyl and ethyl p-tolyl sulfides by recombinant human FMO3 proceeds with little stereochemical preference, whereas sulfoxidation of the n-propyl and n-butyl homologs demonstrated increasing selectivity for formation of the (R)-sulfoxide. In addition, S-oxidation of methyl-p-tolyl sulfide by FMO1 was stereoselective for (R)-sulfoxide formation (Table 4).
Examples of reactions resulting in the formation of toxic metabolites
N-oxygenations
Trimethylamine (an agonist of human TAAR5 (trace amine associated receptor 5))
In humans, FMO3 is polymorphic and can be associated with clinically relevant drug–drug or drug–chemical interactions. FMO3 enzyme polymorphism in humans is related to a rare hereditary disorder of the inability to metabolize trimethylamine. This leads to the accumulation of trimethylamine and to a disorder called trimethylaminuria, which results in a so-called “fish odor” syndrome (Al-Waiz et al. 1987; Dolphin et al. 1997; Phillips et al. 1995).
In humans, trimethylamine is formed mainly from the metabolism of phosphatidylcholine/choline, carnitine, betaine, dimethylglycine, and ergothioneine from food by intestinal microflora in the colon. It is absorbed into the bloodstream and transformed into trimethylamine N-oxide (TMAO) (Fig. 13) by hepatic FMO1 and FMO3 but can be also converted to (mono)methylamine, dimethylamine, and ammonia within the colon. Although the oxidation of trimethylamine to its N-oxide had been known for years, the detrimental effects of TMAO were discovered only recently. Elevated TMAO plasma levels have been correlated with an elevated risk for cardiovascular disease (atherosclerosis and thrombosis) and were implicated in reverse cholesterol transport and glucose and lipid homeostasis. High plasma TMAO levels were also positively associated with the incidence of gallstone disease in humans (Gatarek and Kaluzna-Czaplinska 2021; Papandreou et al. 2020; Schneider et al. 2018; Steel et al. 1988; Zhu et al. 2018). The major enzyme involved in trimethylamine N-oxygenation is FMO3 (Table 5). In some individuals, due to the genetic polymorphism of FMO3, decreased trimethylamine oxidation occurs (Fig. 13) with an accumulation of trimethylamine resulting in “fish odor.” Trimethylamine N-oxide accounts for almost 98% of the administered dose of the parent compound trimethylamine. However, in individuals deficient in the FMO3 the formation of toxic trimethylamine N-oxide is reduced to 80%, with the remainder (i.e., 20%) being present as trimethylamine. This polymorphism in amine metabolism, due to attenuated catalytic activity of FMO3, is heritable (Cashman et al. 2003; Phillips and Shephard 2020; Shimizu et al. 2014).

N-Oxygenation of trimethylamine by FMO3
The ratio of trimethylamine to TMAO in urine is used as an index of FMO3 activity, FMO3 polymorphism, and the occurrence of trimethylaminuria.
Amphetamine (CNS stimulant, anorexic)
Multiple mechanisms are involved and interact to promote neurotoxicity from amphetamine and derivatives, which are widely abused psychostimulant drugs (Carvalho et al. 2012; Yamamoto et al. 2010). Oxygenation of the amino group of amphetamine occurs less in humans because deamination and aromatic hydroxylation predominate, catalyzed by P450 enzymes (Bach et al. 1999; Miranda et al. 2007). N-Oxygenation of amphetamine is catalyzed by FMO3, and reactive and toxic metabolites are formed that can contribute to the toxic effects of amphetamine by participating in the autooxidation of dopamine, norepinephrine, and serotonin (Tables 3, 7).
Potential toxic effects are ascribed to amphetamine hydroxylamine. A proposed mechanism of amphetamine activation is N-oxygenation to a hydroxylamine in the first step, which is then re-oxygenated with FMO3 to form an unstable intermediate that, after spontaneous dehydration, is transformed into a trans-oxime (Cashman et al. 1999b; Szöko et al. 2004) (Fig. 14).

N-Oxygenation of amphetamine
N-Methylamphetamine (CNS psychostimulant)
N-Methylamphetamine (methamphetamine) is an illicit, highly addictive psychostimulant amphetamine derivative that is widely abused. Large doses of the drug are associated with serious neuropsychiatric consequences including agitation, anxiety, hallucinations, paranoia, and psychosis (Jayanthi et al. 2021). N-Methylamphetamine can severely damage the central nervous system and is toxic to the cardiovascular system (Halpin et al. 2014; Tan et al. 2021; Zhao et al. 2021). Metabolism of N-methylamphetamine proceeds with the initial formation of N-methylamphetamine hydroxylamine, and the final product is phenyl propanone (Tables 3, 7) (Fig. 15). The formation of phenyl propanone oxime and the nitrone are proposed as part of an overall detoxication process, with the potentially toxic effects ascribed to N-methylamphetamine hydroxylamine (Cashman et al. 1999b; Szöko et al. 2004).

N-Oxygenation of N-methylamphetamine
Arecoline (tetrahydropyridine alkaloid)
The alkaloid arecoline, a major constituent of areca nuts, has been classified as a Class I carcinogen by the International Agency for Research on Cancer (IARC) (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, 2004). Arecoline is converted to the metabolite arecoline N-oxide by human FMO1 and FMO3, with FMO1 as the major enzyme (Tables 5, 7) (Fig. 16). Arecoline N-oxide was shown to be mutagenic in bacterial tester strains and to induce DNA damage in mammalian test systems, including cultured fibroblasts. The metabolite arecoline N-oxide is further converted to mercapturic acid derivatives in vivo (Das and Giri 2020; Giri et al. 2007; Lin et al. 2011; Oliveira et al. 2021).

N-Oxygenation of arecoline by FMO enzymes
S-Oxygenations
Substances with a sulfur atom can be oxygenated with FMO enzymes to form electrophilic intermediates (e.g., thiols, thioamide, 2-mercaptoimidazole, thiocarbamate, thiocarbamide metabolites). Such electrophilic metabolites can bind to cellular proteins and inactivate enzymes in the endoplasmic reticulum, e.g., P450s (Başaran and Can Eke 2017; Jones and Ballou 1986).
Thiourea and derivatives (organosulfur compounds)
Thiourea (also called thiocarbamide) is a pro-carcinogenic, moderate to a highly toxic substance that is oxidized to carcinogenic products by FMO enzymes. The thiourea moiety is part of chemicals with different applications, including rodenticides, bactericides, components used in the manufacture of rubber, and therapeutic agents. Some derivatives of thiourea are known toxins (e.g., phenylthiourea) (Henderson et al. 2014; Smith and Crespi 2002).
Thiourea is oxygenated via a sulfenic acid to a sulfinic acid by human FMO1, FMO2, and FMO3, with FMO2 as a major enzyme (Tables 4, 7) (Fig. 17). The sulfinic acid formed can be detoxicated in the cells by reaction with glutathione. Similarly, N-substituted derivatives of thiourea (e.g., N′-(4-imidazole-ethyl)thiourea derivatives) exerted cytotoxicity and are activated by oxygenation of the sulfur atom to sulfenic acids (Furnes and Schlenk 2004; Kim and Ziegler 2000; Onderwater et al. 2006; Smith and Crespi 2002).

Oxygenation of thiourea by FMO enzymes
Fenthion (organophosphate, insecticide)
Fenthion, an inhibitor of human acetylcholinesterase, is a substrate in the reaction of S-oxygenation catalyzed by FMO1, FMO3, and FMO5, with FMO1 being the major enzyme. The reaction is characterized by high Km values and by the stereoselective formation of (R)-( +)- sulfoxide (Table 4) (Fig. 18). At lower concentrations, fenthion is predominately metabolized by multiple P450 enzymes, with P450 1A2 as the major one (Furnes and Schlenk 2004, 2005; Gadepalli et al. 2007; Leoni et al. 2008) (Fig. 18).

Oxygenation of fenthion by FMO enzymes
Monoamine oxidase (MAO)
We previously reported that human MAOs participate in ~ 1% of the metabolism of xenobiotic and physiological compounds, including natural products. In the metabolism of general chemicals, MAO enzymes participate in ~ 2%, drugs ~ 1%, and natural and physiological chemicals ~ 1% (Rendić and Guengerich 2015). The previous analysis indicated more extensive participation of MAO enzymes in the metabolism of general chemicals when compared to the metabolism of drugs and natural products and physiological compounds, but this pattern may reflect more basic studies and efforts at drug discovery (Rendić and Guengerich 2012, 2015).
Enzymes
Two MAO enzymes are known (MAO A and MAO B), which are encoded by the MAOA and MAOB genes. The enzymes are primarily involved in the catalytic oxidative deamination of endogenous monoamines (Bach et al. 1988; Benedetti 2001; Bortolato et al. 2008; Bortolato and Shih 2011; Edmondson and Binda 2018; Grimsby et al. 1990; Ramsay 2012; Shih et al. 1990; Strolin Benedetti et al. 2007) The MAOs are mitochondrial, membrane-bound enzymes, and are located in many tissues, of which the most significant may be the brain. The enzymes are present also in the liver, where they catalyze the oxidative deamination of some xenobiotics.
MAO A is present in the brain, small intestine, heart, placenta, liver, portal system, and peripheral adrenergic neurons, and it is selective for the metabolism of norepinephrine and serotonin. MAO B is found in blood platelets, cerebral glial cells, and hepatic cells and is relatively selective for the metabolism of benzylamine and phenylethylamine. Physiological substrates are amines that are oxidized to aldehydes, which may be reduced by aldehyde reductase to alcohols. In vivo inhibition of MAO with either irreversible or nonselective compounds permits the up-take of high concentrations of tyramine and other sympathomimetic molecules into the blood circulation, where they gain access to peripheral adrenergic neurons, trigger catecholamine release, and cause a marked and rapid increase in blood pressure (Lavian et al. 1993).
Substrates
The substrates are nitrogen-containing compounds, including primary, secondary, and tertiary amines (Kalgutkar et al. 2001; Strolin Benedetti et al. 2007) (Tables 8, 9, 10). Substrates have also been synthesized as prodrugs (e.g., dopamine prodrugs synthetized as esters, amides, dimeric amides, carrier-mediated, peptide transport-mediated, cyclic, chemical delivery systems) to enhance their bioavailability in the treatment of Parkinson’s disease (Haddad et al. 2017; Sozio et al. 2012). Endogenous substrates include biogenic and dietary amines, monoamine hormones, and neurotransmitters such as serotonin, dopamine, norepinephrine, and epinephrine, as well as tyramine, tryptamine, 2-phenylethylamine, 5-hydroxytryptamine, monoacetyl putrescine (a precursor to γ-aminobutyric acid (GABA), adrenaline, and metanephrine) (Bortolato and Shih 2011). Similar to FMO enzymes, the substrates of MAO enzymes are often substrates for other drug-metabolizing enzymes as well (e.g., P450 and/or FMO enzymes).
Inhibitors
The inhibitors of MAO enzymes are developed and tested either as selective or nonselective reversible or irreversible inhibitors. Many compounds (drugs, natural products, as well as general chemicals) have been shown to inhibit MAO enzymes. In the clinic, drugs are used either as selective or nonselective MAO inhibitors in the therapy of several neuropsychiatric disorders (mood disorders, Parkinson's disease, Alzheimer’s disease) (Table 11). Tested natural products have shown a variety of activities and some of them were selective and strong as either MAO B (e.g., (−)-maackiain and (−)-medicarpin) or MAO A (e.g., apigenin) inhibitors (Table 12). In addition, extensive work has been done to synthesize derivatives of natural products as MAO inhibitors to be used as CNS drugs (Gulcan and Orhan 2020; Lu et al. 2013; Mathew and Kim 2020) (Table 13).
Activators and inducers
Valproic acid, which has been widely used in clinics for the treatment of multiple neuropsychiatric disorders such as epilepsy and bipolar disorder, exerts its activity by regulating the brain levels of serotonin. The compound was reported to increase MAO A catalytic activity, mRNA levels, and promoter activity (Wu and Shih 2011).
Bavachin, a Psoralea corylifolia L. seed compound, has been also reported to be an activator of the activity (Zarmouh et al. 2015), along with clomipramine (Reid et al. 1988).
Reactions
The general reaction catalyzed by MAO enzymes (Ramsay and Albreht 2018) is shown in Fig. 19.

Typical reaction catalyzed by MAO enzymes, where R denotes part of the molecule
MAO enzymes catalyze oxidative deamination reactions, including cleavage of C–N bonds with the formation of several chemical species with neurotoxic potential, e.g., hydrogen peroxide, ammonia, and aldehydes. As a consequence, prolonged excessive activity of these enzymes can lead to mitochondrial damage and neurodegenerative disorders.
Oxidative deamination reactions are also catalyzed by P450 enzymes. However, the mechanism catalyzed by MAO enzymes differs from the reaction catalyzed by P450s in that one of the products of the overall reaction is hydrogen peroxide (Fig. 19), while in the reactions catalyzed by cytochromes P450 the product is a water molecule, i.e. fully reduced oxygen (Guengerich 2022).
Substrate oxidation by MAO enzymes
The MAO enzymes share similar overall structures, with nearly identical FAD-binding domains, but contain varied substrate binding sites. It should be noted that, in contrast to the FMOs, AOX, XOR, and NADPH-P450 reductase, the MAO enzymes have the flavin covalently attached to the protein via a histidine residue. As flavoprotein oxidases, they catalyze substrate oxidation via two half-reactions. In the reductive half-reaction two hydrogen atoms are transferred to the MAO FAD complex when it accepts a hydride equivalent from the substrate, while in the oxidative step the MAO FADH2 complex is oxidized to form MAO FAD by molecular oxygen (generating H2O2) (Figs. 20, 21). Due to the ability of the flavin prosthetic group to accept either one or two electrons (i.e., as a biological “transformer” (Walsh 1979)), several mechanisms have been proposed for the transfer of electrons from the substrate to the prosthetic group (Behl et al. 2021; Edmondson et al. 2007; Fitzpatrick 2010; Gaweska and Fitzpatrick 2011; Ramsay and Albreht 2018; Scrutton 2004).

Substrate oxidation by MAO enzymes (Edmondson et al. 2007)

Deamination of benzylamines by MAO enzymes
Drugs as substrates of MAO enzymes
Numerous drugs possessing a nucleophilic heteroatom are substrates of MAO enzymes (Table 8). Knowledge of the involvement of either MAO A, MAO B, or both enzymes in the metabolism of a drug allows for the prediction of drug–drug interactions with selective or non-selective MAO inhibitors. It should be emphasized that these are mitochondrial enzymes and that in vitro studies with microsomes will not include these enzymes or evaluate their metabolic potential. The metabolism of a drug that is deaminated by both forms of MAO is not necessarily inhibited in vivo by selective MAO A or MAO B inhibitors. If a drug is metabolized by MAOs, competitive interactions can occur with other drugs that are MAO substrates, e.g., with β-adrenoceptor agonists and antagonists, prodrugs of dopamine, and serotonin 5-HT1-receptor agonists, as well as with primaquine, flurazepam, and citalopram (Benedetti 2001; Masuo et al. 2017).
Drugs or drug metabolites that are substrates for human MAOs include β-blockers (i.e., amines formed by dealkylations of β-blockers), primaquine, β-phenylethylamine, phenelzine (also an irreversible inhibitor), almotriptan, bicifadine, citalopram, and its active metabolite desmethylcitalopram, rizatriptan, and zolmitriptan (Table 8).
The drug ozanimod is oxidatively deaminated to a pharmacologically active metabolite by MAO B, yielding the major circulating active compound. The reaction follows a prior N-dealkylation reaction catalyzed by P450 3A4 (Fig. 22) (Table 8). Also involved in the overall metabolism of ozanimod are P450s 1A1 and 2C8, aldehyde dehydrogenase, and alcohol dehydrogenase, plus reductive metabolism by gut microflora (Surapaneni et al. 2021; Tran et al. 2020).

Ozanimod metabolism by P450 and MAO enzymes
Drugs as MAO inhibitors
In addition to being substrates of MAO enzymes, many nitrogen-containing drugs are also MAO inhibitors (Table 11) and were among the first agents shown to be efficacious in the treatment of clinical depression (Fernandez and Chen 2007; Kalgutkar et al. 2001; Suchting et al. 2021). For instance, the therapeutic effects of some antidepressants, hydrazine derivatives (e.g., iproniazid), and tranylcypromine are based on irreversible inhibition of the MAO enzyme and result in the accumulation of sympathetic amines in adrenergic neurons.
As already mentioned, the drugs used in clinical practice are either nonselective and irreversible MAO enzymes inhibitors or selective inhibitors for either MAO A or MAO B enzymes. Some irreversible inhibitors include rasagiline (MAO A and B, selective MAO B inhibitor), tranylcypromine (MAO A and B, nonselective), iproniazid (MAO A and B, nonselective), phenelzine (MAO A and B, nonselective inhibitor), selegiline (MAO A and B, selective MAO B inhibitor at lower concentrations/doses), pargyline (MAO A and MAO B, partially MAO B selective inhibitor), iproniazid (MAO A and B, nonselective inhibitor), clorgyline (MAO A, MAO B, selective MAO A inhibitor), ladostigil (MAO A and B, non-selective inhibitor), and isocarboxazid (MAO A and B, non-selective inhibitor). Some selective reversible MAO enzyme inhibitors are lazabemide (MAO B selective inhibitor), befloxatone (MAO A and B, selective MAO A inhibitor), toloxatone (MAO A and B, selective MAO A inhibitor), brofaromine (MAO A and B, selective MAO A inhibitor), and moclobemide (MAO A selective inhibitor) (Table 11).
Due to the observed toxic effects, irreversible inhibitors of MAO enzymes have been largely replaced in therapy with selective reversible inhibitors.
Antidepressant drugs, besides inhibiting the active uptake of amines into presynaptic cells (Stahl 1998), also exert inhibitory activity on MAO enzymes with potencies dependent on the model and experimental conditions used. For instance, when testing mitochondrial MAO activity in mouse, rat, dog, and monkey brains with antidepressant drugs (zimeldine, imipramine, maprotiline, and nomifensine) (which inhibit MAO A and MAO B at high concentrations), inhibition was dependent on the species used and experimental conditions applied. Imipramine, for instance, inhibited MAO B more strongly than MAO A activity in mouse and rat brains. When dog and monkey brains were investigated, MAO A activity was inhibited with greater potency than MAO B activity at high concentrations of imipramine; at low concentrations, however, MAO B activity was more strongly inhibited. Also, maprotiline and nomifensine inhibited mouse and rat brain MAO B activity more strongly than MAO A activity, while the inverse was found for dog and monkey brain models (Egashira et al. 1999).
As an example, the non-selective MAO A and MAO B irreversible inhibitor phenelzine (Table 11, Fig. 23) elevates brain levels of the monoamine neurotransmitters 5-hydroxytryptamine (serotonin), noradrenaline, and dopamine. Phenelzine is also a substrate for MAO enzymes, and different metabolites are formed including β-phenylethylamine, phenylacetic acid, p-hydroxyphenyl acetic acid, β-phenylethylidenehydrazine, and phenylethyldiazenephenylethylidenehydrazine. Of these metabolites, neuroprotective/neuro-rescue activity has been suggested for the metabolite β-phenylethylidenehydrazine and irreversible inactivation of MAO enzymes has been ascribed to the formation of phenethyl free radicals (Ortiz de Montellano et al. 1983; Rumyantseva et al. 1991). Thus, besides its MAO inhibiting activity, phenelzine also elevates brain levels of γ-aminobutyric acid (GABA) which may also contribute to its anxiolytic effects, and the effects ascribed to the phenelzine intermediate metabolite β-phenylethylidenehydrazine, a weak MAO inhibitor (Baker et al. 2019; Parent et al. 2002). Phenelzine may also ameliorate the effects of oxidative stress by reducing the formation of reactive metabolites (aldehydes, hydrogen peroxide, ammonia/ammonia derivatives) produced by the interaction of MAO with biogenic amines, as well as by inhibiting primary amine oxidase (Baker et al. 2019; Matveychuk et al. 2021). This example illustrates the complex interactions of the parent drug and its metabolite(s) on the final effects.

Phenelzine oxidation to β-phenylethylidenehydrazine
The first generation of non-selective (iproniazid, tranylcypromine, phenelzine) and irreversible MAO A inhibitors was shown to produce associations with the “cheese reaction,” whereas MAO B inhibitors (used at the recommended selective dosage) did not produce the effect. The cheese reaction occurs because of potentiation of the sympathomimetic activity of ingested tyramine present in cheese and other fermented food. This cheese reaction provoked by inhibition of MAO A may consequently produce a hypertensive crisis due to the increased release of norepinephrine. In contrast to irreversible MAO A inhibitors, reversible MAO A inhibitors (e.g., the antidepressants moclobemide and brofaromine) exert limited tyramine potentiation activity (McCabe 1986; Youdim and Weinstock 2004). In addition, some of the early MAO inhibitors have been withdrawn from the market due to hepatotoxic reactions (e.g., nialamide, pargyline). Because of the observed toxic effects, nonselective irreversible inhibitors of MAO enzymes have been replaced with selective reversible inhibitors in clinical therapy. At present, drugs that are inhibitors of MAO A are used and investigated for the treatment of depression, while selective MAO B inhibitors (e.g., rasagiline, selegiline), are used in the treatment of Parkinson's disease, avoiding severe side effects. It has been suggested that MAO B inhibitor drugs might be effective in the treatment of Alzheimer’s disease (Finberg 2014; Özdemir et al. 2021; Shulman et al. 2013; Szökő et al. 2018; Yamada and Yasuhara 2004; Youdim 1975; Youdim and Bakhle 2006).
Natural products and physiological compounds, derivatives, preparations, and MAO enzymes
The physiological substrates of MAO enzymes are brain neurotransmitters (e.g., serotonin, dopamine, norepinephrine, and epinephrine), as well as trace amines (e.g., tyramine, tryptamine, 2-phenylethylamine, octopamine, 3-iodothyronamine (Table 9). The products of oxidative deamination are aldehydes and H2O2, both of which have some potential toxicity in cells (Tables 9, 14). The formation of potentially toxic metabolites has been associated with neurodegenerative disorders of the central nervous system such as Parkinson’s disease and dementia. Thus, the reactions of physiological compounds catalyzed by MAO enzymes are examples of the bioactivation of non-toxic amines to potentially toxic metabolites. However, in the cells, the aldehydes that are formed are either oxidized to polar carboxylic acids by the activity of aldehyde dehydrogenases (ALDH) or reduced to alcohols or glycols by aldehyde reductases (AKR enzymes). These polar products can often be excreted through kidneys and/or participate in conjugation reactions. Dopamine and norepinephrine can alternatively participate as substrates in methylation reactions catalyzed by catechol O-methyltransferase (COMT) to form 3-methoxytyramine and epinephrine, respectively, or participate in conjugation reactions such as sulfoconjugation (Behl et al. 2021; Buu 1985; Danielczyk et al. 1988; Ji et al. 2005; Rivett et al. 1982).
Some natural products are substrates of MAO enzymes, but there is also a growing interest in testing natural products and compounds as inhibitors of MAO enzymes (Table 12) for possible use in the treatment of Parkinson’s disease (Zarmouh et al. 2016) or to explain possible side effects or their toxicity when ingested.
When using natural products, care should be taken because the same preparation may contain a compound that inhibits the enzymes, as well as compounds that act as enzyme activators. For example, ethanolic extracts of the seeds of Psoralea corylifolia L. contain flavanone bavachinin, which showed competitive MAO A and MAO B inhibition. P. corylifolia L. extracts also contain its analog bavachin, which has stimulatory properties (Zarmouh et al. 2015).
General chemicals and synthetic derivatives of natural products as MAO enzyme inhibitors
In the group of general chemicals as substrates of MAO enzymes, special attention has been focused on the tetrahydropyridine compound MPTP, which is oxidized to the neurotoxic products MPDP+ and MPP+ by MAO B as the major enzyme. During the reaction the enzyme is inactivated (Tables 10, 13, 14, Fig. 26).
Some of the synthetic MAO enzyme inhibitors are compounds with structures based on scaffolds of natural compounds known to be MAO inhibitors, e.g., caffeine, coumarin, piperazine, and chalcone (a structural isomer of coumarin) (Tables 12, 13). Caffeine, an adenosine receptor antagonist (A2A), is a weak and reversible MAO A and MAO B inhibitor, both in vivo and in vitro (Grzelczyk et al. 2021; Haj Ahmed et al. 2020). Its structure has been used to design compounds having both A2A receptor antagonist and MAO A and MAO B inhibition activity. The compounds have been developed with the potential for treating Parkinson's disease. Although structural modifications of caffeine led to strong MAO inhibitors, the MAO inhibitory activity of caffeine itself is not likely to be of pharmacological relevance in typical coffee consumption (Petzer et al. 2013; Petzer and Petzer 2015).
The β-carboline alkaloids, which are also components of coffee (and also present in cigarette smoke), were reported to be reversible, competitive, and strong inhibitors of MAO enzymes and linked to a reported lower incidence of Parkinson’s disease in coffee drinkers and cigarette smokers (Herraiz and Chaparro 2005, 2006). However, the MAO inhibitory activity of natural products may be dependent on and affected by the type of product used (e.g., type of coffee), as well as by the method of preparation of the sample for testing (e.g., light or dark roasted coffee beans) (Grzelczyk et al. 2021) (Table 12).
Coumarin, for instance, exhibited nonselective intermediary MAO A and MAO B inhibitory activity, but some of its natural derivatives exhibited selective strong MAO A (osthenol) or MAO B inhibitory activity (rutamarin) (Table 12). Furthermore, some synthetic coumarin derivatives exerted strong, selective inhibition of either MAO A or MAO B activity. Many other derivatives of various natural compound-based structures (e.g., indoles, chromones, chalcones, carboxamides, benzylamine, sulfonamide, benzofuran, pyrazole, pyrrole, quinazolinone, and others) were synthesized and reported to exhibit strong, selective inhibition of either MAO A or MAO B activity (Table 13) (Patil et al. 2013).
Stereoselective inhibition of MAO enzymes was reported for enantiomers of the 8-aminoquinoline derivative NPC1161. Racemic NPC1161 exerted both MAO A and MAO B inhibitory activity with 3.7-fold selectivity of MAO A compared to MAO B, while the (S)-( +)- enantiomer was shown to be an intermediate (MAO A) and strong (MAO B) mixed-type irreversible inhibitor with about tenfold selectivity for inhibition of MAO B over MAO A. The (R)-(−)-enantiomer was shown to be a mixed-type nonselective intermediate reversible inhibitor (Table 13). Stereoselective MAO inhibition was also observed in the interaction of enantiomers of antimalarial drug primaquine. Racemic primaquine and (R)-( −)-primaquine were weak and very weak inhibitors, respectively, both being nonselective inhibitors. (S)-( +)-Primaquine was also a weak inhibitor but showed 1.5-fold selectivity for inhibition of MAO A over MAO B (Chaurasiya et al. 2021) (Table 11).
Examples of compounds bioactivated to toxic products by MAO enzymes
Examples of compounds that are bioactivated to toxic products by MAO enzymes include physiological compounds (e.g., neurotransmitters dopamine, serotonin, noradrenaline, the biogenic amine kynuramine) that are metabolized to toxic aldehydes, the antidepressant drug nomifensine (by forming dihydroisoquinolinium ions exerting risks of anemia and hepatotoxicity), and the general chemical MPTP, which was shown to be mechanism-based inhibitor inactivating the enzyme and forming 1,4-dihydropyridine adducts. Benzylamine, widely used as a model substrate for MAO B, is converted to toxic benzaldehyde, which is consequently reduced and deactivated to an alcohol by aldehyde dehydrogenase (Table 14).
Examples of metabolic reactions of MAO substrates
Tyramine
The aromatic amine tyramine, which is both a natural product and physiological compound, is oxidatively deaminated preferentially by MAO A. The product of its metabolism is the toxic 4-hydroxyacetaldehyde, which is converted to nontoxic 4-hydroxyphenyl acetic acid by aldehyde dehydrogenase (ALDH) (Tables 9, 14) (Fig. 24). Alternatively, tyramine can be hydroxylated to dopamine by P450 2D6 in a reaction considered as the main elimination/detoxication pathway for tyramine (Niwa et al. 2004). In a minor reaction, tyramine is converted to tyrosol by alcohol dehydrogenase (ADH) and, in human liver microsomes, to a trans-oxime by FMO3 through a hydroxylamine intermediate (Lin and Cashman 1997a; Niwa et al. 2011; Phillips and Shephard 2019). The oxidative deamination reaction can potentially be inhibited by MAO A inhibitors, resulting in an enhanced concentration of other sympathomimetics in peripheral adrenergic neurons and causing a rapid increase in blood pressure and the onset of the cheese reaction (McCabe 1986).

Deamination of tyramine by MAO enzymes
Dopamine and other neurotransmitters
MAO plays a central role in the metabolism of the neurotransmitter dopamine, as well as norepinephrine and serotonin (Table 9). Dopamine metabolism is complex (Meiser et al. 2013) and, in addition to MAO enzymes, dopamine is also a substrate for catechol O-methyl transferase (COMT). 3,4-Dihydroxyphenylacetaldehyde (DOPAL), a product of the MAO-catalyzed deamination reaction, is toxic and is converted to 3,4-dihydroxyphenylacetic acid (DOPAC) by aldehyde dehydrogenase (ALDH), which rapidly exits the neurons and is also a substrate for COMT, producing homovanillic acid (Fig. 25). In addition to DOPAL, the oxidative deamination produces H2O2, which (in the presence of divalent metal atoms) may form hydroxyl radicals (OH⋅). The formation of toxic species from dopamine (and also from other neurotransmitter substrates of MAO enzymes) has been suggested to contribute to catecholaminergic denervation in Parkinson’s disease. Cytoplasmic dopamine levels are maintained at low, non-toxic levels by the combined activity of the vesicular monoamine transporter (VMAT) and MAO and ALDH enzymes (Goldstein 2020; Goldstein et al. 2012).

Activation and deactivation of dopamine
MPTP
MPTP, a selective nigrostriatal neurotoxin, is bioactivated by MAO B (and less effectively by MAO A) to 2,3-MPDP+, and this intermediate undergoes further oxidation to MPP+ by MAOs (Fig. 26). MPTP and its two primary metabolites are competitive and mechanism-based inactivators of MAO A and MAO B enzymes (Trevor et al. 1988, 1987b). To express the selective nigrostriatal neurotoxicity of MPTP, bioactivation by MAO B is required, leading to the formation of the potentially reactive products MPDP+ and (the 4-electron oxidation product) MPP+. The latter product accumulates in brain striatal tissue, is a substrate for dopaminergic active uptake systems, and is an inhibitor of mitochondrial NADH dehydrogenase, a respiratory chain enzyme located in the inner mitochondrial membrane (Peterson et al. 1985; Singer et al. 1988; Trevor et al. 1987a). Both reactions, MPTP activation to MPP+ and its deactivation by N-demethylation, are catalyzed by MAO B and P450s (Fig. 26) (Bajpai et al. 2013; Hanna et al. 2001; Herraiz et al. 2006; Nakamura et al. 2020; Trevor et al. 1987a; Uehara et al. 2015).

Bioactivation and detoxication of MPTP by MAO and P450 enzymes
NAD(P)H-quinone oxidoreductase (NQO) enzymes
NAD(P)H quinone oxidoreductase 1 (NQO1) and NAD(P)H quinone oxidoreductase 2 (NQO2), are homodimeric flavoproteins containing one molecule of non-covalently bound FAD per monomer. These enzymes are members of a larger mammalian quinone oxidoreductase family and catalyze the reduction of quinones and similar molecules possessing quinone-like structures, e.g. quinone imines, benzotriazine oxides, tocopherols (Fig. 27). Nitro groups are also reduced by NQO enzymes. These enzymes use both NADH and NADPH and were termed “DT diaphoras” in the early literature because they use both DPNH (NADH) and TPNH (NADPH) (former names used for these pyridine nucleotides) (Ernster et al. 1962).

Typical reaction catalyzed by NQO enzymes
These enzymes are generally considered to be detoxicating enzymes that protect cells by catalyzing the 2-electron reduction of quinones and thus participate in the protection of cells against toxicity. NQO enzymes are constitutively expressed in a variety of tissues and also in many solid tumors. The latter property has been considered in the context of potential targets for the activation of certain bioreductive anticancer agents (e.g., activation of the anticancer drug mitomycin C in tumor cells) (Siegel et al. 2004; Workman 1994). In our previous reports, human quinone reductase enzymes were classified in the group of “other oxidoreductases” (Rendić and Guengerich 2012, 2015). These enzymes participate in < 1% of the metabolism of xenobiotics and natural products, including drugs. The enzymes were also classified in the group of enzymes participating to the extent of < 4% of the activation of chemical carcinogens.
Changes in the activity of NQO1 are associated with different pathologies (including cancer and cardiovascular and neurodegenerative diseases), and these properties have been considered in the context of potential targets for the treatment of the diseases. Induction or depletion (knockout) of NQO1 was shown to be associated with decreased or increased susceptibilities to oxidative stress, respectively. Human NQO1 is often over-expressed in cancer cells, and the enzyme has been considered as a possible drug target. Two common polymorphic forms of human NQO1, pR139W and pP187S, were found to be associated with an increased risk of several forms of cancer. Dicumarol and some structurally related compounds act as competitive inhibitors of both variants. In addition, NQO1 was reported to be inhibited by nicotinamide, and resveratrol inhibited both NQO1 and NQO2 (Megarity and Timson 2019; Nolan et al. 2012; Pey et al. 2019). On the other hand, quercetin was shown to increase NQO1 transcription in human MCF-7 human breast cells (Valerio et al. 2001), and resveratrol increased NQO1 protein levels in K562 cells (Hsieh et al. 2006).
A review of the literature and examples of drugs, physiological, and environmental compounds that interact with NQO1 and NQO2 enzymes is provided in a recently published article (Rashid et al. 2021). The compounds are presented as being either activated (e.g., mitomycin C, doxorubicin, porfiromycin) or inactivated (e.g., acetaminophen, menadione, amrubicin) by NQO enzymes. One of the physiological substrates of the NQO1 enzyme is the highly unstable DOPA quinone, formed by auto-oxidation of dopamine catechol. DOPA-quinone may induce neuronal damage resulting from the formation of reactive oxygen species, e.g. superoxide radicals and hydrogen peroxide. In a mouse model, that quinone formed up-regulates astroglial NQO, which might reduce the potentially toxic dopamine quinone to more stable hydroquinone, a detoxication reaction catalyzed by NQO enzymes (Drukarch et al. 2001; van Muiswinkel et al. 2000).
Molybdenum-containing hydroxylases
Molybdenum hydroxylases are cytosolic molybdoflavoproteins with a molecular mass of approximately 300 kDa (Hille 2005). Human molybdenum-containing hydroxylase enzymes were classified in the group of “other oxidoreductases” in our previous work (Rendić and Guengerich 2012); (Rendić and Guengerich 2015). According to this classification, as mentioned before, the enzymes from this group participate in < 1% of the metabolism of xenobiotics and natural products, including drugs. The enzymes were also classified in the group of enzymes participating to the extent of < 4% of the activation of chemical carcinogens (Rendić and Guengerich 2012); (Rendić and Guengerich 2015).
Enzymes
The molybdoflavoenzyme family in humans is composed of aldehyde oxidase (AOX), xanthine oxidoreductase (XOR), sulfite oxidase, and an enzyme known as mitochondrial amidoxime-reducing component (Terao et al. 2020) (Tables 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29). This review focuses on AOX and XOR, which are known to play roles in the metabolism of drugs and other xenobiotics (Tables 15, 19, 23). AOX and XOR enzymes also catalyze the metabolism of physiological compounds (Tables 17, 25) and are involved in both detoxications and activation of substrates to toxic/pharmacologically active intermediates or products (Tables 21, 22, 29).
The functional XOR and AOX enzymes are homodimers composed of two identical subunits of approximately 150 kDa, each possessing three cofactor-binding domains connected by flexible linker regions (Terao et al. 2020). The N-terminal domain contains two distinct iron–sulfur (2Fe–2S) redox centers, the central domain binds FAD, and the C-terminal domain houses a molybdenum cofactor (Moco) within the active site. The molybdenum atom of the Moco is coordinated with a sulfido ligand that is essential for catalytic activity. Whereas AOX exists only in a single form, mammalian XOR can interconvert between a dehydrogenase (XDH) and an oxidase (XO) (Battelli et al. 1973; Corte and Stirpe 1972; Della Corte and Stirpe 1968; Stirpe and Della Corte 1969). Accordingly, AOX and the XO utilize molecular oxygen as a final electron acceptor, whereas only XDH can transfer electrons to NAD+. With amino acid sequence identities of approximately 50%, AOX and XOR enzymes possess similarities in substrate specificity (e.g., aromatic azaheterocycles); however, the larger, more anionic active site of AOX is able to accommodate a wider range of substrates relative to XOR (Mahro et al. 2013).
The tissue distribution of both AOX and XOR is species-dependent. AOX expression in humans is distributed across many different tissues, including liver (major), kidneys, lungs, gastrointestinal tract, skin, male reproductive tissues, and endocrine tissues, most notably the adrenal glands (Moriwaki et al. 2001; Terao et al. 2016). Constitutive expression of XOR in human tissues is low, and consequently, XOR activity is primarily present in the liver and gastrointestinal tract, as well as in lactating breast and kidney (Battelli et al. 2016a; Bortolotti et al. 2021). Notably, XOR is located in the vascular endothelium and can also be released into the systemic circulation, e.g., as a consequence of hepatic or intestinal damage (Kumar et al. 2018; Pritsos 2000).
The exact physiological roles of AOX and XOR are not well-defined, particularly with regard to AOX. Of the four human molybdenum hydroxylases, only sulfite oxidase is an essential enzyme (Duran et al. 1978; Shih et al. 1977; Terao et al. 2020; Veldman et al. 2010). XOR is responsible for the conversion of hypoxanthine to xanthine and of xanthine to uric acid (Balis 1976; Krenitsky et al. 1986). Consequently, XOR deficiency leads to the accumulation of xanthine, a condition referred to as xanthinurea (Kumar et al. 2018). Xanthinurea is an autosomal recessive disorder and is categorized as either Type I (Nakamura et al. 2012), which is associated with a deficiency in XOR alone, or Type II, which is associated with a deficiency of both XOR and AOX (Reiter et al. 1990). In addition, XOR is capable of reducing nitrates to nitrites and both AOX and XOR have been shown to reduce nitrites to nitric oxide (Maia and Moura 2018; Maia et al. 2015). Because AOX and XO utilize molecular oxygen as an electron acceptor, both enzymes produce reactive oxygen species (hydrogen peroxide and/or superoxide) as by products in catalyzing the oxidation of substrates. Oxidative damage has been linked to the development of cancer (Oberley 2002), and both AOX and XOR have been implicated in tumor growth and development (Kusano et al. 2019; Qiao et al. 2020; Takeuchi et al. 2018).
Reactions
Molybdenum hydroxylases catalyze the transfer of an oxygen atom, ultimately derived from water, to a substrate in a two-electron redox reaction (Fig. 28) (Kisker et al. 1997). The enzymes oxidize carbon atoms of a number of different aldehyde and heteroaromatic rings. In general, aromatic azaheterocyclic compounds are better substrates of molybdenum-hydroxylases than aldehydes.

General reaction catalyzed by molybdenum hydroxylases
The catalytic mechanism of molybdenum hydroxylases used to oxidize aromatic azaheterocycles and aldehydes involves the oxidation of an electrophilic carbon, typically located adjacent to a nitrogen in heterocyclic substrates (Alfaro and Jones 2008). The process begins with deprotonation of a hydroxyl group on the Moco by a conserved glutamate residue, followed by a nucleophilic attack on the electron deficient carbon atom of the heteroaromatic substrate. Hydride transfer from the electrophilic carbon of the substrate to the sulfur of the Moco then follows, resulting in a reduction of the molybdenum from Mo(VI) to Mo(IV). While the reaction could proceed via a tetrahedral intermediate in a step-wise mechanism, it is believed to proceed via a concerted mechanism (Fig. 29). The reaction intermediate is hydrolyzed, releasing the oxidized product, and a water molecule replaces the lost hydroxyl ligand on the molybdenum. The reducing equivalents are shuttled from the Moco to FAD via the iron-sulfur clusters. FADH2 is then reoxidized by molecular oxygen via a one or two-electron transfer, generating superoxide ion or H2O2, respectively. The oxidized products are structurally similar to those generated by P450 enzymes. However, the oxygen molecule used to oxidize substrates of molybdenum hydroxylases is derived from water (Garattini and Terao 2012), unlike P450s which use molecular oxygen as the source of the oxygen in the product (Guengerich 2001). Accordingly, the inclusion of H218O in incubations with molybdenum hydroxylases is utilized as a reaction phenotyping strategy for these enzymes.

Catalytic cycle and proposed mechanism for the oxidation of aromatic heterocycles and aldehydes by the molybdenum hydroxylases using quinazoline as an example (glutamate numbering represents human AOX) (Alfaro and Jones 2008)
In addition to oxidation reactions, AOX and XOR are capable of catalyzing reduction reactions. Both AOX and XOR have been demonstrated to reduce nitrite to nitric oxide (Fig. 30), an important signaling molecule involved in numerous physiological functions, including vasodilation, platelet aggregation, and immune response (Godber et al. 2000; Li et al. 2009; Maia et al. 2015). AOX is also known to reduce a variety of other functional groups, including N- and S-oxides, heterocycles, and nitro groups (Amano et al. 2018; Cashman et al. 2020; Dalvie and Di 2019; Ogiso et al. 2018; Pryde et al. 2010; Sung et al. 2020).

General reaction of nitrite reduction catalyzed by molybdenum hydroxylases
While reductive reactions and mechanisms have received less attention relative to oxidation reactions catalyzed by molybdenum hydroxylases, Maia and Moura have described the mechanism of nitrite reduction to nitric oxide (Fig. 31), which takes place at the Moco center (Maia and Moura 2018). A reducing substrate, such as an aldehyde or aromatic heterocycle, is required to reduce the Moco from Mo(VI) to Mo(IV) as previously described in Fig. 29. The nitrite reduction then proceeds via sequential one electron transfer to two molecules of nitrite, reoxidizing the Moco from Mo(IV) to Mo(V) and then back to Mo(VI). Maia and Moura also demonstrated that the reaction is independent of the FAD center with experiments using an FAD inhibitor or enzyme lacking FAD.

Proposed mechanism for reduction of nitrite to nitric oxide in the presence of a reducing substrate by the molybdenum hydroxylases (Maia and Moura 2018)
Aldehyde oxidase 1 (AOX1)
Mammalian aldehyde oxidases (AOXs) are cytosolic molybdoflavoenzymes involved in the metabolism of drugs, natural and physiological compounds, and general chemicals (Tables 15, 17, 19). The enzymes participate not only in the detoxication of toxic metabolites endogenously formed by other enzymes such as P450s (e.g., aldehyde intermediates) but also in the production of toxic and therapeutically active metabolites (Tables 21, 22), and the generation of reactive oxygen species (ROS) as a byproduct of their enzymatic activity.
Enzymes
Different animal species are characterized by a different complement of aldehyde oxidase genes clustering at a short distance on the same chromosome (chromosome 2 in humans) (Terao et al. 2016). Humans contain a single active gene, AOX1, and two pseudogenes, while rodents are characterized by four active genes. Both AOX1 and AOX3 are major enzymes present in rodent liver (with the exception of guinea pigs, which only express AOX1 in the liver). The mouse Aox1 enzyme bears 85% sequence identity with human AOX1, whereas mouse Aox3 is only 65% identical to human AOX1 (Garattini et al. 2008). Primates, like humans, have only a single functional AOX enzyme (AOX1) in the liver, which bears 96% sequence identity with the human enzyme (Hoshino et al. 2007). Accordingly, marked species differences in AOX-mediated metabolism are common, and these differences present in a substrate-dependent manner (Beedham et al. 1987; Choughule et al. 2015; Crouch et al. 2018; Dalvie et al. 2013; Diamond et al. 2010; Hutzler et al. 2014; Sahi et al. 2008). However, AOX catalytic activity generally tends to be highest in monkeys and humans and lowest in mice and rats, whereas rabbits and guinea pigs tend to fall somewhere in between. Dog liver is completely devoid of an active AOX enzyme (Terao et al. 2016).
The human AOX1 protein has been reported in many tissues, including liver, pancreas, kidney, adrenal gland, thyroid gland, prostate, bladder, gastrointestinal tract, testis, bronchi, uterus, and skin (Moriwaki et al. 2001). The liver contains the highest concentration of AOX1 protein, though substantial quantities are also present in the adrenal glands. AOX1 mRNA expression has been found in many human tissues as well (Terao et al. 2016).
Humans have functionally inactive AOX1 allelic variants as well as variants encoding enzymes with different catalytic activities (i.e., slow and rapid metabolizers) (Foti et al. 2016; Hartmann et al. 2016; Mota et al. 2019). In addition, single nucleotide polymorphisms affecting the FAD binding site have been demonstrated to increase the rate of superoxide production (Foti et al. 2017). The clinical relevance of these variants has yet to be established. Garrido and Leimkühler demonstrated that the L438V variant, which produces a higher ratio of superoxide/H2O2 relative to the wild-type enzyme, is more extensively inactivated over time (i.e. inactivated by ROS production) relative to the wild type enzyme (Garrido and Leimkühler 2021). The L438V variant, which bears a single nucleotide polymorphism affecting the FAD binding site, produces superoxide at a rate of 75% compared to the amount of H2O2 produced, whereas the wild-type enzyme produces only 10% superoxide in comparison to H2O2.
As the name suggests, AOX catalyzes not only the aldehydes but can also catalyze the oxidation of aromatic azaheterocycles, as well as reductive reactions. AOX is best understood for its role in xenobiotic metabolism, but the enzyme may have additional physiological functions. Species differences in the expression and activity of AOX present a challenge in defining the physiological role(s) of human AOX. The list of endogenous substrates of AOX includes N-methylnicotinamide, pyridoxal, and all-trans-retinaldehyde (Table 17) (Johns 1967; Zhong et al. 2021). AOX has also been shown to be capable of reducing nitrites to nitric oxide and has been proposed to be involved in adipogenesis (Heid et al. 2020; Maia et al. 2015; Weigert et al. 2008). Due to its function in producing ROS, AOX may also contribute to pathological conditions resulting from oxidative stress (e.g., cancer). However, the exact physiological/pathological roles of AOX remain poorly understood.
Substrates
AOX is characterized by broad substrate specificity, in contrast to XOR, which has a specificity more limited to purine-like compounds. AOX has been most frequently reported to oxidize aromatic azaheterocycles, e.g. substituted pyrroles, pyridines, pyrimidines, purines, pteridines, and quinolines, among others (Dalvie and Di 2019; Garattini and Terao 2012; Kitamura et al. 2006; Manevski et al. 2019; Pryde et al. 2010). In addition, compounds containing iminium ions (often intermediate metabolites generated by P450s or MAOs) are relatively common AOX substrates. Aliphatic and aromatic aldehydes (which also often arise as intermediate metabolites) are oxidized by AOX as well. However, compounds containing aldehydes tend to be more efficiently oxidized by ALDH. AOX substrates for reduction include nitro-containing compounds, sulfoxides, N-oxides, and nitrites, as well as heterocycles such as isoxazoles and isothiazoles. More recently, AOX has also been demonstrated to be capable of hydrolyzing amides (Sodhi et al. 2015). While AOX and P450s have somewhat opposing substrate preferences due to different catalytic mechanisms (e.g. P450s prefer to oxidize electron-rich carbon atoms, whereas AOXs prefer electron deficient carbon atoms), they can sometimes share heterocyclic substrates and produce the same hydroxylated products (e.g., idelalisib) (Jin et al. 2015).
Inhibitors
A number of drugs, natural compounds, and general chemicals have been reported to inhibit human AOX (Tables 16, 18, 20). The strongest known inhibitor of AOX is the selective estrogen receptor modulator, raloxifene (IC50 2.9 nM), while several other drugs have also been also reported to strongly inhibit the enzyme, particularly phenothiazines (e.g. perphenazine, IC50 33 nM) (Obach et al. 2004). 17 β-Estradiol and 17α-ethinylestradiol are also strong inhibitors. In addition to a number of general chemical compounds (e.g., phenothiazines, dibenzazepines, flavonoids, purines, and pyrimidines) (Table 20), a number of diet-derived natural products have been found to inhibit AOX activity, including various catechins, flavones, flavonoids, flavonols, and flavanonols (Table 18). The compounds most commonly used to phenotype AOX reactions are raloxifene, menadione, and the time-dependent inhibitor hydralazine. Each of these three compounds is selective for AOX over XOR, but they exhibit varying degrees of inhibition toward various microsomal enzymes (Zientek and Youdim 2015). As the inhibition kinetics of AOX is complex (and substrate-dependent), the use of multiple probe substrates is recommended when results are used to assess the potential for drug–drug and/or drug–chemical interactions (Barr and Jones 2013). Clinically significant drug–drug interactions involving AOX inhibition have not been reported, with the exception of zaleplon and cimetidine. Zaleplon inhibits not only the AOX-mediated metabolism (of zaleplon) but also the CYP3A-mediated pathway, which represent approximately 70% and 30% of the fractional metabolism of zaleplon, respectively (Renwick et al. 2002).
Inducers
Limited reports are available on the regulation of AOX expression, particularly regarding the human enzyme. Maeda et al. demonstrated regulation of the human AOX gene involving the Nrf2 pathway (Maeda et al. 2012). In addition, Zhou et al. reported increased AOX-mediated metabolism of methotrexate, increased AOX protein levels, and a minimal increase in AOX mRNA following treatment of human hepatocytes with the nonsteroidal anti-inflammatory drug nimesulide (Zhou et al. 2020). Others have demonstrated induction of AOX activity, mRNA, and/or protein in animals by various compounds (e.g. phthalazine, dioxin) (Johnson et al. 1984; Rivera et al. 2005). Notably, androgens have been reported to increase AOX expression in rodents, while estrogens reduced expression, which is consistent with sex-dependent AOX activity observed in rodents (Al-Salmy 2001; Beedham 1985; Garattini and Terao 2012). Humans, however, appear to exhibit no sex-dependent differences in AOX activity, although estrogens are known to inhibit human AOX in vitro (Obach 2004).
Reactions
AOX catalyzes the oxidation of aromatic and aliphatic aldehydes into the corresponding carboxylic acids, hydroxylation of electrophilic carbon atoms on heteroaromatic rings, oxidation of iminium ion intermediates to the corresponding lactams, and hydrolysis of amides. AOX is also reported to catalyze the reductive metabolism of nitro groups, N-oxides, sulfoxides, isoxazoles, isothiazoles, benzisoxazoles, nitrite, and hydroxamic acids. Certain reductive transformations (e.g., nitro-reduction) have the potential to cause toxicity due to the formation of reactive metabolites. Reductive reactions may be accelerated in the presence of a reducing substrate, as was demonstrated for the reduction of dantrolene in the presence of N-methylnicotinamide (Amano et al. 2018).
AOX catalyzes the oxidation of heteroaromatic rings, iminium ions, and aldehydes. During the oxidation of the substrate, the enzyme is reduced and reoxidized with molecular oxygen, therefore behaving as an oxidase (Fig. 32). For both AOX and the XO form of XOR, the reduction of molecular oxygen produces the reactive oxygen species (ROS), hydrogen peroxide and superoxide anion, with AOX favoring the production of hydrogen peroxide (Foti et al. 2017). However, it has been suggested that AOX may produce more than 20-fold higher amounts of superoxide versus XOR, based on the enzymatic activities of the two enzymes in the human liver (Krenitsky et al. 1972; Kundu et al. 2007). Although NAD+ is the preferred electron acceptor for the XDH form of the XOR enzyme, it is capable of transferring electrons to O2 and thus producing ROS by acting as an NADH oxidase (Sanders et al. 1997).

Typical reaction catalyzed by AOX and XO enzymes
Garrido and Leimkühler recently reported that AOX is inactivated in a substrate-dependent manner by ROS production, with a high turnover substrate inactivating the enzyme more rapidly than a low turnover substrate (Garrido and Leimkühler 2021). Alternatively, because the enzyme inactivation was not prevented by ROS scavengers (catalase and superoxide dismutase) in their studies, Abbasi et al. reported that the enzyme inactivation results, not from ROS production, but rather from the rate-limiting transfer of electrons to O2, which is required to reoxidize the enzyme (Abbasi et al. 2019). Similar to the results obtained by Garrido and Leimkühler with AOX, Lynch and Fridovich previously reported that XO is autoinactivated by ROS production (Lynch and Fridovich 1979). Differences in incubation conditions were cited as a possible explanation for the discrepancy between the findings in the two AOX studies.
Other compounds can also serve as electron acceptors for AOX as well. While some accept electrons at the FAD site (e.g. 5-nitroquinoline) (Abbasi et al. 2019), where O2 accepts electrons, others may directly receive electrons from the Moco site (e.g. 2,6-dichlorophenolindophenol) (Foti et al. 2017; Garrido and Leimkühler 2021).
Examples of substrates and reactions resulting in the formation of nontoxic metabolites
Oxidation of aromatic heterocycles
Phthalazine (general chemical)
Phthalazine is a bicyclic heteroaromatic compound that is rapidly oxidized by AOX to phthalazone (Beedham et al. 1987, 1995) (Fig. 33). Phthalazine is commonly used as an AOX probe substrate (Table 19). The time-dependent AOX inhibitor hydralazine is a derivative of phthalazine. Because phthalazine and phthalazone are metabolites of hydralazine, it is not a suitable inhibitor to evaluate the AOX-mediated metabolism of phthalazine.

Oxidation of phthalazine by AOX
DACA (antineoplastic, DNA intercalating dual topoisomerase I/II poison)
While most heterocyclic substrates of AOX are oxidized on an electrophilic carbon atom adjacent to a nitrogen atom, in some cases the oxidation may occur para to the nitrogen, as is the case for the antineoplastic agent DACA (Fig. 34). DACA is oxidized to an acridone product by AOX on the carbon atom opposite the nitrogen in the acridine moiety (Schofield et al. 2000) (Table 15).

Oxidation of DACA by AOX
Lapatinib (antineoplastic, EGFR inhibitor)
Lapatinib, an anticancer agent known to be associated with hepatotoxicity, is debenzylated by P450 3A enzymes to a metabolite that subsequently undergoes metabolic activation via P450 3A enzymes (Castellino et al. 2012) (Fig. 35). Lapatinib and its debenzylated metabolite are also oxidized by AOX to AO-M1 and M3, respectively (Dick 2018), which may serve as a detoxication pathway in opposition to the bioactivation pathway (Table 15).

Bioactivation of lapatinib by P450 3A enzymes and oxidation by AOX
Zaleplon (nonbenzodiazepine sedative hypnotic)
Zaleplon is a dual AOX and P450 substrate, undergoing oxidation by AOX to 5-oxo-zaleplon (Table 15, Fig. 36) and N-dealkylation by P450 3A4 to desethyl-zaleplon. Species differences are noted in the fractional metabolism by AOX and P450, with 5-oxo-zaleplon representing the major metabolite in humans and monkeys (approximate fm,AO (fraction of metabolism due to AOX)) = 0.7 in humans) and desethyl-zaleplon representing the major metabolite in rodents (Crouch et al. 2018; Kawashima et al. 1999; Strelevitz et al. 2012). In addition, an interaction between cimetidine and zaleplon is one of the only known clinically relevant drug–drug interactions associated with AOX metabolism (Renwick et al. 2002). However, cimetidine inhibits not only the AOX metabolism pathway but also the P450 3A4 pathway as well.

Oxidation and N-deethylation of zaleplon by AOX and P450 3A enzymes
Idelalisib (antineoplastic, phosphatidylinositol 3-kinase inhibitor)
Despite differences in catalytic mechanism and general substrate preference, some metabolites may be produced by both AOX and P450 enzymes, as is the case for idelalisib. Both AOX and P450 3A4 catalyze the oxidation of idelalisib to the inactive metabolite GS-563117, which is a mechanism-based inactivator of P450 3A (Jin et al. 2015; Ramanathan et al. 2016) (Table 15) (Fig. 37).

Oxidation of idelalisib by AOX and P450 3A enzymes
Oxidation of aldehydes
All-trans-retinaldehyde (vitamin A derivative)
Retinal is the most well-studied endogenous substrate of AOX (Ambroziak et al. 1999; Zhong et al. 2021). The aldehyde undergoes oxidation to produce the active metabolite retinoic acid, a reaction that is catalyzed by both AOX and ALDH (Table 17) (Fig. 38). Zhong and coworkers recently reported that ALDH1A1 serves as the low Km, low kcat enzyme contributing to the biosynthesis of retinoic acid in the human liver, while AOX serves as the high Km, high kcat enzyme (Zhong et al. 2021).

Oxidation of retinaldehyde to retinoic acid by AOX
Citalopram aldehyde (metabolite of SSRI citalopram)
Aldehydes are relatively uncommon in parent drug molecules. However, they are often generated by enzymes such as P450s. Citalopram is demethylated and converted to an aldehyde metabolite by a combination of P450 and MAO enzymes (Rochat et al. 1998) (Table 8) (Fig. 39). The aldehyde metabolite undergoes subsequent oxidation by AOX to the carboxylic acid (Table 15).

Oxidation of citalopram by P450, MAO, and AOX enzymes to citalopram propionic acid
Oxidation of iminium ions
Like aldehyde intermediates, AOX has been found to oxidize iminium ion intermediates generated by P450s or MAOs. As iminium ions have the potential to produce toxic effects, AOX-mediated oxidation of iminium ion intermediates may serve as a detoxication pathway.
KW-2449 (multikinase inhibitor)
KW-2449 is a multikinase inhibitor that was previously under investigation for the treatment of leukemia. The drug, which has been discontinued from further development, displayed unexpected rapid metabolism to an oxo-piperazine metabolite in clinical trials (Hosogi et al. 2018). The pharmacologically active metabolite was determined to be generated via sequential metabolism by MAO B to an iminium ion intermediate, followed by AOX-mediated oxidation to the oxo-piperazine metabolite (Table 15, Fig. 40). In addition, the iminium ion intermediate was found to be a time-dependent inhibitor of AO, reducing exposure of the active metabolite following repeat dosing (Table 16). Interspecies differences in both MAO B and AOX likely contributed to the failure to recognize this metabolic pathway prior to clinical trials.

Oxidation of KW-2449 to an oxo-piperazine metabolite by MAO B and AOX
Reduction of N-oxides
Oxycodone N-oxide (metabolite of the opioid analgesic oxycodone)
Oxycodone is extensively metabolized by P450 enzymes, but it is also converted to oxycodone N-oxide by FMO3 (Cashman et al. 2020). The N-oxide metabolite was found to be retro-reduced back to oxycodone by AOX, quinone reductase, and hemoglobin (Table 15) (Fig. 41). Consequently, interindividual variability in AOX activity could potentially influence the duration of action and toxicity of oxycodone across patients.

N-Oxidation of oxycodone by FMO and retro-reduction by AOX and other enzymes
Reduction of S-oxides
Sulindac (NSAID)
Fewer examples of AOX-mediated S-oxidation are available in the literature relative to N-oxidation. The reduction of the nonsteroidal anti-inflammatory drug sulindac to its pharmacologically active sulfide metabolite represents one example of this reaction (Sung et al. 2020) (Table 15). Alternatively, the sulfide can be oxidized back to sulindac by FMO enzymes, which also oxidize sulindac to an inactive sulfone metabolite (Table 3, Fig. 42).

S-Oxidation of sulindac by FMO and sulfoxide reduction by AOX
Reduction of heterocycles
Ziprasidone (second generation antipsychotic)
Ziprasidone represents an example of a drug containing a heterocycle that is reduced by AOX (Miao et al. 2005; Prakash et al. 1997). The drug is extensively metabolized to multiple metabolites, including several oxidative P450 metabolites. However, the major circulating metabolite results from AOX-mediated reductive cleavage of the benzisothiazole ring, followed by methylation of the thiol (Table 15, Fig. 43).

Benzisothiazole reduction and thiol methylation of ziprasidone by AOX and thiol methyltransferase
Toxic effects of drugs as substrates of AO catalyzed reactions
Several examples of therapeutically successful candidate drugs tested in animal models have been removed from further testing due to differences in the formation of a toxic metabolite in preclinical species relative to humans (e.g., SGX523) (Hutzler et al. 2013; Manevski et al. 2019). Some examples of drugs converted into toxic metabolites by AOX are listed in Table 22. Alternatively, examples of drugs that are converted to active metabolites by AOX are listed in Table 21, which includes metabolites possessing desirable cytotoxic properties (e.g., anti-cancer agents).
Reduction of nitro-groups
Drugs containing nitro groups have been associated with mutagenicity and genotoxicity. AOX is capable of reducing nitro-groups to their corresponding amines, producing a hydroxylamine intermediate in the process. As hydroxylamines are reactive species that have the potential to produce toxic effects, the toxicities (e.g., hepatotoxicity) associated with nitro-aromatic containing drugs such as nimesulide, nitrazepam, dantrolene, and others (Table 22) may result, at least in part, from AOX-mediated nitro-reduction of these drugs.
Nitrazepam (benzodiazepine)
Koneshi et al. demonstrated that AOX participates in the reduction of nitrazepam to hydroxylaminonitrazepam and aminonitrazepam (Konishi et al. 2017). The aminonitrazepam metabolite is further metabolized by N-acetyltransferases (NATs) to N-acetylaminonitrazepam (Table 15, Fig. 44).

Nitro reduction and N-acetylation of nitrazepam by AOX and NAT
Dantrolene (skeletal muscle relaxant)
Dantrolene, like nitrazepam, contains a nitro-group that is reduced to a hydroxylamine, followed by a second reduction to aminodantrolene (Amano et al. 2018; Ogiso et al. 2018) (Table 22). Aminodantrolene is also N-acetylated by NAT2 (Fig. 45). Dantrolene carries a black box warning for severe hepatotoxicity, which is attributed to the formation of the hydroxylamine intermediate.

Nitro reduction and N-acetylation of dantrolene by AOX and NAT
Oxidation of heterocycles to poorly soluble lactam metabolites
SGX523 (antineoplastic, c-MET inhibitor)
SGX-523 is an anticancer agent that was discontinued in clinical trials due to renal toxicity that went undetected in toxicity studies conducted in rats and dogs (Infante et al. 2013). Diamond et al. determined that the toxicity likely resulted from precipitation of an AOX metabolite in the renal tubules, which was not observed in animals studies due to species differences in AO activity (Diamond et al. 2010) (Tables 15, 22). The 2-quinolinone (Fig. 46) metabolite was undetected in dog liver post-mitochondrial supernatant fraction (S9) and only trace amounts were produced in rat S9, unlike human and monkey S9, in which the 2-quinolinone was a major metabolite. Following administration of SGX-523 to cynomologus monkeys, the 2-quinolinone metabolite was present in urine at concentrations 70-fold higher than the parent drug and its solubility was only 3% of the parent solubility. A structural analog of SGX-523, JNJ-38877605, was also discontinued during clinical trials due to renal toxicity, which was presumed to occur via the same AOX-mediated mechanism (Lolkema et al. 2015). JNJ-38877605 has a difluoro-substituted ether linkage rather than a thioether linkage and is otherwise structurally identical to SGX-523.

Oxidation of SGX-523 to a poorly soluble lactam metabolite by AOX
Methotrexate (antineoplastic, antirheumatic, antifolate)
Methotrexate is also known to cause renal toxicity, particularly when administered in high doses (Jordan et al. 1999). As with SGX-523 and JNJ-38877605, renal toxicity is believed to be associated with AOX-mediated oxidation to 7-hydroxymethotrexate, a poorly soluble metabolite (Table 22, Fig. 47).

Oxidation of methotrexate to a poorly soluble lactam metabolite by AOX
Hydrolysis of anilides
GDC-0834 (antirheumatic, Bruton’s tyrosine kinase inhibitor)
GDC-0834 was previously under investigation for the treatment of rheumatoid arthritis but was discontinued from further development due to rapid hydrolysis of the anilide moiety, producing an aniline metabolite (Liu et al. 2011; Sodhi et al. 2015) (Fig. 48). While neither preclinical nor clinical toxicity was reported as a concern or reason for discontinuation of GDC-0834, anilines are known to have the potential to produce toxic effects, if for no reason than guilt by association. Lepri et al. evaluated a series of anilide-containing compounds for their susceptibility to AOX-mediated amide hydrolysis and found several of the compounds to be AOX substrates, thus highlighting this reaction as a potential source of toxicity for anilide-containing drugs (Lepri et al. 2017) (Table 15).

Hydrolysis of GDC-0834 to an aniline metabolite by AOX
Sodhi et al. have proposed a mechanism for the hydrolysis of GDC-0834, on the basis of in silico modeling, that involves a nucleophilic reaction between a hydroxyl group of the Moco and the carbonyl group of the anilide (Sodhi et al. 2015). The authors speculated that this reaction would be more likely to take place with the Moco in the reduced state (Fig. 49) due to the higher electron density, which would require the presence of a reducing substrate. Once the enzyme has initially been reduced, the proposed hydrolysis mechanism does not require any transfer of electrons in order to complete the catalytic cycle, meaning that the entire process would take place at the Moco center without the involvement of the FAD and 2Fe-2S centers.

Proposed mechanism of hydrolysis of GDC-0834 by AOX
Xanthine oxidoreductase (XOR)
Xanthine oxidoreductase (XOR), perhaps the better-known molybdenum hydroxylase, plays an important role in the catabolism of endogenous purines and pyrimidines in humans, as well as drugs such as thiopurines and methyl-xanthine compounds (Tables 23, 25, 27). XOR is less promiscuous than AOX, preferring substrates that are more purine-like, although some compounds are substrates for both enzymes.
Enzymes
Unlike AOX, XOR can exist in two interconvertible forms, as a dehydrogenase (XDH), which prefers NAD+ as an electron acceptor, or an oxidase (XO), which can only transfer electrons to O2. Based upon studies with rat liver and bovine milk XOR, the XDH form can be postranslationally modified to the XO form either irreversibly through limited proteolysis, or reversibly through the formation of two disulfide bonds involving four cysteine residues (Battelli et al. 1973; Corte and Stirpe 1972; Della Corte and Stirpe 1968; Stirpe and Della Corte 1969). In both cases, either reversible or irreversible conversion from XDH to XO, the modification takes place within a peptide that links the Moco- and FAD-containing domains.
Tissue distribution of XOR is species-dependent, with lower constitutive expression in humans relative to other mammals, presumably due to promotor suppression (Xu et al. 2000). Human XOR enzymes have been found in the lactating mammary gland, intestine, liver, lungs, kidneys, and vascular endothelium, with the highest specific activity in the liver and intestine (Linder et al. 1999; Moriwaki et al. 1996). In addition, relative levels of an inactive enzyme (for example, de-molybdo and/or de-sulfo forms of the enzyme) may contribute to species differences in the tissue distribution of XOR activity (Battelli et al. 2014). For example, in human milk, active XOR was found to account for < 2% of the total enzyme content, and the xanthine oxidizing activity in human milk was found to be 2–3 orders of magnitude lower than bovine milk, despite similar total XOR enzyme content (Abadeh et al. 1992).
XOR has been detected in the vascular endothelial cells of various human tissues (Kooij et al. 1992; Linder et al. 1999; Moriwaki et al. 1993). While XOR is a cytosolic enzyme like AOX, it has also been detected on the outer surface of bovine and porcine endothelial cells (Vickers et al. 1998). XOR is also present (in the XO form) in circulating plasma, although the constitutive presence in plasma is species-dependent (Al-Khalidi and Chaglassian 1965). For example, studies evaluating the plasma stability of a quinoxaline-containing compound within a series of cyanopyridine derivatives revealed a species-specific oxidation of the quinaoxaline mediated by plasma XOR (Sharma et al. 2011). The quinoxaline-containing compound was rapidly degraded in rat, mouse, and guinea pig plasma but not in dog, monkey, or human plasma. The lactam metabolite was also detected in human liver cytosol; however, a further evaluation to distinguish whether the metabolite was produced by human hepatic XOR vs AOX was not reported. Though plasma levels of XOR in healthy humans is low, increased levels of circulating plasma XOR have been associated with various pathological conditions, primarily related to hepatic injury, including viral hepatitis, toxic agents, transplantation, hypoxia, and ischemia/reperfusion (Battelli et al. 2014). Other conditions that have been associated with elevated circulating XOR include pneumonia, type 2 diabetes, post-surgical procedures, and sickle cell disease, among others (Battelli et al. 2014).
Products of XOR catalyzed reactions have been associated with both beneficial and toxic effects and elevated XOR activity has been connected to different pathological conditions causing tissue damage and cell necrosis (Battelli et al. 2016b; Bortolotti et al. 2021; Harrison 2002). Uric acid, the product of xanthine oxidation, and NO, the product of nitrite reduction, play a role in blood pressure regulation and vascular tone. In addition, uric acid has anti-oxidant activity, contributes to the inflammatory response, and promotes gluconeogenesis and fat accumulation. Consequently, XOR may play a role in the pathogenesis of metabolic syndrome and insulin resistance. While ROS produced by XOR can contribute to pathological conditions associated with oxidative stress such as cancer, uric acid plays a role in preventing these pathological conditions. Beneficial effects are also derived from ROS production, for example, via their bactericidal action.
Substrates
Similar to AOX, XOR utilizes a variety of heterocycles and some aldehydes as substrates. However, XOR has a narrower specificity than AOX, generally preferring substrates that are more purine-like (Tables 23, 25, 27). Oxidation of purines occurs at C-atoms in positions C2-, C6-, and C8- (Okamoto et al. 2013) (Fig. 50). The affinity of C-atoms for oxidation increases with the number of adjacent N-atoms.

Oxygenation of purine compounds by XOR enzymes
The substrate specificity for AOX and XOR does sometimes overlap. For example, 6-mercaptopurine is oxidized by both AOX and XOR to 6-thioxanthine, whereas oxidation of 6-thioxanthine to 6-thiouric acid is catalyzed by XOR alone (Choughule et al. 2014). In some cases, a compound may be a dual substrate for XOR and AOX, but the site of metabolism may differ. Examples of this observation in the literature, however, have been determined using non-human sources of XOR and/or AOX. For example, 6-deoxyacyclovir was reported to be oxidized to the active metabolite acyclovir by bovine milk XOR and to the inactive metabolite 8-hydroxy-6-deoxyacyclovir by rabbit liver AOX (Krenitsky et al. 1984). In fact, many compounds not reported in this review are substrates for mammalian XOR or AOX but have not been confirmed to be substrates of the human enzymes. XOR studies in particular have been frequently carried out using a non-human enzyme source, most often bovine milk XOR owing to its wide availability and low cost. Due to species differences in AOX/XOR substrate specificities, it cannot be assumed that a substrate for a non-human enzyme is also a substrate for the human enzyme. For example, using enzyme-selective inhibitors in multiple species of S9 preparations, the 6-oxopyrimidine metabolite of VU424238, for example, was found to be oxidized to a 2,6-oxopyrimidine metabolite by either AOX or XOR in a species-dependent manner (Crouch et al. 2017).
Inhibitors
As previously mentioned, XOR is responsible for the conversion of hypoxanthine to xanthine and xanthine to uric acid, the accumulation of which is associated with the pathophysiology of gout. Consequently, inhibition of XOR is a therapeutic strategy in the treatment of gout, and multiple drugs that inhibit XOR are on the market (allopurinol, febuxostat, and topiroxostat). Allopurinol has been in use since the 1960s, whereas febuxostat only received FDA approval in 2009. Topiroxostat is not available in the United States but was approved for use in Japan in 2013. Allopurinol is a substrate of both XOR and AOX and is converted to the active metabolite oxypurinol, which forms a covalent bond with Mo(IV) and strongly inhibits XOR (Okamoto et al. 2008). Febuxostat is characterized as a structure-based inhibitor and can bind to the enzyme regardless of the Moco oxidation state (Okamoto et al. 2003). Topiroxostat initially displays competitive inhibition, followed by a covalent type of inhibition, based on studies with bovine milk XOR (Matsumoto et al. 2011). Several additional purine and nonpurine-like compounds have been found to inhibit XOR but are not utilized as clinical XOR inhibitors (Tables 24, 26, 28).
Inducers
Constitutive activity of XOR in human tissues is relatively low in comparison with other mammals (Harrison 2002). Increased activity and/or expression of XOR in various tissues has been associated with several compounds, including interferon, 3-methylcholanthrene, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), prolactin, and cortisol in mice (Ghezzi et al. 1984; McManaman et al. 2000; Sugihara et al. 2001), and sodium metabisulfite and phenytoin in rats (Ercan et al. 2015; Ekaidem et al. 2014). TCDD and 3-methylcholanthrene also increase AOX activity in mouse liver, and these effects were diminished in aryl hydrocarbon receptor-null mice (Sugihara et al. 2001). Cytokines have also been shown to increase XOR activity on a transcriptional and/or post-translational level in rodent, bovine and human cultured cells of various tissues (Dupont et al. 1992; Page et al. 1998; Pfeffer et al. 1994). Menadione, an inhibitor of AOX, stimulates human XOR activity, presumably by functioning as an electron acceptor to reoxidize the enzyme following substrate oxidation (Ferreira Antunes et al. 2016).
Examples of substrates and reactions resulting in the formation of nontoxic metabolites
Hypoxanthine and xanthine (endogenous purines)
XOR catalyzes the oxidation of hypoxanthine into xanthine and further oxidizes xanthine into uric acid (Fig. 51) (Table 22) (Okamoto et al. 2013). In the purine metabolism, hydroxylation of hypoxanthine (6-hydroxypurine) initially takes place at the 2-position, yielding xanthine (2,6-dihydroxypurine). The next hydroxylation occurs at the 8-position, affording uric acid (2,6,8-trihydroxypurine). Studies using bovine milk XOR indicated that xanthine accumulates prior to conversion into uric acid, suggesting that oxidation of the 2-position influences oxidation of the 8-position (Okamoto et al. 2013). In addition, 6,8-dihydroxypurine was not detected. Dimethylated (theophylline, theobromine) and trimethylated xanthine derivatives (caffeine) are better substrates of cytochrome P450 enzymes than XOR.

Oxidation of hypoxanthine to xanthine and uric acid by XOR
Allopurinol and oxypurinol (antigout, XOR inhibitor)
Allopurinol is a substrate and specific XOR inhibitor. An analog of hypoxanthine, allopurinol has a nitrogen atom in 8-position with a carbon atom in the 7-position. Both AOX and XOR can metabolize allopurinol to the active metabolite oxypurinol (Fig. 52) (Tables 15, 23), although this reaction is probably mediated primarily by AOX. Allopurinol is both a competitive (at lower concentrations), and uncompetitive inhibitor (at higher concentrations) of hypoxanthine and xanthine oxidations, catalyzed by XOR (Table 24). Oxypurinol is an uncompetitive inhibitor and covalently binds to the reduced form of XOR (Okamoto et al. 2008; Spector 1988; Spector et al. 1986) (Table 24). Allopurinol/oxypurinol inhibit the conversion of hypoxanthine and xanthine to uric acid, thus regulating blood urate levels and is used to treat gout and hyperuricemia. In addition, it was suggested that allopurinol, by suppression of XOR activity, ameliorates myocardial inefficiency and poor vascular flow, and accordingly, may present an innovative contribution to the future treatment of ischemia and reperfusion (I/R) injury in heart failure patients (Harzand et al. 2012; Lee et al. 2009a, b). Allopurinol is also associated with potentially life-threatening severe cutaneous adverse reactions (Table 29) for which the HLA-B*5801 allele has been identified as a genetic risk factor (Hung et al. 2005).

Oxidation of allopurinol to oxypurinol by AOX and XOR enzymes
When human liver tissue is harvested, it is commonly perfused with a solution containing allopurinol to prevent XOR-related oxidative damage. Barr et al. reported the presence of both allopurinol and oxypurinol at micromolar concentrations in cytosolic human liver fractions obtained from livers perfused with an allopurinol-containing solution, with a corresponding lack of XOR activity in these samples (Barr et al. 2014). Importantly, the authors noted that commercial liver fractions are likely to contain residual allopurinol and/or oxypurinol and should be screened prior to use in metabolism studies.
Acyclovir (prodrug, antiviral, antiherpetic)
6-Deoxyacyclovir is an example of a prodrug that is activated by the catalytic activity of XOR (Fig. 53). 6-Deoxyacyclovir is converted into the active drug acyclovir via 6-oxidation (Krenitsky et al. 1986; Rees et al. 1986) (Table 29). Rabbit liver AOX was found to oxidize both 6-deoxyacyclovir and acyclovir at the 8-position to inactive metabolites (Krenitsky et al. 1984), but whether or not this deactivating reaction is catalyzed by human AOX has not been reported.

Oxidation of 6-deoxyacyclovir to the active metabolite acyclovir by XOR
6-Mercaptopurine (antineoplastic)
6-Mercaptopurine, a thiopurine drug, can be oxidized to 6-thioxanthine (6TX) and 6-thiouric acid (6TUA) through 6TX as an intermediate (Fig. 54). Both AOX and XOR are found to be involved in the formation of the 6TX intermediate, whereas only XOR was responsible for the conversion of 6TX to 6TUA (Choughule et al. 2014) (Tables 15, 23). In addition, both the xanthine dehydrogenase (XDH) and xanthine oxidase (XO) forms of XOR were evaluated and found to contribute to the formation of 6TX and 6TUA in studies with human liver cytosol in the presence and absence of NAD+, the preferential electron acceptor for XDH.

Oxidation of 6-mercaptopurine to 6-thiouric acid by AOX, XO, and XDH
Toxic effects of drugs as substrates of XOR catalyzed reactions
Pyrazinamide (antituburculosis prodrug)
Pyrazinamide is a prodrug used to treat tuberculosis, but it is associated with dose-related hepatotoxicity. Pyrazinamide is converted into the active metabolite pyrazioic acid by amidases, and it can also be oxidized by XOR to 5-hydroxypyrazinamide (5-OH-PZN) (Fig. 55) (Lacroix et al. 1989; Yamamoto et al. 1987) (Table 23). Both metabolites can undergo further conversion to 5-hydroxypyrazinoic acid (5-OH-PA) via the action of XOR on pyrazinoic acid or amidases on 5-hydroxypyrazinamide. The 5-OH-PA metabolite is proposed to be primarily responsible for the hepatotoxicity associated with pyrazinamide, as inhibition of amidase activity decreased pyrazinamide-induced hepatotoxicity, but did not prevent pyrazinoic acid-induced hepatotoxicity in rats (Shih et al. 2013). These data were also supported by in vitro studies demonstrating increased toxicity of pyrazinoic acid and 5-OH-PA relative to pyrazinamide in HepG2 cells. In addition, greater hepatotoxicity was observed in tuberculosis patients receiving pyrazinamide who had higher urine ratios of pyrazinoic acid/pyrazinamide and 5-OH-PA/pyrazinamide.

Metabolism of pyrazinamide to active and toxic metabolites via amidase and XOR catalyzed reactions
Concluding remarks
We have presented an overview of the metabolic reactions of drugs, natural products, physiological compounds, and other (general) chemicals catalyzed by the major non-P450 human oxidoreductase enzymes, i.e., FMOs, MAOs, NQOs, and molybdenum hydroxylases (AOX and XOR). All of these enzymes, in addition to their roles of facilitating excretion of exogenous and endogenous compounds, also catalyze reactions producing toxic products from both physiological compounds (e.g., bioactivation of neurotransmitters by MAO enzymes activity), as well as from xenobiotic compounds under specific conditions (e.g., supra-physiological substrate concentrations, anaerobic vs aerobic conditions, presence of specific inhibitors, presence/absence of cofactors, enzyme polymorphism). The participation of non-P450 oxidoreductases in the activation reactions forming toxic products is relatively low, compared to P450 enzymes (Rendić and Guengerich 2012, 2015). However, important therapeutic agents (including antibiotics, antibacterial, antitubercular, and CNS stimulants) (Table 7) are substrates in some bioactivation reactions catalyzed by FMO enzymes. An important role of MAO inhibitors is as drugs that are used in the clinic to treat depression (Table 11), and potential roles exist for natural products and their derivatives (Tables 12, 13). In addition, potential roles for toxic/reactive metabolites in the MAO-catalyzed metabolic reactions have to be considered with neurotransmitters as substrates (i.e., formation of aldehydes and H2O2) (Table 14). The toxic products can be eliminated by detoxication reactions catalyzed by aldehyde dehydrogenases and aldehyde reductases. Another example of detoxication is illustrated by the deactivation of highly reactive DOPA quinone, which might be formed by oxidation of dopamine or 3,4-l-DOPA by the catalytic activity of tyrosine oxidase (Asanuma et al. 2003; Ito et al. 2020). DOPA quinone might be deactivated by NQO enzymes or by conjugation with glutathione. These examples illustrate the multiple factors that can affect bioactivation/detoxication reactions and the outcome of the metabolic reactions with a particular compound as substrate. The literature also indicates that a number of compounds that are substrates of non-P450 oxidoreductases are also substrates of one or more P450 or other enzymes, and they might also interact with drug transporters in addition. Thus, multiple metabolic properties of a compound/drug have to be considered when drug–drug metabolic interactions or toxicity caused by a compound is evaluated.
Availability of data and materials
All the data are available in the text and tables of the review.
Code availability
Not applicable.
References
Abadeh S, Killacky J, Benboubetra M, Harrison R (1992) Purification and partial characterization of xanthine oxidase from human milk. Biochim Biophys Acta 1117(1):25–32. https://doi.org/10.1016/0304-4165(92)90157-p
Abbasi A, Paragas EM, Joswig-Jones CA, Rodgers JT, Jones JP (2019) Time course of aldehyde oxidase and why it is nonlinear. Drug Metab Dispos 47(5):473–483. https://doi.org/10.1124/dmd.118.085787
Abbasi A, Joswig-Jones CA, Jones JP (2020) Site-directed mutagenesis at the molybdenum pterin cofactor site of the human aldehyde oxidase: Interrogating the kinetic differences between human and cynomolgus monkey. Drug Metab Dispos 48(12):1364–1371. https://doi.org/10.1124/dmd.120.000187
Acheampong AA, Chien DS, Lam S et al (1996) Characterization of brimonidine metabolism with rat, rabbit, dog, monkey and human liver fractions and rabbit liver aldehyde oxidase. Xenobiotica 26(10):1035–1055. https://doi.org/10.3109/00498259609167421
Adali O, Carver GC, Philpot RM (1998) Modulation of human flavin-containing monooxygenase 3 activity by tricyclic antidepressants and other agents: importance of residue 428. Arch Biochem Biophys 358(1):92–97. https://doi.org/10.1006/abbi.1998.0835
Adali O, Carver GC, Philpot RM (1999) The effect of arginine-428 mutation on modulation of activity of human liver flavin monooxygenase 3 (FMO3) by imipramine and chlorpromazine. Exp Toxicol Pathol 51(4–5):271–276. https://doi.org/10.1016/s0940-2993(99)80004-9
Adusumalli S, Jamwal R, Obach RS, Ryder TF, Leggio L, Akhlaghi F (2019) Role of molybdenum-containing enzymes in the biotransformation of the novel ghrelin receptor inverse agonist PF-5190457: A reverse translational bed-to-bench approach. Drug Metab Dispos 47(8):874–882. https://doi.org/10.1124/dmd.119.087015
Akabane T, Tanaka K, Irie M, Terashita S, Teramura T (2011) Case report of extensive metabolism by aldehyde oxidase in humans: pharmacokinetics and metabolite profile of FK3453 in rats, dogs, and humans. Xenobiotica 41(5):372–384. https://doi.org/10.3109/00498254.2010.549970
Al-Khalidi UA, Chaglassian TH (1965) The species distribution of xanthine oxidase. Biochem J 97(1):318–320. https://doi.org/10.1042/bj0970318
Al-Salmy HS (2001) Individual variation in hepatic aldehyde oxidase activity. IUBMB Life 51(4):249–253. https://doi.org/10.1080/152165401753311799
Al-Waiz M, Ayesh R, Mitchell SC, Idle JR, Smith RL (1987) A genetic polymorphism of the N-oxidation of trimethylamine in humans. Clin Pharmacol Therapeut 42:588–594
Alfaro JF, Jones JP (2008) Studies on the mechanism of aldehyde oxidase and xanthine oxidase. J Org Chem 73(23):9469–9472. https://doi.org/10.1021/jo801053u
Alfaro JF, Joswig-Jones CA, Ouyang W, Nichols J, Crouch GJ, Jones JP (2009) Purification and mechanism of human aldehyde oxidase expressed in Escherichia coli. Drug Metab Dispos 37(12):2393–2398. https://doi.org/10.1124/dmd.109.029520
Amano T, Fukami T, Ogiso T et al (2018) Identification of enzymes responsible for dantrolene metabolism in the human liver: A clue to uncover the cause of liver injury. Biochem Pharmacol 151:69–78. https://doi.org/10.1016/j.bcp.2018.03.002
Ambroziak W, Izaguirre G, Pietruszko R (1999) Metabolism of retinaldehyde and other aldehydes in soluble extracts of human liver and kidney. J Biol Chem 274(47):33366–33673. https://doi.org/10.1074/jbc.274.47.33366
Anastassova N, Aluani D, Kostadinov A et al (2021) Evaluation of the combined activity of benzimidazole arylhydrazones as new anti-Parkinsonian agents: monoamine oxidase-B inhibition, neuroprotection and oxidative stress modulation. Neural Regen Res 16(11):2299–2309. https://doi.org/10.4103/1673-5374.309843
Anderson LW, Collins JM, Klecker RW et al (2005) Metabolic profile of XK469 (2(R)-[4-(7-chloro-2-quinoxalinyl)oxyphenoxy]-propionic acid; NSC698215) in patients and in vitro: low potential for active or toxic metabolites or for drug–drug interactions. Cancer Chemother Pharmacol 56(4):351–357. https://doi.org/10.1007/s00280-004-0962-3
Apenova N, Peng H, Hecker M, Brinkmann M (2018) A rapid and sensitive fluorometric method for determination of aldehyde oxidase activity. Toxicol Appl Pharmacol 341:30–37. https://doi.org/10.1016/j.taap.2018.01.006
Asano D, Shibayama T, Shiozawa H et al (2021) Evaluation of species differences in the metabolism of the selective Na(V)1.7 inhibitor DS-1971a, a mixed substrate of cytochrome P450 and aldehyde oxidase. Xenobiotica 51(9):1060–1070. https://doi.org/10.1080/00498254.2021.1963009
Asanuma M, Miyazaki I, Ogawa N (2003) Dopamine- or L-DOPA-induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox Res 5(3):165–176. https://doi.org/10.1007/bf03033137
Attar M, Dong D, Ling KH, Tang-Liu DD (2003) Cytochrome P450 2C8 and flavin-containing monooxygenases are involved in the metabolism of tazarotenic acid in humans. Drug Metab Dispos 31(4):476–481. https://doi.org/10.1124/dmd.31.4.476
Bach AW, Lan NC, Johnson DL et al (1988) cDNA cloning of human liver monoamine oxidase A and B: molecular basis of differences in enzymatic properties. Proc Natl Acad Sci USA 85(13):4934–4938. https://doi.org/10.1073/pnas.85.13.4934
Bach MV, Coutts RT, Baker GB (1999) Involvement of CYP2D6 in the in vitro metabolism of amphetamine, two N-alkylamphetamines and their 4-methoxylated derivatives. Xenobiotica 29(7):719–732. https://doi.org/10.1080/004982599238344
Baek SC, Lee HW, Ryu HW et al (2018a) Selective inhibition of monoamine oxidase A by hispidol. Bioorg Med Chem Lett 28(4):584–588. https://doi.org/10.1016/j.bmcl.2018.01.049
Baek SC, Ryu HW, Kang MG et al (2018b) Selective inhibition of monoamine oxidase A by chelerythrine, an isoquinoline alkaloid. Bioorg Med Chem Lett 28(14):2403–2407. https://doi.org/10.1016/j.bmcl.2018.06.023
Baek SC, Kang MG, Park JE et al (2019a) Osthenol, a prenylated coumarin, as a monoamine oxidase A inhibitor with high selectivity. Bioorg Med Chem Lett 29(6):839–843. https://doi.org/10.1016/j.bmcl.2019.01.016
Baek SC, Park MH, Ryu HW et al (2019b) Rhamnocitrin isolated from Prunus padus var. seoulensis: a strong and selective reversible inhibitor of human monoamine oxidase A. Bioorg Chem 83:317–325. https://doi.org/10.1016/j.bioorg.2018.10.051
Bajpai P, Sangar MC, Singh S et al (2013) Metabolism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by mitochondrion-targeted cytochrome P450 2D6: implications in Parkinson disease. J Biol Chem 288(6):4436–4451. https://doi.org/10.1074/jbc.M112.402123
Baker GB, Urichuk LJ, McKenna KF, Kennedy SH (1999) Metabolism of monoamine oxidase inhibitors. Cell Mol Neurobiol 19(3):411–426. https://doi.org/10.1023/a:1006901900106
Baker G, Matveychuk D, MacKenzie EM, Holt A, Wang Y, Kar S (2019) Attenuation of the effects of oxidative stress by the MAO-inhibiting antidepressant and carbonyl scavenger phenelzine. Chem Biol Interact 304:139–147. https://doi.org/10.1016/j.cbi.2019.03.003
Balis ME (1976) Uric acid metabolism in man. Adv Clin Chem 18:213–246. https://doi.org/10.1016/s0065-2423(08)60299-1
Balis FM, Holcenberg JS, Zimm S et al (1987) The effect of methotrexate on the bioavailability of oral 6-mercaptopurine. Clin Pharmacol Ther 41(4):384–387. https://doi.org/10.1038/clpt.1987.45
Ballard JE, Prueksaritanont T, Tang C (2007) Hepatic metabolism of MK-0457, a strong aurora kinase inhibitor: interspecies comparison and role of human cytochrome P450 and flavin-containing monooxygenase. Drug Metab Dispos 35(9):1447–1451. https://doi.org/10.1124/dmd.107.015438
Barr JT, Choughule KV, Nepal S et al (2014) Why do most human liver cytosol preparations lack xanthine oxidase activity? Drug Metab Dispos 42(4):695–699. https://doi.org/10.1124/dmd.113.056374
Barr JT, Jones JP (2011) Inhibition of human liver aldehyde oxidase: implications for potential drug–drug interactions. Drug Metab Dispos 39(12):2381–2386. https://doi.org/10.1124/dmd.111.041806
Barr JT, Jones JP (2013) Evidence for substrate-dependent inhibition profiles for human liver aldehyde oxidase. Drug Metab Dispos 41(1):24–29. https://doi.org/10.1124/dmd.112.048546
Barr JT, Jones JP, Joswig-Jones CA, Rock DA (2013) Absolute quantification of aldehyde oxidase protein in human liver using liquid chromatography-tandem mass spectrometry. Mol Pharmacol 10(10):3842–3849. https://doi.org/10.1021/mp4003046
Barr JT, Jones JP, Oberlies NH, Paine MF (2015) Inhibition of human aldehyde oxidase activity by diet-derived constituents: structural influence, enzyme-ligand interactions, and clinical relevance. Drug Metab Dispos 43(1):34–41. https://doi.org/10.1124/dmd.114.061192
Başaran R, Can Eke B (2017) Flavin containing monooxygenases and metabolism of xenobiotics. Turk J Pharm Sci 14(1):90–94. https://doi.org/10.4274/tjps.30592
Battelli MG, Lorenzoni E, Stripe F (1973) Milk xanthine oxidase type D (dehydrogenase) and type O (oxidase). Purification, interconversion and some properties. Biochem J 131(2):191–198. https://doi.org/10.1042/bj1310191
Battelli MG, Bolognesi A, Polito L (2014) Pathophysiology of circulating xanthine oxidoreductase: new emerging roles for a multi-tasking enzyme. Biochim Biophys Acta 1842(9):1502–1517. https://doi.org/10.1016/j.bbadis.2014.05.022
Battelli MG, Polito L, Bortolotti M, Bolognesi A (2016a) Xanthine oxidoreductase in cancer: more than a differentiation marker. Cancer Med 5(3):546–557. https://doi.org/10.1002/cam4.601
Battelli MG, Polito L, Bortolotti M, Bolognesi A (2016b) Xanthine oxidoreductase in drug metabolism: beyond a role as a detoxifying enzyme. Curr Med Chem 23(35):4027–4036. https://doi.org/10.2174/0929867323666160725091915
Battelli MG, Bortolotti M, Polito L, Bolognesi A (2018) The role of xanthine oxidoreductase and uric acid in metabolic syndrome. Biochim Biophys Acta Mol Basis Dis 1864(8):2557–2565. https://doi.org/10.1016/j.bbadis.2018.05.003
Beckett AH, Navas GE, Hutt AJ (1988) Metabolism of chlorpromazine and promazine in vitro: isolation and characterization of N-oxidation products. Xenobiotica 18(1):61–74. https://doi.org/10.3109/00498258809055137
Beedham C (1985) Molybdenum hydroxylases as drug-metabolizing enzymes. Drug Metab Rev 16(1–2):119–156. https://doi.org/10.3109/03602538508991432
Beedham C, Bruce SE, Critchley DJ, al-Tayib Y, Rance DJ, (1987) Species variation in hepatic aldehyde oxidase activity. Eur J Drug Metab Pharmacokinet 12(4):307–310. https://doi.org/10.1007/bf03189919
Beedham C, Critchley DJ, Rance DJ (1995) Substrate specificity of human liver aldehyde oxidase toward substituted quinazolines and phthalazines: a comparison with hepatic enzyme from guinea pig, rabbit, and baboon. Arch Biochem Biophys 319(2):481–490. https://doi.org/10.1006/abbi.1995.1320
Behera D, Pattem R, Gudi G (2014) Effect of commonly used organic solvents on aldehyde oxidase-mediated vanillin, phthalazine and methotrexate oxidation in human, rat and mouse liver subcellular fractions. Xenobiotica 44(8):722–733. https://doi.org/10.3109/00498254.2014.889332
Behl T, Kaur D, Sehgal A, et al. (2021) Role of monoamine oxidase activity in Alzheimer's disease: an insight into the therapeutic potential of inhibitors. Molecules 26(12). https://doi.org/10.3390/molecules26123724
Benedetti MS (2001) Biotransformation of xenobiotics by amine oxidases. Fundam Clin Pharmacol 15(2):75–84. https://doi.org/10.1046/j.1472-8206.2001.00011.x
Binda C, Wang J, Pisani L et al (2007) Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: safinamide and coumarin analogs. J Med Chem 50(23):5848–5852. https://doi.org/10.1021/jm070677y
Bissada JE, Truong V, Abouda AA, Wines KJ, Crouch RD, Jackson KD (2019) Interindividual variation in CYP3A activity influences lapatinib bioactivation. Drug Metab Dispos 47(11):1257–1269. https://doi.org/10.1124/dmd.119.088823
Bloom AJ, Murphy SE, Martinez M, von Weymarn LB, Bierut LJ, Goate A (2013) Effects upon in-vivo nicotine metabolism reveal functional variation in FMO3 associated with cigarette consumption. Pharmacogenet Genomics 23(2):62–68. https://doi.org/10.1097/FPC.0b013e32835c3b48
Borroni E, Bohrmann B, Grueninger F et al (2017) Sembragiline: a novel, selective monoamine oxidase type B inhibitor for the treatment of Alzheimer’s disease. J Pharmacol Exp Ther 362(3):413–423. https://doi.org/10.1124/jpet.117.241653
Bortolato M, Shih JC (2011) Behavioral outcomes of monoamine oxidase deficiency: Preclinical and clinical evidence. Int Rev Neurobiol 100:13–42. https://doi.org/10.1016/b978-0-12-386467-3.00002-9
Bortolato M, Chen K, Shih JC (2008) Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev 60(13–14):1527–1533. https://doi.org/10.1016/j.addr.2008.06.002
Bortolotti M, Polito L, Battelli MG, Bolognesi A (2021) Xanthine oxidoreductase: one enzyme for multiple physiological tasks. Redox Biol 41:101882. https://doi.org/10.1016/j.redox.2021.101882
Bove M, Cicero AFG, Borghi C (2017) The effect of xanthine oxidase inhibitors on blood pressure and renal function. Curr Hypertens Rep 19(12):95. https://doi.org/10.1007/s11906-017-0793-3
Bredemeier M, Lopes LM, Eisenreich MA et al (2018) Xanthine oxidase inhibitors for prevention of cardiovascular events: a systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc Disord 18(1):24. https://doi.org/10.1186/s12872-018-0757-9
Brossi A, Millet P, Landau I, Bembenek ME, Abell CW (1987) Antimalarial activity and inhibition of monoamine oxidases A and B by exo-erythrocytic antimalarials. Optical isomers of primaquine, N-acylated congeners, primaquine metabolites and 5-phenoxy-substituted analogues. FEBS Lett 214(2):291–294. https://doi.org/10.1016/0014-5793(87)80072-8
Brunelle A, Bi YA, Lin J et al (1997) Characterization of two human flavin-containing monooxygenase (form 3) enzymes expressed in Escherichia coli as maltose binding protein fusions. Drug Metab Dispos 25(8):1001–1007
Burton RD, Hieronymus T, Chamem T et al (2018) Assessment of the biotransformation of low-turnover drugs in the HµREL human hepatocyte coculture model. Drug Metab Dispos 46(11):1617–1625. https://doi.org/10.1124/dmd.118.082867
Buu NT (1985) Relationship between catechol-O-methyltransferase and phenolsulfotransferase in the metabolism of dopamine in the rat brain. J Neurochem 45(5):1612–1619. https://doi.org/10.1111/j.1471-4159.1985.tb07234.x
Buur-Rasmussen B, Brøsen K (1999) Cytochrome P450 and therapeutic drug monitoring with respect to clozapine. Eur Neuropsychopharmacol 9(6):453–459. https://doi.org/10.1016/s0924-977x(99)00033-4
Capi M, Curto M, Lionetto L et al (2016) Eletriptan in the management of acute migraine: an update on the evidence for efficacy, safety, and consistent response. Ther Adv Neurol Disord 9(5):414–423. https://doi.org/10.1177/1756285616650619
Cardillo C, Kilcoyne CM, Cannon RO 3rd, Quyyumi AA, Panza JA (1997) Xanthine oxidase inhibition with oxypurinol improves endothelial vasodilator function in hypercholesterolemic but not in hypertensive patients. Hypertension 30(1 Pt 1):57–63. https://doi.org/10.1161/01.hyp.30.1.57
Carradori S, D’Ascenzio M, Chimenti P, Secci D, Bolasco A (2014) Selective MAO-B inhibitors: a lesson from natural products. Mol Divers 18(1):219–243. https://doi.org/10.1007/s11030-013-9490-6
Carvalho M, Carmo H, Costa VM et al (2012) Toxicity of amphetamines: an update. Arch Toxicol 86(8):1167–1231. https://doi.org/10.1007/s00204-012-0815-5
Cashman JR (1998) Stereoselectivity in S- and N-oxygenation by the mammalian flavin-containing and cytochrome P-450 monooxygenases. Drug Metab Rev 30(4):675–707. https://doi.org/10.3109/03602539808996327
Cashman JR (2000) Human flavin-containing monooxygenase: substrate specificity and role in drug metabolism. Curr Drug Metab 1(2):181–191. https://doi.org/10.2174/1389200003339135
Cashman JR (2002a) Human and plant flavin-containing monooxygenase N-oxygenation of amines: detoxication vs. bioactivation. Drug Metab Rev 34(0360–2532(Print)):513–521
Cashman JR (2002b) Human flavin-containing monooxygenase (form 3): polymorphisms and variations in chemical metabolism. Pharmacogenomics 3(3):325–339. https://doi.org/10.1517/14622416.3.3.325
Cashman JR (2004) The implications of polymorphisms in mammalian flavin-containing monooxygenases in drug discovery and development. Drug Discov Today 9(13):574–581. https://doi.org/10.1016/s1359-6446(04)03136-8
Cashman JR, Zhang J (2002) Interindividual differences of human flavin-containing monooxygenase 3: Genetic polymorphisms and functional variation. Drug Metab Dispos 30(10):1043–1052. https://doi.org/10.1124/dmd.30.10.1043
Cashman JR, Zhang J (2006) Human flavin-containing monooxygenases. Annu Rev Pharmacol Toxicol 46:65–100. https://doi.org/10.1146/annurev.pharmtox.46.120604.141043
Cashman JR, Park SB, Yang ZC, Wrighton SA, Jacob P 3rd, Benowitz NL (1992) Metabolism of nicotine by human liver microsomes: stereoselective formation of trans-nicotine N′-oxide. Chem Res Toxicol 5(5):639–646. https://doi.org/10.1021/tx00029a008
Cashman JR, Park SB, Yang ZC et al (1993a) Chemical, enzymatic, and human enantioselective S-oxygenation of cimetidine. Drug Metab Dispos 21(4):587–597
Cashman JR, Yang Z, Yang L, Wrighton SA (1993b) Stereo- and regioselective N- and S-oxidation of tertiary amines and sulfides in the presence of adult human liver microsomes. Drug Metab Dispos 21(3):492–501
Cashman JR, Park SB, Berkman CE, Cashman LE (1995) Role of hepatic flavin-containing monooxygenase 3 in drug and chemical metabolism in adult humans. Chem-Biol Interact 96(1):33–46. https://doi.org/10.1016/0009-2797(94)03581-r
Cashman JR, Xiong Y, Lin J et al (1999a) In vitro and in vivo inhibition of human flavin-containing monooxygenase form 3 (FMO3) in the presence of dietary indoles. Biochem Pharmacol 58(6):1047–1055. https://doi.org/10.1016/s0006-2952(99)00166-5
Cashman JR, Xiong YN, Xu L, Janowsky A (1999b) N-Oxygenation of amphetamine and methamphetamine by the human flavin-containing monooxygenase (form 3): role in bioactivation and detoxication. J Pharmacol Exp Ther 288(3):1251–1260
Cashman JR, Akerman BR, Forrest SM, Treacy EP (2000) Population-specific polymorphisms of the human FMO3 gene: significance for detoxication. Drug Metab Dispos 28(2):169–173
Cashman JR, Camp K, Fakharzadeh SS et al (2003) Biochemical and clinical aspects of the human flavin-containing monooxygenase form 3 (FMO3) related to trimethylaminuria. Curr Drug Metab 4(2):151–170. https://doi.org/10.2174/1389200033489505
Cashman JR, Gohdes M, de Kater A, Schoenhard G (2020) N-Oxygenation of oxycodone and retro-reduction of oxycodone N-oxide. Drug Metab Dispos 48(2):106–115. https://doi.org/10.1124/dmd.119.089300
Castellino S, O’Mara M, Koch K, Borts DJ, Bowers GD, MacLauchlin C (2012) Human metabolism of lapatinib, a dual kinase inhibitor: implications for hepatotoxicity. Drug Metab Dispos 40(1):139–150. https://doi.org/10.1124/dmd.111.040949
Catucci G, Occhipinti A, Maffei M, Gilardi G, Sadeghi SJ (2013) Effect of human flavin-containing monooxygenase 3 polymorphism on the metabolism of aurora kinase inhibitors. Int J Mol Sci 14(2):2707–2716. https://doi.org/10.3390/ijms14022707
Catucci G, Polignano I, Cusumano D, Medana C, Gilardi G, Sadeghi SJ (2017) Identification of human flavin-containing monooxygenase 3 substrates by a colorimetric screening assay. Anal Biochem 522:46–52. https://doi.org/10.1016/j.ab.2017.01.024
Catucci G, Bortolussi S, Rampolla G, Cusumano D, Gilardi G, Sadeghi SJ (2018) Flavin-containing monooxygenase 3 polymorphic variants significantly affect clearance of tamoxifen and clomiphene. Basic Clin Pharmacol Toxicol 123(6):687–691. https://doi.org/10.1111/bcpt.13089
Catucci G, Gilardi G, Sadeghi SJ (2020) Production of drug metabolites by human FMO3 in Escherichia coli. Microb Cell Fact 19(1):74. https://doi.org/10.1186/s12934-020-01332-1
Cesura AM, Galva MD, Imhof R, Da Prada M (1987) Binding of [3H]Ro 16–6491, a reversible inhibitor of monoamine oxidase type B, to human brain mitochondria and platelet membranes. J Neurochem 48(1):170–176. https://doi.org/10.1111/j.1471-4159.1987.tb13143.x
Cesura AM, Imhof R, Takacs B, Galva MD, Picotti GB, Da Prada M (1988) [3H]-Ro 16–6491, a selective probe for affinity labelling of monoamine oxidase type B in human brain and platelet membranes. J Neurochem 50(4):1037–1043. https://doi.org/10.1111/j.1471-4159.1988.tb10570.x
Cesura AM, Galva MD, Imhof R, Kyburz E, Picotti GB, Da Prada M (1989) [3H]-Ro 19–6327: a reversible ligand and affinity labelling probe for monoamine oxidase-B. Eur J Pharmacol 162(3):457–465. https://doi.org/10.1016/0014-2999(89)90336-1
Cesura AM, Bös M, Galva MD, Imhof R, Da Prada M (1990a) Characterization of the binding of [3H]Ro 41–1049 to the active site of human monoamine oxidase-A. Mol Pharmacol 37(3):358–366
Cesura AM, Muggli-Maniglio D, Lang G, Imhof R, Da Prada M (1990b) Monoamine oxidase inhibition by moclobemide and 2-amino-ethyl carboxamide derivatives: mode of action and kinetic characteristics. J Neural Transm Suppl 32:165–170. https://doi.org/10.1007/978-3-7091-9113-2_24
Chalmers AH, Knight PR, Atkinson MR (1969) 6-Thiopurines as substrates and inhibitors of purine oxidases: a pathway for conversion of azathioprine into 6-thiouric acid without release of 6-mercaptopurine. Aust J Exp Biol Med Sci 47(2):263–273. https://doi.org/10.1038/icb.1969.27
Chang CN, Doong SL, Cheng YC (1992) Conversion of 5-iodo-2-pyrimidinone-2′-deoxyribose to 5-iodo-deoxyuridine by aldehyde oxidase. Implication in hepatotropic drug design. Biochem Pharmacol 43(10):2269–2273. https://doi.org/10.1016/0006-2952(92)90186-m
Chaurasiya ND, Ibrahim MA, Muhammad I, Walker LA, Tekwani BL (2014) Monoamine oxidase inhibitory constituents of propolis: kinetics and mechanism of inhibition of recombinant human MAO-A and MAO-B. Molecules 19(11):18936–18952. https://doi.org/10.3390/molecules191118936
Chaurasiya ND, Gogineni V, Elokely KM et al (2016) Isolation of acacetin from Calea urticifolia with inhibitory properties against human monoamine oxidase-A and -B. J Nat Prod 79(10):2538–2544. https://doi.org/10.1021/acs.jnatprod.6b00440
Chaurasiya ND, Zhao J, Pandey P, Doerksen RJ, Muhammad I, Tekwani BL (2019) Selective inhibition of human monoamine oxidase B by acacetin 7-methyl ether isolated from Turnera diffusa (Damiana). Molecules 24(4). https://doi.org/10.3390/molecules24040810
Chaurasiya ND, Liu H, Doerksen RJ, Nanayakkara NPD, Walker LA, Tekwani BL (2021) Enantioselective interactions of anti-infective 8-aminoquinoline therapeutics with human monoamine oxidases A and B. Pharmaceuticals (Basel) 14(5). https://doi.org/10.3390/ph14050398
Chen S, Austin-Muttitt K, Zhang LH, Mullins JGL, Lau AJ (2019) In vitro and in silico analyses of the inhibition of human aldehyde oxidase by bazedoxifene, lasofoxifene, and structural analogues. J Pharmacol Exp Ther 371(1):75–86. https://doi.org/10.1124/jpet.119.259267
Chen JJ, Swope DM, Dashtipour K (2007) Comprehensive review of rasagiline, a second-generation monoamine oxidase inhibitor, for the treatment of Parkinson’s disease. Clin Ther 29(9):1825–1849. https://doi.org/10.1016/j.clinthera.2007.09.021
Cherrington NJ, Cao Y, Cherrington JW, Rose RL, Hodgson E (1998) Physiological factors affecting protein expression of flavin-containing monooxygenases 1, 3 and 5. Xenobiotica 28(7):673–682. https://doi.org/10.1080/004982598239254
Cherrington B, Englich U, Niruntari S, Grant W, Hodgman M (2020) Monoamine oxidase A inhibition by toxic concentrations of metaxalone. Clin Toxicol (phila) 58(5):383–387. https://doi.org/10.1080/15563650.2019.1648815
Chetty M, Moodley SV, Miller R (1994) Important metabolites to measure in pharmacodynamic studies of chlorpromazine. Ther Drug Monit 16(1):30–36. https://doi.org/10.1097/00007691-199402000-00004
Chimenti F, Secci D, Bolasco A et al (2009) Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J Med Chem 52(7):1935–1942. https://doi.org/10.1021/jm801496u
Chládek J, Martínková J, Sispera L (1997) An in vitro study on methotrexate hydroxylation in rat and human liver. Physiol Res 46(5):371–379
Choughule KV, Barnaba C, Joswig-Jones CA, Jones JP (2014) In vitro oxidative metabolism of 6-mercaptopurine in human liver: insights into the role of the molybdoflavoenzymes aldehyde oxidase, xanthine oxidase, and xanthine dehydrogenase. Drug Metab Dispos 42(8):1334–1340. https://doi.org/10.1124/dmd.114.058107
Choughule KV, Joswig-Jones CA, Jones JP (2015) Interspecies differences in the metabolism of methotrexate: an insight into the active site differences between human and rabbit aldehyde oxidase. Biochem Pharmacol 96(3):288–295. https://doi.org/10.1016/j.bcp.2015.05.010
Chowdhury S, Kumar S (2021) Inhibition of BACE1, MAO-B, cholinesterase enzymes, and anti-amyloidogenic potential of selected natural phytoconstituents: multi-target-directed ligand approach. J Food Biochem 45(1):e13571. https://doi.org/10.1111/jfbc.13571
Chung WG, Kang JH, Park CS, Cho MH, Cha YN (2000a) Effect of age and smoking on in vivo CYP1A2, flavin-containing monooxygenase, and xanthine oxidase activities in Koreans: determination by caffeine metabolism. Clin Pharmacol Ther 67(3):258–266. https://doi.org/10.1067/mcp.2000.104617
Chung WG, Park CS, Roh HK, Lee WK, Cha YN (2000b) Oxidation of ranitidine by isozymes of flavin-containing monooxygenase and cytochrome P450. Jpn J Pharmacol 84(2):213–220. https://doi.org/10.1254/jjp.84.213
Cicero AFG, Fogacci F, Cincione RI, Tocci G, Borghi C (2021) Clinical effects of xanthine oxidase inhibitors in hyperuricemic patients. Med Princ Pract 30(2):122–130. https://doi.org/10.1159/000512178
Clarke SE, Harrell AW, Chenery RJ (1995) Role of aldehyde oxidase in the in vitro conversion of famciclovir to penciclovir in human liver. Drug Metab Dispos 23(2):251–254
Coelho C, Foti A, Hartmann T, Santos-Silva T, Leimkühler S, Romão MJ (2015) Structural insights into xenobiotic and inhibitor binding to human aldehyde oxidase. Nat Chem Biol 11(10):779–783. https://doi.org/10.1038/nchembio.1895
Corte ED, Stirpe F (1972) The regulation of rat liver xanthine oxidase. Involvement of thiol groups in the conversion of the enzyme activity from dehydrogenase (type D) into oxidase (type O) and purification of the enzyme. Biochem J 126(3):739–745. https://doi.org/10.1042/bj1260739
Costa C, Catania S, Silvari V (2003) [Genotoxicity and activation of organophosphate and carbamate pesticides by cytochrome P450 2D6]. G Ital Med Lav Ergon 25 Suppl(3):81–82 [Italian]
Crouch RD, Morrison RD, Byers FW, Lindsley CW, Emmitte KA, Daniels JS (2016) Evaluating the disposition of a mixed aldehyde oxidase/cytochrome P450 substrate in rats with attenuated P450 activity. Drug Metab Dispos 44(8):1296–1303. https://doi.org/10.1124/dmd.115.068338
Crouch RD, Blobaum AL, Felts AS, Conn PJ, Lindsley CW (2017) Species-specific involvement of aldehyde oxidase and xanthine oxidase in the metabolism of the pyrimidine-containing mglu5-negative allosteric modulator VU0424238 (Auglurant). Drug Metab Dispos 45(12):1245–1259. https://doi.org/10.1124/dmd.117.077552
Crouch RD, Hutzler JM, Daniels JS (2018) A novel in vitro allometric scaling methodology for aldehyde oxidase substrates to enable selection of appropriate species for traditional allometry. Xenobiotica 48(3):219–231. https://doi.org/10.1080/00498254.2017.1296208
Cruciani G, Valeri A, Goracci L, Pellegrino RM, Buonerba F, Baroni M (2014) Flavin monooxygenase metabolism: why medicinal chemists should matter. J Med Chem 57(14):6183–6196. https://doi.org/10.1021/jm5007098
Curet O, Damoiseau G, Aubin N, Sontag N, Rovei V, Jarreau FX (1996) Befloxatone, a new reversible and selective monoamine oxidase-A inhibitor. I. Biochemical profile. J Pharmacol Exp Ther 277(1):253–264. https://doi.org/10.1163/2211730x96x00144
Da Prada M, Kettler R, Keller HH et al (1990) From moclobemide to Ro 19–6327 and Ro 41–1049: the development of a new class of reversible, selective MAO-A and MAO-B inhibitors. J Neural Transm Suppl 29:279–292. https://doi.org/10.1007/978-3-7091-9050-0_27
Dalmadi B, Leibinger J, Szeberényi S, Borbás T et al (2003) Identification of metabolic pathways involved in the biotransformation of tolperisone by human microsomal enzymes. Drug Metab Dispos 31(5):631–636. https://doi.org/10.1124/dmd.31.5.631(0090-9556(Print))
Dalvie D, Di L (2019) Aldehyde oxidase and its role as a drug metabolizing enzyme. Pharmacol Ther 201:137–180. https://doi.org/10.1016/j.pharmthera.2019.05.011
Dalvie DK, O’Connell TN (2004) Characterization of novel dihydrothienopyridinium and thienopyridinium metabolites of ticlopidine in vitro: role of peroxidases, cytochromes P450, and monoamine oxidases. Drug Metab Dispos 32(1):49–57. https://doi.org/10.1124/dmd.32.1.49
Dalvie D, Cosker T, Boyden T, Zhou S, Schroeder C, Potchoiba MJ (2008) Metabolism distribution and excretion of a matrix metalloproteinase-13 inhibitor, 4-[4-(4-fluorophenoxy)-benzenesulfonylamino]tetrahydropyran-4-carboxylic acid hydroxyamide (CP-544439), in rats and dogs: assessment of the metabolic profile of CP-544439 in plasma and urine of humans. Drug Metab Dispos 36(9):1869–1883. https://doi.org/10.1124/dmd.108.022566
Dalvie D, Zhang C, Chen W, Smolarek T, Obach RS, Loi CM (2010) Cross-species comparison of the metabolism and excretion of zoniporide: contribution of aldehyde oxidase to interspecies differences. Drug Metab Dispos 38(4):641–654. https://doi.org/10.1124/dmd.109.030783
Dalvie D, Sun H, Xiang C, Hu Q, Jiang Y, Kang P (2012) Effect of structural variation on aldehyde oxidase-catalyzed oxidation of zoniporide. Drug Metab Dispos 40(8):1575–1587. https://doi.org/10.1124/dmd.112.045823
Dalvie D, Xiang C, Kang P, Zhou S (2013) Interspecies variation in the metabolism of zoniporide by aldehyde oxidase. Xenobiotica 43(5):399–408. https://doi.org/10.3109/00498254.2012.727499
Danielczyk W, Streifler M, Konradi C, Riederer P, Moll G (1988) Platelet MAO-B activity and the psychopathology of Parkinson’s disease, senile dementia and multi-infarct dementia. Acta Psychiatr Scand 78(6):730–736. https://doi.org/10.1111/j.1600-0447.1988.tb06412.x
Dannan GA, Guengerich FP (1982) Immunochemical comparison and quantitation of microsomal flavin-containing monooxygenases in various hog, mouse, rat, rabbit, dog, and human tissues. Mol Pharmacol 22(3):787–794
Das A, Giri S (2020) A review on role of arecoline and its metabolites in the molecular pathogenesis of oral lesions with an insight into current status of its metabolomics. Prague Med Rep 121(4):209–235. https://doi.org/10.14712/23362936.2020.19
Day RO, Miners J, Birkett DJ, Graham GG, Whitehead A (1988) Relationship between plasma oxipurinol concentrations and xanthine oxidase activity in volunteers dosed with allopurinol. Br J Clin Pharmacol 26(4):429–434. https://doi.org/10.1111/j.1365-2125.1988.tb03402.x
Della Corte E, Stirpe F (1968) The regulation of rat-liver xanthine oxidase: activation by proteolytic enzymes. FEBS Lett 2(2):83–84. https://doi.org/10.1016/0014-5793(68)80107-3
Delogu G, Picciau C, Ferino G et al (2011) Synthesis, human monoamine oxidase inhibitory activity and molecular docking studies of 3-heteroarylcoumarin derivatives. Eur J Med Chem 46(4):1147–1152. https://doi.org/10.1016/j.ejmech.2011.01.033
Deng P, Zhong D, Yu K, Zhang Y, Wang T, Chen X (2013) Pharmacokinetics, metabolism, and excretion of the antiviral drug arbidol in humans. Antimicrob Agents Chemother 57(4):1743–1755. https://doi.org/10.1128/aac.02282-12
Diamond S, Boer J, Maduskuie TP Jr, Falahatpisheh N, Li Y, Yeleswaram S (2010) Species-specific metabolism of SGX523 by aldehyde oxidase and the toxicological implications. Drug Metab Dispos 38(8):1277–1285. https://doi.org/10.1124/dmd.110.032375
Dick RA (2018) Refinement of in vitro methods for identification of aldehyde oxidase substrates reveals metabolites of kinase inhibitors. Drug Metab Dispos 46(6):846–859. https://doi.org/10.1124/dmd.118.080960
Dixon M, Webb EC (1964) Enzymes, 2nd edn. Longman's, Green and Co Ltd, London
Dixon CM, Park GR, Tarbit MH (1994) Characterization of the enzyme responsible for the metabolism of sumatriptan in human liver. Biochem Pharmacol 47(7):1253–1257. https://doi.org/10.1016/0006-2952(94)90397-2
Dolan ME, Roy SK, Fasanmade AA, Paras PR, Schilsky RL, Ratain MJ (1998) O6-Benzylguanine in humans: metabolic, pharmacokinetic, and pharmacodynamic findings. J Clin Oncol 16(5):1803–1810. https://doi.org/10.1200/jco.1998.16.5.1803
Dolphin CT, Janmohamed A, Smith RL, Shephard EA, Phillips IR (1997) Missense mutation in flavin-containing mono-oxygenase 3 gene, FMO3, underlies fish-odour syndrome. Nat Genet 17:491–494
Donnelly CH, Murphy DL (1977) Substrate- and inhibitor-related characteristics of human platelet monoamine oxidase. Biochem Pharmacol 26(9):853–858. https://doi.org/10.1016/0006-2952(77)90398-7
Drukarch B, Jongenelen CA, van Muiswinkel FL (2001) NAD(P)H:quinone oxidoreductase (NQO1) protects astroglial cells against L-DOPA toxicity. Adv Exp Med Biol 500:237–240. https://doi.org/10.1007/978-1-4615-0667-6_35
Dupont GP, Huecksteadt TP, Marshall BC, Ryan US, Michael JR, Hoidal JR (1992) Regulation of xanthine dehydrogenase and xanthine oxidase activity and gene expression in cultured rat pulmonary endothelial cells. J Clin Invest 89(1):197–202. https://doi.org/10.1172/jci115563
Duran M, Beemer FA, van de Heiden C et al (1978) Combined deficiency of xanthine oxidase and sulphite oxidase: a defect of molybdenum metabolism or transport? J Inherit Metab Dis 1(4):175–178. https://doi.org/10.1007/bf01805591
Edmondson DE, Binda C (2018) Monoamine oxidases. Subcell Biochem 87:117–139. https://doi.org/10.1007/978-981-10-7757-9_5
Edmondson DE, Binda C, Mattevi A (2007) Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch Biochem Biophys 464(2):269–276. https://doi.org/10.1016/j.abb.2007.05.006
Egashira T, Takayama F, Yamanaka Y (1999) The inhibition of monoamine oxidase activity by various antidepressants: differences found in various mammalian species. Jpn J Pharmacol 81(1):115–121. https://doi.org/10.1254/jjp.81.115
Ekaidem IS, Usoh IF, Akpanabiatu MI, Uboh FE, Akpan HD (2014) Urate synthesis and oxidative stress in phenytoin hepatotoxicity: the role of antioxidant vitamins. Pak J Biol Sci 17(11):1179–1184. https://doi.org/10.3923/pjbs.2014.1179.1184
El-Hawary SS, Sayed AM, Issa MY et al (2021) Anti-Alzheimer chemical constituents of Morus macroura Miq.: chemical profiling, in silico and in vitro investigations. Food Funct 12(17):8078–8089. https://doi.org/10.1039/d1fo01177d
Elfarra AA (1995) Potential role of the flavin-containing monooxygenases in the metabolism of endogenous compounds. Chem-Biol Interact 96(0009–2797):47–55
Elfarra AA, Krause RJ (2005) Potential roles of flavin-containing monooxygenases in sulfoxidation reactions of L-methionine, N-acetyl-l-methionine and peptides containing l-methionine. Biochim Biophys Acta 1703(2):183–189. https://doi.org/10.1016/j.bbapap.2004.11.011
Entzeroth M, Ratty KK (2017) Monoamine oxidase inhibitors—revisiting a therapeutic principle. Open J Depression 6(2). https://doi.org/10.4236/ojd.2017.62004
Ercan S, Kencebay C, Basaranlar G, Derin N, Aslan M (2015) Induction of xanthine oxidase activity, endoplasmic reticulum stress and caspase activation by sodium metabisulfite in rat liver and their attenuation by ghrelin. Food Chem Toxicol 76:27–32. https://doi.org/10.1016/j.fct.2014.11.021
Erickson DA, Hollfelder S, Tenge J, Gohdes M, Burkhardt JJ, Krieter PA (2007) In vitro metabolism of the analgesic bicifadine in the mouse, rat, monkey, and human. Drug Metab Dispos 35(12):2232–2241. https://doi.org/10.1124/dmd.107.016055
Ernst ME, Fravel MA (2009) Febuxostat: a selective xanthine-oxidase/xanthine-dehydrogenase inhibitor for the management of hyperuricemia in adults with gout. Clin Ther 31(11):2503–2518. https://doi.org/10.1016/j.clinthera.2009.11.033
Ernster L, Danielson L, Ljunggren M (1962) DT diaphorase. I. Purification from the soluble fraction of rat-liver cytoplasm, and properties. Biochim Biophys Acta 58:171–188. https://doi.org/10.1016/0006-3002(62)90997-6
Falls JG, Cherrington NJ, Clements KM et al (1997) Molecular cloning, sequencing, and expression in Escherichia coli of mouse flavin-containing monooxygenase 3 (FMO3): COMPARISON with the human isoform. Arch Biochem Biophys 347(1):9–18. https://doi.org/10.1006/abbi.1997.0322
Fang J, Coutts RT, McKenna KF, Baker GB (1998) Elucidation of individual cytochrome P450 enzymes involved in the metabolism of clozapine. Naunyn Schmiedebergs Arch Pharmacol 358(5):592–599. https://doi.org/10.1007/pl00005298
Fang J, Yu PH, Gorrod JW, Boulton AA (1995) Inhibition of monoamine oxidases by haloperidol and its metabolites: pharmacological implications for the chemotherapy of schizophrenia. Psychopharmacology (berlin) 118(2):206–212. https://doi.org/10.1007/bf02245841
Fedejko-Kap B, Niemira M, Radominska-Pandya A, Mazerska Z (2011) Flavin monooxygenases, FMO1 and FMO3, not cytochrome P450 isoenzymes, contribute to metabolism of anti-tumour triazoloacridinone, C-1305, in liver microsomes and HepG2 cells. Xenobiotica 41(12):1044–1055. https://doi.org/10.3109/00498254.2011.604743
Fernandez HH, Chen JJ (2007) Monamine oxidase inhibitors: current and emerging agents for Parkinson disease. Clin Neuropharmacol 30(3):150–168. https://doi.org/10.1097/01.wnf.0000240956.49315.be
Ferreira Antunes M, Eggimann FK, Kittelmann M et al (2016) Human xanthine oxidase recombinant in E. coli: a whole cell catalyst for preparative drug metabolite synthesis. J Biotechnol 235:3–10. https://doi.org/10.1016/j.jbiotec.2016.03.045
Fierro A, Osorio-Olivares M, Cassels BK, Edmondson DE, Sepúlveda-Boza S, Reyes-Parada M (2007) Human and rat monoamine oxidase-A are differentially inhibited by (S)-4-alkylthioamphetamine derivatives: insights from molecular modeling studies. Bioorg Med Chem 15(15):5198–5206. https://doi.org/10.1016/j.bmc.2007.05.021
Finberg JP (2014) Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: focus on modulation of CNS monoamine neurotransmitter release. Pharmacol Ther 143(2):133–152. https://doi.org/10.1016/j.pharmthera.2014.02.010
Finberg JP, Youdim MB (1983) Selective MAO A and B inhibitors: their mechanism of action and pharmacology. Neuropharmacology 22(3 Spec No):441–446. https://doi.org/10.1016/0028-3908(83)90194-6
Fiorentini F, Geier M, Binda C et al (2016) Biocatalytic characterization of human FMO5: unearthing Baeyer-Villiger reactions in humans. ACS Chem Biol 11(4):1039–1048. https://doi.org/10.1021/acschembio.5b01016
Fiorentini F, Romero E, Fraaije MW, Faber K, Hall M, Mattevi A (2017) Baeyer-Villiger monooxygenase FMO5 as entry point in drug metabolism. ACS Chem Biol 12(9):2379–2387. https://doi.org/10.1021/acschembio.7b00470
Fitzpatrick PF (2010) Oxidation of amines by flavoproteins. Arch Biochem Biophys 493(1):13–25. https://doi.org/10.1016/j.abb.2009.07.019
Flanagan S, Bartizal K, Minassian SL, Fang E, Prokocimer P (2013) In vitro, in vivo, and clinical studies of tedizolid to assess the potential for peripheral or central monoamine oxidase interactions. Antimicrob Agents Chemother 57(7):3060–3066. https://doi.org/10.1128/aac.00431-13
Food and Drug Administration (U.S.) (2021) Drug development and drug interactions. Table of substrates, inhibitors and inducers. FDA
Foti A, Hartmann T, Coelho C, Santos-Silva T, Romao MJ, Leimkühler S (2016) Optimization of the expression of human aldehyde oxidase for investigations of single-nucleotide polymorphisms. Drug Metab Dispos 44(8):1277–1285. https://doi.org/10.1124/dmd.115.068395
Foti A, Dorendorf F, Leimkühler S (2017) A single nucleotide polymorphism causes enhanced radical oxygen species production by human aldehyde oxidase. PLoS ONE 12(7):e0182061. https://doi.org/10.1371/journal.pone.0182061
Fox AW (2010) Subcutaneous sumatriptan pharmacokinetics: delimiting the monoamine oxidase inhibitor effect. Headache 50(2):249–255. https://doi.org/10.1111/j.1526-4610.2009.01568.x
Francois AA, Nishida CR, de Montellano PR, Phillips IR, Shephard EA (2009) Human flavin-containing monooxygenase 2.1 catalyzes oxygenation of the antitubercular drugs thiacetazone and ethionamide. Drug Metab Dispos 37(1):178–186. https://doi.org/10.1124/dmd.108.024158
Frischer H, Mellovitz RL, Ahmad T, Nora MV (1991) The conversion of primaquine into primaquine-aldehyde, primaquine-alcohol, and carboxyprimaquine, a major plasma metabolite. J Lab Clin Med 117(6):468–476
Fritz RR, Abell CW, Patel NT, Gessner W, Brossi A (1985) Metabolism of the neurotoxin in MPTP by human liver monoamine oxidase B. FEBS Lett 186(2):224–228. https://doi.org/10.1016/0014-5793(85)80713-4
Fuchs P, Haefeli WE, Ledermann HR, Wenk M (1999) Xanthine oxidase inhibition by allopurinol affects the reliability of urinary caffeine metabolic ratios as markers for N-acetyltransferase 2 and CYP1A2 activities. Eur J Clin Pharmacol 54(11):869–876. https://doi.org/10.1007/s002280050569
Fujino C, Tamura Y, Tange S et al (2016) Metabolism of methiocarb and carbaryl by rat and human livers and plasma, and effect on their PXR, CAR and PPARα activities. J Toxicol Sci 41(5):677–691. https://doi.org/10.2131/jts.41.677
Fukami T, Iida A, Konishi K, Nakajima M (2016) Human arylacetamide deacetylase hydrolyzes ketoconazole to trigger hepatocellular toxicity. Biochem Pharmacol 116:153–161. https://doi.org/10.1016/j.bcp.2016.07.007
Furnes B, Schlenk D (2004) Evaluation of xenobiotic N- and S-oxidation by variant flavin-containing monooxygenase 1 (FMO1) enzymes. Toxicol Sci 78(2):196–203. https://doi.org/10.1093/toxsci/kfh079
Furnes B, Schlenk D (2005) Extrahepatic metabolism of carbamate and organophosphate thioether compounds by the flavin-containing monooxygenase and cytochrome P450 systems. Drug Metab Dispos 33(2):214–218. https://doi.org/10.1124/dmd.104.000984
Gadepalli RS, Rimoldi JM, Fronczek FR et al (2007) Synthesis of fenthion sulfoxide and fenoxon sulfoxide enantiomers: effect of sulfur chirality on acetylcholinesterase activity. Chem Res Toxicol 20(2):257–262. https://doi.org/10.1021/tx060153l
Gao C, Zheng T (2020) Expression of concern to: drug metabolite synthesis by immobilized human FMO3 and whole cell catalysts. Microb Cell Fact 19(1):78. https://doi.org/10.1186/s12934-020-01327-y
Gao C, Catucci G, Gilardi G, Sadeghi SJ (2018) Binding of methimazole and NADP(H) to human FMO3: in vitro and in silico studies. Int J Biol Macromol 118(Pt A):460–468. https://doi.org/10.1016/j.ijbiomac.2018.06.104
Garattini E, Terao M (2012) The role of aldehyde oxidase in drug metabolism. Expert Opin Drug Metab Toxicol 8(4):487–503. https://doi.org/10.1517/17425255.2012.663352
Garattini E, Fratelli M, Terao M (2008) Mammalian aldehyde oxidases: Genetics, evolution and biochemistry. Cell Mol Life Sci 65(7–8):1019–1048. https://doi.org/10.1007/s00018-007-7398-y
Garrido C, Leimkühler S (2021) The inactivation of human aldehyde oxidase 1 by hydrogen peroxide and superoxide. Drug Metab Dispos 49(9):729–735. https://doi.org/10.1124/dmd.121.000549
Gatarek P, Kaluzna-Czaplinska J (2021) Trimethylamine N-oxide (TMAO) in human health. Excli J 20:301–319. https://doi.org/10.17179/excli2020-3239
Gaweska H, Fitzpatrick PF (2011) Structures and mechanism of the monoamine oxidase family. Biomol Concepts 2(5):365–377. https://doi.org/10.1515/bmc.2011.030
Geha RM, Rebrin I, Chen K, Shih JC (2001) Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. J Biol Chem 276(13):9877–9882. https://doi.org/10.1074/jbc.M006972200
Gerlach M, Youdim MB, Riederer P (1996) Pharmacology of selegiline. Neurology 47(6 Suppl 3):S137–S145. https://doi.org/10.1212/wnl.47.6_suppl_3.137s
Ghelardoni S, Chiellini G, Frascarelli S, Saba A, Zucchi R (2014) Uptake and metabolic effects of 3-iodothyronamine in hepatocytes. J Endocrinol 221(1):101–110. https://doi.org/10.1530/joe-13-0311
Ghezzi P, Bianchi M, Mantovani A, Spreafico F, Salmona M (1984) Enhanced xanthine oxidase activity in mice treated with interferon and interferon inducers. Biochem Biophys Res Commun 119(1):144–149. https://doi.org/10.1016/0006-291x(84)91630-9
Gidaro MC, Astorino C, Petzer A et al (2016) Kaempferol as selective human MAO-A inhibitor: analytical detection in calabrian red wines, biological and molecular modeling studies. J Agric Food Chem 64(6):1394–1400. https://doi.org/10.1021/acs.jafc.5b06043
Giller E, Hall H, Reubens L, Wojciechoswki J (1984) Haloperidol inhibition of monoamine oxidase in vivo and in vitro. Biol Psychiatry 19(4):517–523
Giller E Jr, Jatlow P, Bialos D, Harkness L, Docherty JP (1980) Platelet MAO and amitriptyline treatment. Psychiatry Res 2(3):259–265. https://doi.org/10.1016/0165-1781(80)90018-9
Giri S, Krausz KW, Idle JR, Gonzalez FJ (2007) The metabolomics of (+/−)-arecoline 1-oxide in the mouse and its formation by human flavin-containing monooxygenases. Biochem Pharmacol 73(4):561–573. https://doi.org/10.1016/j.bcp.2006.10.017
Giri P, Naidu S, Patel N, Patel H, Srinivas NR (2017) Evaluation of in vitro cytochrome P450 inhibition and in vitro fate of structurally diverse N-oxide metabolites: case studies with clozapine, levofloxacin, roflumilast, voriconazole and zopiclone. Eur J Drug Metab Pharmacokinet 42(4):677–688. https://doi.org/10.1007/s13318-016-0385-7
Giri P, Gupta L, Naidu S et al (2018) In vitro drug–drug interaction potential of sulfoxide and/or sulfone metabolites of albendazole, triclabendazole, aldicarb, methiocarb, montelukast and ziprasidone. Drug Metab Lett 12(2):101–116. https://doi.org/10.2174/1872312812666180816164626
Glaenzel U, Jin Y, Hansen R et al (2020) Absorption, distribution, metabolism, and excretion of capmatinib (INC280) in healthy male volunteers and in vitro aldehyde oxidase phenotyping of the major metabolite. Drug Metab Dispos 48(10):873–885. https://doi.org/10.1124/dmd.119.090324
Glover V, Gibb C, Sandler M (1986) Monoamine oxidase B (MAO-B) is the major catalyst for 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) oxidation in human brain and other tissues. Neurosci Lett 64(2):216–220. https://doi.org/10.1016/0304-3940(86)90103-5
Godber BL, Doel JJ, Sapkota GP et al (2000) Reduction of nitrite to nitric oxide catalyzed by xanthine oxidoreductase. J Biol Chem 275(11):7757–7763. https://doi.org/10.1074/jbc.275.11.7757
Goldberg MR, Sciberras D, De Smet M et al (2001) Influence of b-adrenoceptor antagonists on the pharmacokinetics of rizatriptan, a 5-HT1B/1D agonist: differential effects of propranolol, nadolol and metoprolol. Br J Clin Pharmacol 52(1):69–76. https://doi.org/10.1046/j.0306-5251.2001.01417.x
Goldstein DS (2020) The catecholaldehyde hypothesis: where MAO fits in. J Neural Transm (vienna) 127(2):169–177. https://doi.org/10.1007/s00702-019-02106-9
Goldstein DS, Sullivan P, Cooney A et al (2012) Vesicular uptake blockade generates the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde in PC12 cells: relevance to the pathogenesis of Parkinson’s disease. J Neurochem 123(6):932–943. https://doi.org/10.1111/j.1471-4159.2012.07924.x
Grimsby J, Lan NC, Neve R, Chen K, Shih JC (1990) Tissue distribution of human monoamine oxidase A and B mRNA. J Neurochem 55(4):1166–1169. https://doi.org/10.1111/j.1471-4159.1990.tb03121.x
Grothusen A, Hardt J, Bräutigam L, Lang D, Böcker R (1996) A convenient method to discriminate between cytochrome P450 enzymes and flavin-containing monooxygenases in human liver microsomes. Arch Toxicol 71(1–2):64–71. https://doi.org/10.1007/s002040050359
Grzelczyk J, Budryn G, Peña-García J et al (2021) Evaluation of the inhibition of monoamine oxidase A by bioactive coffee compounds protecting serotonin degradation. Food Chem 348:129108. https://doi.org/10.1016/j.foodchem.2021.129108
Guay DR (2006) Rasagiline (TVP-1012): a new selective monoamine oxidase inhibitor for Parkinson’s disease. Am J Geriatr Pharmacother 4(4):330–346. https://doi.org/10.1016/j.amjopharm.2006.12.001
Guengerich FP (2001) Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol 14(6):611–650. https://doi.org/10.1021/tx0002583
Guengerich FP (2022) Cytochrome P450. In: Riddick DS (ed) Pharmacokinetics, vol X of Comprehensive pharmacology (still in press)
Guengerich FP, Yoshimoto FK (2018) Formation and cleavage of C-C bonds by enzymatic oxidation-reduction reactions. Chem Rev 118(14):6573–6655. https://doi.org/10.1021/acs.chemrev.8b00031
Gulcan HO, Orhan IE (2020) A recent look into natural products that have potential to inhibit cholinesterases and monoamine oxidase B: update for 2010–2019. Comb Chem High Throughput Screen 23(9):862–876. https://doi.org/10.2174/1386207323666200127145246
Haddad F, Sawalha M, Khawaja Y, Najjar A, Karaman R (2017) Dopamine and levodopa prodrugs for the treatment of Parkinson's disease. Molecules 23(1). https://doi.org/10.3390/molecules23010040
Hai X, Adams E, Hoogmartens J, Van Schepdael A (2009) Enantioselective in-line and off-line CE methods for the kinetic study on cimetidine and its chiral metabolites with reference to flavin-containing monooxygenase genetic isoforms. Electrophoresis 30(7):1248–1257. https://doi.org/10.1002/elps.200800604
Hai X, Nauwelaers T, Busson R, Adams E, Hoogmartens J, Van Schepdael A (2010) A rapid and sensitive CE method with field-enhanced sample injection and in-capillary derivatization for selenomethionine metabolism catalyzed by flavin-containing monooxygenases. Electrophoresis 31(19):3352–3361. https://doi.org/10.1002/elps.201000248
Haining RL, Hunter AP, Sadeque AJ, Sadeque AJ, Philpot RM, Rettie AE (1997) Baculovirus-mediated expression and purification of human FMO3: catalytic, immunochemical, and structural characterization. Drug Metab Dispos 25(7):790–797
Haj Ahmed W, Peiro C, Fontaine J et al (2020) Methylxanthines inhibit primary amine oxidase and monoamine oxidase activities of human adipose tissue. Medicines (Basel) 7(4). https://doi.org/10.3390/medicines7040018
Hakooz NM (2009) Caffeine metabolic ratios for the in vivo evaluation of CYP1A2, N-acetyltransferase 2, xanthine oxidase and CYP2A6 enzymatic activities. Curr Drug Metab 10(4):329–338. https://doi.org/10.2174/138920009788499003
Halpin LE, Collins SA, Yamamoto BK (2014) Neurotoxicity of methamphetamine and 3,4-methylenedioxymethamphetamine. Life Sci 97(1):37–44. https://doi.org/10.1016/j.lfs.2013.07.014
Hamman MA, Haehner-Daniels BD, Wrighton SA, Rettie AE, Hall SD (2000) Stereoselective sulfoxidation of sulindac sulfide by flavin-containing monooxygenases. Comparison of human liver and kidney microsomes and mammalian enzymes. Biochem Pharmacol 60(1):7–17. https://doi.org/10.1016/s0006-2952(00)00301-4
Hanioka N, Saito K, Isobe T, Ohkawara S, Jinno H, Tanaka-Kagawa T (2021) Favipiravir biotransformation in liver cytosol: species and sex differences in humans, monkeys, rats, and mice. Biopharm Drug Dispos 42(5):218–225. https://doi.org/10.1002/bdd.2275
Hanna IH, Krauser JA, Cai H, Kim M-S, Guengerich FP (2001) Diversity in mechanisms of substrate oxidation by cytochrome P450 2D6. Lack of an allosteric role of NADPH-cytochrome P450 reductase in catalytic regioselectivity. J Biol Chem 276(43):39553–39561. https://doi.org/10.1074/jbc.M106841200
Hanson KL, VandenBrink BM, Babu KN, Allen KE, Nelson WL, Kunze KL, Kunze KL (2010) Sequential metabolism of secondary alkyl amines to metabolic-intermediate complexes: opposing roles for the secondary hydroxylamine and primary amine metabolites of desipramine, (S)-fluoxetine, and N-desmethyldiltiazem. Drug Metab Dispos 38(6):963–972
Harrell AW, Wheeler SM, East P, Clarke SE, Chenery RJ (1994) Use of rat and human in vitro systems to assess the effectiveness and enzymology of deoxy-guanine analogues as prodrugs of an antiviral agent. Drug Metab Dispos 22(1):124–128
Harrison R (2002) Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med 33(6):774–797. https://doi.org/10.1016/s0891-5849(02)00956-5
Hartmann T, Terao M, Garattini E et al (2012) The impact of single nucleotide polymorphisms on human aldehyde oxidase. Drug Metab Dispos 40(5):8568–8564. https://doi.org/10.1124/dmd.111.043828 and (2016) Correction to “The impact of single nucleotide polymorphisms on human aldehyde oxidase”. Drug Metab Dispos 44(3):365. https://doi.org/10.1124/dmd.112.043828err
Harzand A, Tamariz L, Hare JM (2012) Uric acid, heart failure survival, and the impact of xanthine oxidase inhibition. Congest Heart Fail 18(3):179–182. https://doi.org/10.1111/j.1751-7133.2011.00262.x
He X, Chen YY, Shi JB et al (2014) New coumarin derivatives: design, synthesis and use as inhibitors of hMAO. Bioorg Med Chem 22(14):3732–3738. https://doi.org/10.1016/j.bmc.2014.05.002
Heid H, Zimbelmann R, Dörflinger Y, Rickelt S (2020) Formation and degradation of lipid droplets in human adipocytes and the expression of aldehyde oxidase (AOX). Cell Tissue Res 379(1):45–62. https://doi.org/10.1007/s00441-019-03152-1
Henderson MC, Krueger SK, Siddens LK, Stevens JF, Williams DE (2004a) S-Oxygenation of the thioether organophosphate insecticides phorate and disulfoton by human lung flavin-containing monooxygenase 2. Biochem Pharmacol 68(5):959–967. https://doi.org/10.1016/j.bcp.2004.05.051
Henderson MC, Krueger SK, Stevens JF, Williams DE (2004b) Human flavin-containing monooxygenase form 2 S-oxygenation: sulfenic acid formation from thioureas and oxidation of glutathione. Chem Res Toxicol 17(5):633–640. https://doi.org/10.1021/tx034253s
Henderson MC, Siddens LK, Morré JT, Krueger SK, Williams DE (2008) Metabolism of the anti-tuberculosis drug ethionamide by mouse and human FMO1, FMO2 and FMO3 and mouse and human lung microsomes. Toxicol Appl Pharmacol 233(3):420–427. https://doi.org/10.1016/j.taap.2008.09.017
Henderson MC, Siddens LK, Krueger SK et al (2014) Flavin-containing monooxygenase S-oxygenation of a series of thioureas and thiones. Toxicol Appl Pharmacol 278(2):91–99. https://doi.org/10.1016/j.taap.2014.04.002
Herraiz T (2012) Evaluation of the oxidation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) to toxic pyridinium cations by monoamine oxidase (MAO) enzymes and its use to search for new MAO inhibitors and protective agents. J Enzyme Inhib Med Chem 27(6):810–817. https://doi.org/10.3109/14756366.2011.616946
Herraiz T, Chaparro C (2005) Human monoamine oxidase is inhibited by tobacco smoke: b-Carboline alkaloids act as strong and reversible inhibitors. Biochem Biophys Res Commun 326(2):378–386. https://doi.org/10.1016/j.bbrc.2004.11.033
Herraiz T, Chaparro C (2006) Human monoamine oxidase enzyme inhibition by coffee and b-carbolines norharman and harman isolated from coffee. Life Sci 78(8):795–802. https://doi.org/10.1016/j.lfs.2005.05.074
Herraiz T, Guillén H (2011) Inhibition of the bioactivation of the neurotoxin MPTP by antioxidants, redox agents and monoamine oxidase inhibitors. Food Chem Toxicol 49(8):1773–1781. https://doi.org/10.1016/j.fct.2011.04.026
Herraiz T, Guillén H, Arán VJ, Idle JR, Gonzalez FJ (2006) Comparative aromatic hydroxylation and N-demethylation of MPTP neurotoxin and its analogs, N-methylated b-carboline and isoquinoline alkaloids, by human cytochrome P450 2D6. Toxicol Appl Pharmacol 216(3):387–398. https://doi.org/10.1016/j.taap.2006.06.003
Hille R (2005) Molybdenum-containing hydroxylases. Arch Biochem Biophys 433(1):107–116. https://doi.org/10.1016/j.abb.2004.08.012
Hines RN (2006) Developmental and tissue-specific expression of human flavin-containing monooxygenases 1 and 3. Expert Opin Drug Metab Toxicol 2(1):41–49. https://doi.org/10.1517/17425255.2.1.41
Hisamuddin IM, Yang VW (2007) Genetic polymorphisms of human flavin-containing monooxygenase 3: implications for drug metabolism and clinical perspectives. Pharmacogenomics 8(6):635–643. https://doi.org/10.2217/14622416.8.6.635
Hodgson E, Levi PE (1992) The role of the flavin-containing monooxygenase (EC 1.14.13.8) in the metaolism and mode of action of agricultural chemicals. Xenobiotica 22:1175–1183
Hodgson E, Rose RL, Cao Y, Dehal SS, Kupfer D (2000) Flavin-containing monooxygenase isoform specificity for the N-oxidation of tamoxifen determined by product measurement and NADPH oxidation. J Biochem Mol Toxicol 14(2):118–120. https://doi.org/10.1002/(sici)1099-0461(2000)14:2%3c118::aid-jbt8%3e3.0.co;2-t
Hong YK, Kim YH, Lee JM, Yoo HH, Choi SO, Kang MS (2021) Characterization of in vitro Phase I metabolites of methamnetamine in human liver microsomes by liquid chromatography-quadrupole time-of-flight mass spectrometry. Int J Legal Med 135(4):1471–1476. https://doi.org/10.1007/s00414-021-02594-z
Hoon M, Petzer JP, Viljoen F, Petzer A (2017) The design and evaluation of an L-DOPA-lazabemide prodrug for the treatment of Parkinson's disease. Molecules 22(12). https://doi.org/10.3390/molecules22122076
Hoshino K, Itoh K, Masubuchi A et al (2007) Cloning, expression, and characterization of male cynomolgus monkey liver aldehyde oxidase. Biol Pharm Bull 30(7):1191–1198. https://doi.org/10.1248/bpb.30.1191
Hoskins J, Shenfield G, Murray M, Gross A (2001) Characterization of moclobemide N-oxidation in human liver microsomes. Xenobiotica 31(7):387–397. https://doi.org/10.1080/00498250110055488
Hosogi J, Ohashi R, Maeda H et al (2017) Monoamine oxidase B oxidizes a novel multikinase inhibitor KW-2449 to its iminium ion and aldehyde oxidase further converts it to the oxo-piperazine form in human. Drug Metab Pharmacokinet 32(5):255–264. https://doi.org/10.1016/j.dmpk.2017.06.002
Hosogi J, Ohashi R, Maeda H et al (2018) An iminium ion metabolite hampers the production of the pharmacologically active metabolite of a multikinase inhibitor KW-2449 in primates: Irreversible inhibition of aldehyde oxidase and covalent binding with endogenous proteins. Biopharm Drug Dispos 39(3):164–174. https://doi.org/10.1002/bdd.2123
Hou X, Zhou J, Yu S et al (2018) Differences in the in vivo and in vitro metabolism of imrecoxib in humans: Formation of the rate-limiting aldehyde intermediate. Drug Metab Dispos 46(9):1320–1328. https://doi.org/10.1124/dmd.118.081182
Hsieh TC, Lu X, Wang Z, Wu JM (2006) Induction of quinone reductase NQO1 by resveratrol in human K562 cells involves the antioxidant response element ARE and is accompanied by nuclear translocation of transcription factor Nrf2. Med Chem 2(3):275–285. https://doi.org/10.2174/157340606776930709
Huang S, Howington MB, Dobry CJ, Evans CR, Leiser SF (2021) Flavin-containing monooxygenases are conserved regulators of stress resistance and metabolism. Front Cell Dev Biol 9:630188. https://doi.org/10.3389/fcell.2021.630188
Huebert ND, Dulery BD, Schoun J, Schwach V, Hinze C, Haegele KD (1994) Kinetics and metabolism of p-tyramine during monoamine oxidase inhibition by mofegiline. Clin Pharmacol Ther 56(5):537–542. https://doi.org/10.1038/clpt.1994.175
Hummel MA, Dickmann LJ, Rettie AE, Haining RL, Tracy TS (2004) Differential activation of CYP2C9 variants by dapsone. Biochem Pharmacol 67(10):1831–1841. https://doi.org/10.1016/j.bcp.2004.01.017
Hung SI, Chung WH, Liou LB et al (2005) HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA 102(11):4134–4139. https://doi.org/10.1073/pnas.0409500102
Hutzler JM, Yang YS, Albaugh D, Fullenwider CL, Schmenk J, Fisher MB (2012) Characterization of aldehyde oxidase enzyme activity in cryopreserved human hepatocytes. Drug Metab Dispos 40(2):267–275. https://doi.org/10.1124/dmd.111.042861
Hutzler JM, Obach RS, Dalvie D, Zientek MA (2013) Strategies for a comprehensive understanding of metabolism by aldehyde oxidase. Expert Opin Drug Metab Toxicol 9(2):153–168. https://doi.org/10.1517/17425255.2013.738668
Hutzler JM, Cerny MA, Yang YS et al (2014) Cynomolgus monkey as a surrogate for human aldehyde oxidase metabolism of the EGFR inhibitor BIBX1382. Drug Metab Dispos 42(10):1751–1760. https://doi.org/10.1124/dmd.114.059030
Indra R, Pompach P, Martínek V, et al. (2019) Identification of human enzymes oxidizing the anti-thyroid-cancer drug vandetanib and explanation of the high efficiency of cytochrome P450 3A4 in its oxidation. Int J Mol Sci 20(14). https://doi.org/10.3390/ijms20143392
Indra R, Pompach P, Vavrová K et al (2020) Cytochrome P450 and flavin-containing monooxygenase enzymes are responsible for differential oxidation of the anti-thyroid-cancer drug vandetanib by human and rat hepatic microsomal systems. Environ Toxicol Pharmacol 74:103310. https://doi.org/10.1016/j.etap.2019.103310
Infante JR, Rugg T, Gordon M et al (2013) Unexpected renal toxicity associated with SGX523, a small molecule inhibitor of MET. Invest New Drugs 31(2):363–369. https://doi.org/10.1007/s10637-012-9823-9
Innocenti F, Danesi R, Di Paolo A et al (1996) Clinical and experimental pharmacokinetic interaction between 6-mercaptopurine and methotrexate. Cancer Chemother Pharmacol 37(5):409–414. https://doi.org/10.1007/s002800050405
Inoue K, Mizuo H, Kawaguchi S, Fukuda K, Kusano K, Yoshimura T (2014) Oxidative metabolic pathway of lenvatinib mediated by aldehyde oxidase. Drug Metab Dispos 42(8):1326–1333. https://doi.org/10.1124/dmd.114.058073
Isobe T, Ohta M, Kaneko Y, Kawai H (2016) Species differences in metabolism of ripasudil (K-115) are attributed to aldehyde oxidase. Xenobiotica 46(7):579–590. https://doi.org/10.3109/00498254.2015.1096981
Itagaki K, Carver GT, Philpot RM (1996) Expression and characterization of a modified flavin-containing monooxygenase 4 from humans. J Biol Chem 271(33):20102–20107. https://doi.org/10.1074/jbc.271.33.20102
Ito S, Sugumaran M, Wakamatsu K (2020) Chemical reactivities of ortho-quinones produced in living organisms: fate of quinonoid products formed by tyrosinase and phenoloxidase action on phenols and catechols. Int J Mol Sci 21(17). https://doi.org/10.3390/ijms21176080
Itoh K, Yamamura M, Muramatsu S et al (2005) Stereospecific oxidation of the (S)-enantiomer of RS-8359, a selective and reversible monoamine oxidase A (MAO-A) inhibitor, by aldehyde oxidase. Xenobiotica 35(6):561–573. https://doi.org/10.1080/00498250500202106
Iwasa T, Sano H, Sugiura A et al (2003) An in vitro interethnic comparison of monoamine oxidase activities between Japanese and Caucasian livers using rizatriptan, a serotonin receptor 1B/1D agonist, as a model drug. Br J Clin Pharmacol 56(5):537–544. https://doi.org/10.1046/j.1365-2125.2003.01922.x
Jacobsen W, Christians U, Benet LZ (2000) In vitro evaluation of the disposition of a novel cysteine protease inhibitor. Drug Metab Dispos 28(11):1343–1351
Janssens de Varebeke P, Cavalier R, David-Remacle M, Youdim MB (1988) Formation of the neurotransmitter glycine from the anticonvulsant milacemide is mediated by brain monoamine oxidase B. J Neurochem 50(4):1011–1016. https://doi.org/10.1111/j.1471-4159.1988.tb10566.x
Jaworski TJ, Hawes EM, McKay G, Midha KK (1990) The metabolism of chlorpromazine N-oxide in man and dog. Xenobiotica 20(1):107–115. https://doi.org/10.3109/00498259009046817
Jayanthi S, Daiwile AP, Cadet JL (2021) Neurotoxicity of methamphetamine: main effects and mechanisms. Exp Neurol 344:113795. https://doi.org/10.1016/j.expneurol.2021.113795
Jensen KG, Jacobsen AM, Bundgaard C et al (2017) Lack of exposure in a first-in-man study due to aldehyde oxidase metabolism: investigated by use of 14C-microdose, humanized mice, monkey pharmacokinetics, and in vitro methods. Drug Metab Dispos 45(1):68–75. https://doi.org/10.1124/dmd.116.072793
Ji Y, Salavaggione OE, Wang L et al (2005) Human phenylethanolamine N-methyltransferase pharmacogenomics: gene re-sequencing and functional genomics. J Neurochem 95(6):1766–1776. https://doi.org/10.1111/j.1471-4159.2005.03453.x
Jin F, Robeson M, Zhou H, Hisoire G, Ramanathan S (2015) The pharmacokinetics and safety of idelalisib in subjects with moderate or severe hepatic impairment. J Clin Pharmacol 55(8):944–952. https://doi.org/10.1002/jcph.504
Johns DG (1967) Human liver aldehyde oxidase: differential inhibition of oxidation of charged and uncharged substrates. J Clin Invest 46(9):1492–1505. https://doi.org/10.1172/jci105641
Johnson C, Stubley-Beedham C, Stell JG (1984) Elevation of molybdenum hydroxylase levels in rabbit liver after ingestion of phthalazine or its hydroxylated metabolite. Biochem Pharmacol 33(22):3699–3705. https://doi.org/10.1016/0006-2952(84)90159-x
Jones KC, Ballou DP (1986) Reactions of the 4a-hydroperoxide of liver microsomal flavin-containing monooxygenase with nucleophilic and electrophilic substrates. J Biol Chem 261(6):2553–2559
Jones BC, Srivastava A, Colclough N et al (2017) An investigation into the prediction of in vivo clearance for a range of flavin-containing monooxygenase substrates. Drug Metab Dispos 45(10):1060–1067. https://doi.org/10.1124/dmd.117.077396
Joo J, Wu Z, Lee B et al (2015) In vitro metabolism of an estrogen-related receptor γ modulator, GSK5182, by human liver microsomes and recombinant cytochrome P450s. Biopharm Drug Dispos 36(3):163–173. https://doi.org/10.1002/bdd.1929
Jordan CG, Rashidi MR, Laljee H, Clarke SE, Brown JE, Beedham C (1999) Aldehyde oxidase-catalysed oxidation of methotrexate in the liver of guinea-pig, rabbit and man. J Pharm Pharmacol 51(4):411–418
Jung HA, Roy A, Jung JH, Choi JS (2017) Evaluation of the inhibitory effects of eckol and dieckol isolated from edible brown alga Eisenia bicyclis on human monoamine oxidases A and B. Arch Pharm Res 40(4):480–491. https://doi.org/10.1007/s12272-017-0904-3
Kajita J, Inano K, Fuse E, Kuwabara T, Kobayashi H (2002) Effects of olopatadine, a new antiallergic agent, on human liver microsomal cytochrome P450 activities. Drug Metab Dispos 30(12):1504–1511. https://doi.org/10.1124/dmd.30.12.1504
Kalgutkar AS, Dalvie DK, Castagnoli N Jr, Taylor TJ (2001) Interactions of nitrogen-containing xenobiotics with monoamine oxidase (MAO) isozymes A and B: SAR studies on MAO substrates and inhibitors. Chem Res Toxicol 14(9):1139–1162. https://doi.org/10.1021/tx010073b
Kalow W, Tang BK (1991) Use of caffeine metabolite ratios to explore CYP1A2 and xanthine oxidase activities. Clin Pharmacol Ther 50(5 Pt 1):508–519. https://doi.org/10.1038/clpt.1991.176
Kang JH, Chung WG, Lee KH et al (2000) Phenotypes of flavin-containing monooxygenase activity determined by ranitidine N-oxidation are positively correlated with genotypes of linked FM03 gene mutations in a Korean population. Pharmacogenetics 10(1):67–78. https://doi.org/10.1097/00008571-200002000-00009
Kawashima K, Hosoi K, Naruke T, Shiba T, Kitamura M, Watabe T (1999) Aldehyde oxidase-dependent marked species difference in hepatic metabolism of the sedative-hypnotic, zaleplon, between monkeys and rats. Drug Metab Dispos 27(3):422–428
Kaye B, Rance DJ, Waring L (1985) Oxidative metabolism of carbazeran in vitro by liver cytosol of baboon and man. Xenobiotica 15(3):237–242. https://doi.org/10.3109/00498258509045354
Keller S, Polanski WH, Enzensperger C, Reichmann H, Hermann A, Gille G (2020) 9-Methyl-β-carboline inhibits monoamine oxidase activity and stimulates the expression of neurotrophic factors by astrocytes. J Neural Transm (vienna) 127(7):999–1012. https://doi.org/10.1007/s00702-020-02189-9
Kim YM, Ziegler DM (2000) Size limits of thiocarbamides accepted as substrates by human flavin-containing monooxygenase 1. Drug Metab Dispos 28(8):1003–1006
Kinsella TJ, Vielhuber KA, Kunugi KA, Schupp J, Davis TW, Sands H (2000) Preclinical toxicity and efficacy study of a 14-day schedule of oral 5-iodo-2-pyrimidinone-2′-deoxyribose as a prodrug for 5-iodo-2’-deoxyuridine radiosensitization in U251 human glioblastoma xenografts. Clin Cancer Res 6(4):1468–1475
Kisker C, Schindelin H, Rees DC (1997) Molybdenum-cofactor-containing enzymes: structure and mechanism. Annu Rev Biochem 66:233–267. https://doi.org/10.1146/annurev.biochem.66.1.233
Kitamura S, Sugihara K, Ohta S (2006) Drug-metabolizing ability of molybdenum hydroxylases. Drug Metab Pharmacokinet 21(2):83–98. https://doi.org/10.2133/dmpk.21.83
Kitamura S, Nitta K, Tayama Y et al (2008) Aldehyde oxidase-catalyzed metabolism of N1-methylnicotinamide in vivo and in vitro in chimeric mice with humanized liver. Drug Metab Dispos 36(7):1202–1205. https://doi.org/10.1124/dmd.107.019075
Kitchen BJ, Moser A, Lowe E et al (1999) Thioguanine administered as a continuous intravenous infusion to pediatric patients is metabolized to the novel metabolite 8-hydroxy-thioguanine. J Pharmacol Exp Ther 291(2):870–874
Klecker RW, Cysyk RL, Collins JM (2006) Zebularine metabolism by aldehyde oxidase in hepatic cytosol from humans, monkeys, dogs, rats, and mice: influence of sex and inhibitors. Bioorg Med Chem 14(1):62–66. https://doi.org/10.1016/j.bmc.2005.07.053
Kong LD, Cheng CH, Tan RX (2004) Inhibition of MAO A and B by some plant-derived alkaloids, phenols and anthraquinones. J Ethnopharmacol 91(2–3):351–355. https://doi.org/10.1016/j.jep.2004.01.013
Konishi K, Fukami T, Gotoh S, Nakajima M (2017) Identification of enzymes responsible for nitrazepam metabolism and toxicity in human. Biochem Pharmacol 140:150–160. https://doi.org/10.1016/j.bcp.2017.06.114
Kooij A, Schijns M, Frederiks WM, Van Noorden CJ, James J (1992) Distribution of xanthine oxidoreductase activity in human tissues–a histochemical and biochemical study. Virchows Archiv B, Cell Pathol (incl Mol Pathol) 63(1):17–23
Kosel M, Amey M, Aubert AC, Baumann P (2001) In vitro metabolism of citalopram by monoamine oxidase B in human blood. Eur Neuropsychopharmacol 11(1):75–78. https://doi.org/10.1016/s0924-977x(00)00128-0
Kosel M, Gnerre C, Voirol P et al (2002) In vitro biotransformation of the selective serotonin reuptake inhibitor citalopram, its enantiomers and demethylated metabolites by monoamine oxidase in rat and human brain preparations. Mol Psychiatry 7(2):181–188. https://doi.org/10.1038/sj.mp.4000946
Koukouritaki SB, Poch MT, Henderson MC et al (2007) Identification and functional analysis of common human flavin-containing monooxygenase 3 genetic variants. J Pharmacol Exp Ther 320(1):266–273. https://doi.org/10.1124/jpet.106.112268
Koukouritaki SB, Simpson P, Yeung CK, Rettie AE, Hines RN (2002) Human hepatic flavin-containing monooxygenases 1 (FMO1) and 3 (FMO3) developmental expression. Pediatr Res 51(2):236–243. https://doi.org/10.1203/00006450-200202000-00018
Kousba A, Soll R, Yee S, Martin M (2007) Cyclic conversion of the novel Src kinase inhibitor [7-(2,6-dichloro-phenyl)-5-methyl-benzo[1,2,4]triazin-3-yl]-[4-(2-pyrrolidin-1-yl-ethoxy)-phenyl]-amine (TG100435) and Its N-oxide metabolite by flavin-containing monoxygenases and cytochrome P450 reductase. Drug Metab Dispos 35(12):2242–2251. https://doi.org/10.1124/dmd.107.017384
Kozhemiakin LA, Antonov VG, Kalikanov SA, Bondarenko IG, Pastushenkov VL (1992) [Xanthine oxidase activity in mononuclear cells of human blood]. Biull Eksp Biol Med 113(2):138–139 [Russian]
Kozioł E, Luca SV, Ağalar HG et al (2020) Rutamarin: Efficient liquid-liquid chromatographic isolation from Ruta graveolens L. and evaluation of its in vitro and in silico MAO-B inhibitory activity. Molecules 25(11). https://doi.org/10.3390/molecules25112678
Krause RJ, Lash LH, Elfarra AA (2003) Human kidney flavin-containing monooxygenases and their potential roles in cysteine S-conjugate metabolism and nephrotoxicity. J Pharmacol Exp Ther 304(1):185–191. https://doi.org/10.1124/jpet.102.042911
Krause RJ, Glocke SC, Sicuri AR, Ripp SL, Elfarra AA (2006) Oxidative metabolism of seleno-L-methionine to L-methionine selenoxide by flavin-containing monooxygenases. Chem Res Toxicol 19(12):1643–1649. https://doi.org/10.1021/tx0601915
Krenitsky TA, Neil SM, Elion GB, Hitchings GH (1972) A comparison of the specificities of xanthine oxidase and aldehyde oxidase. Arch Biochem Biophys 150(2):585–599. https://doi.org/10.1016/0003-9861(72)90078-1
Krenitsky TA, Hall WW, de Miranda P, Beauchamp LM, Schaeffer HJ, Whiteman PD (1984) 6-Deoxyacyclovir: a xanthine oxidase-activated prodrug of acyclovir. Proc Natl Acad Sci USA 81(10):3209–3213. https://doi.org/10.1073/pnas.81.10.3209
Krenitsky TA, Spector T, Hall WW (1986) Xanthine oxidase from human liver: purification and characterization. Arch Biochem Biophys 247(1):108–119. https://doi.org/10.1016/0003-9861(86)90539-4
Krueger SK, Williams DE (2005) Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther 106(3):357–387. https://doi.org/10.1016/j.pharmthera.2005.01.001
Krueger SK, Martin SR, Yueh MF, Pereira CB, Williams DE (2002a) Identification of active flavin-containing monooxygenase isoform 2 in human lung and characterization of expressed protein. Drug Metab Dispos 30(1):34–41. https://doi.org/10.1124/dmd.30.1.34
Krueger SK, Williams DE, Yueh MF et al (2002b) Genetic polymorphisms of flavin-containing monooxygenase (FMO). Drug Metab Rev 34(3):523–532. https://doi.org/10.1081/dmr-120005653
Krueger SK, Siddens LK, Henderson MC et al (2005) Haplotype and functional analysis of four flavin-containing monooxygenase isoform 2 (FMO2) polymorphisms in Hispanics. Pharmacogenet Genomics 15(4):245–256. https://doi.org/10.1097/01213011-200504000-00008
Krueger SK, Vandyke JE, Williams DE, Hines RN (2006) The role of flavin-containing monooxygenase (FMO) in the metabolism of tamoxifen and other tertiary amines. Drug Metab Rev 38(1–2):139–147. https://doi.org/10.1080/03602530600569919
Krueger SK, Henderson MC, Siddens LK et al (2009) Characterization of sulfoxygenation and structural implications of human flavin-containing monooxygenase isoform 2 (FMO2.1) variants S195L and N413K. Drug Metab Dispos 37(8):1785–1791. https://doi.org/10.1124/dmd.109.027201
Kumar R, Joshi G, Kler H, Kalra S, Kaur M, Arya R (2018) Toward an understanding of structural insights of xanthine and aldehyde oxidases: an overview of their inhibitors and role in various diseases. Med Res Rev 38(4):1073–1125. https://doi.org/10.1002/med.21457
Kundu TK, Hille R, Velayutham M, Zweier JL (2007) Characterization of superoxide production from aldehyde oxidase: an important source of oxidants in biological tissues. Arch Biochem Biophys 460(1):113–121. https://doi.org/10.1016/j.abb.2006.12.032
Kurajoh M, Fukumoto S, Emoto M et al (2020) Independent association of plasma xanthine oxidoreductase activity with serum uric acid level based on stable isotope-labeled xanthine and liquid chromatography/triple quadrupole mass spectrometry: MedCity21 health examination registry. Clin Chem Lab Med 58(5):780–786. https://doi.org/10.1515/cclm-2019-0199
Kurzawski M, Dziewanowski K, Safranow K, Drozdzik M (2012) Polymorphism of genes involved in purine metabolism (XDH, AOX1, MOCOS) in kidney transplant recipients receiving azathioprine. Ther Drug Monit 34(3):266–274. https://doi.org/10.1097/FTD.0b013e31824aa681
Kusano T, Ehirchiou D, Matsumura T et al (2019) Targeted knock-in mice expressing the oxidase-fixed form of xanthine oxidoreductase favor tumor growth. Nat Commun 10(1):4904. https://doi.org/10.1038/s41467-019-12565-z
Kyritsi K, Chen L, O’Brien A et al (2020) Modulation of the tryptophan hydroxylase 1/monoamine oxidase-A/5-hydroxytryptamine/5-hydroxytryptamine receptor 2A/2B/2C axis regulates biliary proliferation and liver fibrosis during cholestasis. Hepatology 71(3):990–1008. https://doi.org/10.1002/hep.30880
Lacroix C, Hoang TP, Nouveau J et al (1989) Pharmacokinetics of pyrazinamide and its metabolites in healthy subjects. Eur J Clin Pharmacol 36(4):395–400. https://doi.org/10.1007/bf00558302
Lai WG, Farah N, Moniz GA, Wong YN (2011) A Baeyer-Villiger oxidation specifically catalyzed by human flavin-containing monooxygenase 5. Drug Metab Dispos 39(1):61–70. https://doi.org/10.1124/dmd.110.035360
Lake BG, Ball SE, Kao J, Renwick AB, Price RJ, Scatina JA (2002) Metabolism of zaleplon by human liver: evidence for involvement of aldehyde oxidase. Xenobiotica 32(10):835–847. https://doi.org/10.1080/00498250210158915
Lambert DM, Mamer OA, Akerman BR et al (2001) In vivo variability of TMA oxidation is partially mediated by polymorphisms of the FMO3 gene. Mol Genet Metab 73(3):224–229. https://doi.org/10.1006/mgme.2001.3189
Lang DH, Rettie AE (2000) In vitro evaluation of potential in vivo probes for human flavin-containing monooxygenase (FMO): Metabolism of benzydamine and caffeine by FMO and P450 isoforms. Br J Clin Pharmacol 50(4):311–314. https://doi.org/10.1046/j.1365-2125.2000.00265.x
Lang DH, Yeung CK, Peter RM et al (1998) Isoform specificity of trimethylamine N-oxygenation by human flavin-containing monooxygenase (FMO) and P450 enzymes: Selective catalysis by FMO3. Biochem Pharmacol 56(8):1005–1012. https://doi.org/10.1016/s0006-2952(98)00218-4
Larit F, Elokely KM, Chaurasiya ND et al (2018) Inhibition of human monoamine oxidase A and B by flavonoids isolated from two Algerian medicinal plants. Phytomedicine 40:27–36. https://doi.org/10.1016/j.phymed.2017.12.032
Lavian G, Finberg JP, Youdim MB (1993) The advent of a new generation of monoamine oxidase inhibitor antidepressants: pharmacologic studies with moclobemide and brofaromine. Clin Neuropharmacol 16(Suppl 2):S1-7
Lee SA, Hwang JS, Han XH et al (2008) Methylpiperate derivatives from Piper longum and their inhibition of monoamine oxidase. Arch Pharm Res 31(6):679–683. https://doi.org/10.1007/s12272-001-1212-7
Lee BE, Toledo AH, Anaya-Prado R, Roach RR, Toledo-Pereyra LH (2009a) Allopurinol, xanthine oxidase, and cardiac ischemia. J Investig Med 57(8):902–909. https://doi.org/10.2310/JIM.0b013e3181bca50c
Lee SK, Kang MJ, Jin C, In MK, Kim DH, Yoo HH (2009b) Flavin-containing monooxygenase 1-catalysed N,N-Dimethylamphetamine N-Oxidation. Xenobiotica 39(9):680–686. https://doi.org/10.1080/00498250902998699
Lee S, Yoo HH, In MK, Jin C, Kim DH (2013) Stereoselectivity in the cytochrome P450-dependent N-demethylation and flavin monooxygenase-dependent N-oxidation of N,N-Dimethylamphetamine. Arch Pharm Res 36(11):1385–1391. https://doi.org/10.1007/s12272-013-0137-z
Lee HW, Ryu HW, Kang MG, Park D, Oh SR, Kim H (2016) Strong selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg Med Chem Lett 26(19):4714–4719. https://doi.org/10.1016/j.bmcl.2016.08.044
Lee HW, Ryu HW, Baek SC et al (2017a) Strong inhibitions of monoamine oxidase A and B by acacetin and its 7-O-(6-O-malonylglucoside) derivative from Agastache rugosa. Int J Biol Macromol 104(Pt A):547–553. https://doi.org/10.1016/j.ijbiomac.2017.06.076
Lee HW, Ryu HW, Kang MG et al (2017b) Strong inhibition of monoamine oxidase A by decursin from Angelica gigas Nakai and by wogonin from Scutellaria baicalensis Georgi. Int J Biol Macromol 97:598–605. https://doi.org/10.1016/j.ijbiomac.2017.01.080
Lee HW, Ryu HW, Kang MG, Park D, Oh SR, Kim H (2017c) Selective inhibition of monoamine oxidase A by purpurin, an anthraquinone. Bioorg Med Chem Lett 27(5):1136–1140. https://doi.org/10.1016/j.bmcl.2017.01.085
Legoabe LJ, Petzer A, Petzer JP (2012a) Inhibition of monoamine oxidase by selected C6-substituted chromone derivatives. Eur J Med Chem 49:343–353. https://doi.org/10.1016/j.ejmech.2012.01.037
Legoabe LJ, Petzer A, Petzer JP (2012b) Selected chromone derivatives as inhibitors of monoamine oxidase. Bioorg Med Chem Lett 22(17):5480–5484. https://doi.org/10.1016/j.bmcl.2012.07.025
Lennard L (1992) The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol 43(4):329–339. https://doi.org/10.1007/bf02220605
Leoni C, Buratti FM, Testai E (2008) The participation of human hepatic P450 isoforms, flavin-containing monooxygenases and aldehyde oxidase in the biotransformation of the insecticide fenthion. Toxicol Appl Pharmacol 233(2):343–352. https://doi.org/10.1016/j.taap.2008.09.004
Lepri S, Ceccarelli M, Milani N et al (2017) Structure-metabolism relationships in human-AOX: chemical insights from a large database of aza-aromatic and amide compounds. Proc Natl Acad Sci USA 114(16):E3178-e3187. https://doi.org/10.1073/pnas.1618881114
Leung K (2004) (S)-5-Methoxymethyl-3-[6-(4,4,4-trifluorobutoxy)benzo[d]isoxazol-3-yl]-oxazolidin-2-[(11)C]one, in molecular imaging and contrast agent database (MICAD). National Center for Biotechnology Information (US), Bethesda (MD)
Levi PE, Hodgson E (1988) Stereospecificity in the oxidation of phorate and phorate sulphoxide by purified FAD-containing mono-oxygenase and cytochrome P-450 isozymes. Xenobiotica 18(1):29–39. https://doi.org/10.3109/00498258809055134
Lewinsohn R, Glover V, Sandler M (1980) b-Phenylethylamine and benzylamine as substrates for human monoamine oxidase A: a source of some anomalies? Biochem Pharmacol 29(5):777–781. https://doi.org/10.1016/0006-2952(80)90556-0
Li XQ, Björkman A, Andersson TB, Gustafsson LL, Masimirembwa CM (2003) Identification of human cytochrome P(450)s that metabolise anti-parasitic drugs and predictions of in vivo drug hepatic clearance from in vitro data. Eur J Clin Pharmacol 59(5–6):429–442. https://doi.org/10.1007/s00228-003-0636-9
Li H, Kundu TK, Zweier JL (2009) Characterization of the magnitude and mechanism of aldehyde oxidase-mediated nitric oxide production from nitrite. J Biol Chem 284(49):33850–33858. https://doi.org/10.1074/jbc.M109.019125
Li Y, Lai WG, Whitcher-Johnstone A et al (2012a) Metabolic switching of BILR 355 in the presence of ritonavir. I. Identifying an unexpected disproportionate human metabolite. Drug Metab Dispos 40(6):1122–1129. https://doi.org/10.1124/dmd.111.044354
Li Y, Xu J, Lai WG, Whitcher-Johnstone A, Tweedie DJ (2012b) Metabolic switching of BILR 355 in the presence of ritonavir. II. Uncovering novel contributions by gut bacteria and aldehyde oxidase. Drug Metab Dispos 40(6):1130–1137. https://doi.org/10.1124/dmd.111.044362
Lin J, Cashman JR (1997a) Detoxication of tyramine by the flavin-containing monooxygenase: stereoselective formation of the trans oxime. Chem Res Toxicol 10(8):842–852. https://doi.org/10.1021/tx970030o
Lin J, Cashman JR (1997b) N-Oxygenation of phenethylamine to the trans-oxime by adult human liver flavin-containing monooxygenase and retroreduction of phenethylamine hydroxylamine by human liver microsomes. J Pharmacol Exp Ther 282(3):1269–1279
Lin J, Berkman CE, Cashman JR (1996) N-Oxygenation of primary amines and hydroxylamines and retroreduction of hydroxylamines by adult human liver microsomes and adult human flavin-containing monooxygenase 3. Chem. Res. Toxicol. 9(7):1183–1193 (0893-228X)
Lin KH, Lin CY, Liu CC, Chou MY, Lin JK (2011) Arecoline N-oxide: its mutagenicity and possible role as ultimate carcinogen in areca oral carcinogenesis. J Agric Food Chem 59(7):3420–3428. https://doi.org/10.1021/jf104831n
Linder N, Rapola J, Raivio KO (1999) Cellular expression of xanthine oxidoreductase protein in normal human tissues. Lab Invest 79(8):967–974
Liu X, Lin WM, Yan XH, Chen XH, Hoidal JR, Xu P (2003) Improved method for measurement of human plasma xanthine oxidoreductase activity. J Chromatogr B Analyt Technol Biomed Life Sci 785(1):101–114. https://doi.org/10.1016/s1570-0232(02)00860-7
Liu L, Halladay JS, Shin Y et al (2011) Significant species difference in amide hydrolysis of GDC-0834, a novel strong and selective Bruton’s tyrosine kinase inhibitor. Drug Metab Dispos 39(10):1840–1849. https://doi.org/10.1124/dmd.111.040840
Liu L, Chen Y, Zeng RF et al (2021a) Design and synthesis of novel 3,4-dihydrocoumarins as strong and selective monoamine oxidase-B inhibitors with the neuroprotection against Parkinson’s disease. Bioorg Chem 109:104685. https://doi.org/10.1016/j.bioorg.2021.104685
Liu X, Sun H, Zhang Y et al (2021b) Clozapine affects the pharmacokinetics of risperidone and inhibits its metabolism and P-glycoprotein-mediated transport in vivo and in vitro: a safety attention to antipsychotic polypharmacy with clozapine and risperidone. Toxicol Appl Pharmacol 422:115560. https://doi.org/10.1016/j.taap.2021.115560
Lolkema MP, Bohets HH, Arkenau HT et al (2015) The c-Met tyrosine kinase inhibitor JNJ-38877605 causes renal toxicity through species-specific insoluble metabolite formation. Clin Cancer Res 21(10):2297–2304. https://doi.org/10.1158/1078-0432.Ccr-14-3258
Lomri N, Yang Z, Cashman JR (1993) Regio- and stereoselective oxygenations by adult human liver flavin-containing monooxygenase 3. Comparison with forms 1 and 2. Chem Res Toxicol 6(6):800–807. https://doi.org/10.1021/tx00036a008
LoRusso PM, Prakash S, Wozniak A et al (2002) Phase I clinical trial of 5-fluoro-pyrimidinone (5FP), an oral prodrug of 5-fluorouracil (5FU). Invest New Drugs 20(1):63–71. https://doi.org/10.1023/a:1014430216434
Lotufo-Neto F, Trivedi M, Thase ME (1999) Meta-analysis of the reversible inhibitors of monoamine oxidase type A moclobemide and brofaromine for the treatment of depression. Neuropsychopharmacology 20(3):226–247. https://doi.org/10.1016/s0893-133x(98)00075-x
Lu X, Li C, Fleisher D (1998) Cimetidine sulfoxidation in small intestinal microsomes. Drug Metab Dispos 26(9):940–942
Lu C, Zhou Q, Yan J, Du Z, Huang L, Li X (2013) A novel series of tacrine-selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer’s disease. Eur J Med Chem 62:745–753. https://doi.org/10.1016/j.ejmech.2013.01.039
Lynch RE, Fridovich I (1979) Autoinactivation of xanthine oxidase: the role of superoxide radical and hydrogen peroxide. Biochim Biophys Acta 571(2):195–200. https://doi.org/10.1016/0005-2744(79)90090-1
Maeda K, Ohno T, Igarashi S, Yoshimura T, Yamashiro K, Sakai M (2012) Aldehyde oxidase 1 gene is regulated by Nrf2 pathway. Gene 505(2):374–378. https://doi.org/10.1016/j.gene.2012.06.010
Magyar K, Szende B (2004) (-)-Deprenyl, a selective MAO-B inhibitor, with apoptotic and anti-apoptotic properties. Neurotoxicology 25(1–2):233–242. https://doi.org/10.1016/s0161-813x(03)00102-5
Mahasneh S, Sharab A, Al Shhab M, Rashid M, Zihlif M (2020) AOX1 and XDH enzymes genotyping and its effect on clinical response to azathioprine in inflammatory bowel disease patients among Jordanian population. Curr Drug Metab 21(2):140–144. https://doi.org/10.2174/1389200221666200413125011
Mahro M, Brás NF, Cerqueira NM et al (2013) Identification of crucial amino acids in mouse aldehyde oxidase 3 that determine substrate specificity. PLoS ONE 8(12):e82285. https://doi.org/10.1371/journal.pone.0082285
Maia LB, Moura JJG (2018) Putting xanthine oxidoreductase and aldehyde oxidase on the NO metabolism map: nitrite reduction by molybdoenzymes. Redox Biol 19:274–289. https://doi.org/10.1016/j.redox.2018.08.020
Maia LB, Pereira V, Mira L, Moura JJ (2015) Nitrite reductase activity of rat and human xanthine oxidase, xanthine dehydrogenase, and aldehyde oxidase: evaluation of their contribution to NO formation in vivo. Biochemistry 54(3):685–710. https://doi.org/10.1021/bi500987w
Mandel S, Weinreb O, Amit T, Youdim MB (2005) Mechanism of neuroprotective action of the anti-Parkinson drug rasagiline and its derivatives. Brain Res Rev 48(2):379–387. https://doi.org/10.1016/j.brainresrev.2004.12.027
Manevski N, Balavenkatraman KK, Bertschi B et al (2014) Aldehyde oxidase activity in fresh human skin. Drug Metab Dispos 42(12):2049–2057. https://doi.org/10.1124/dmd.114.060368
Manevski N, King L, Pitt WR, Lecomte F, Toselli F (2019) Metabolism by aldehyde oxidase: drug design and complementary approaches to challenges in drug discovery. J Med Chem 62(24):10955–10994. https://doi.org/10.1021/acs.jmedchem.9b00875
Mao Z, Wu Y, Li Q, Wang X, Liu Y, Di X (2018) Aldehyde oxidase-dependent species difference in hepatic metabolism of fasudil to hydroxyfasudil. Xenobiotica 48(2):170–177. https://doi.org/10.1080/00498254.2017.1292016
Martini A, Bonollo L, Nicolis FB, Sega R, Palermo A (1981a) Effects of caroxazone, a reversible monoamine oxidase inhibitor, on the pressor response to oral tyramine in man. Br J Clin Pharmacol 11(6):611–615. https://doi.org/10.1111/j.1365-2125.1981.tb01178.x
Martini A, Bonollo L, Nicolis FB, Sega R, Palermo A, Braibanti E (1981b) Effects of caroxazone, a reversible monoamine oxidase inhibitor, on the pressor response to intravenous tyramine in man. Br J Clin Pharmacol 11(6):605–610. https://doi.org/10.1111/j.1365-2125.1981.tb01177.x
Masuo Y, Nagamori S, Hasegawa A et al (2017) Utilization of liver microsomes to estimate hepatic intrinsic clearance of monoamine oxidase substrate drugs in humans. Pharm Res 34(6):1233–1243. https://doi.org/10.1007/s11095-017-2140-4
Mathew B, Kim H (2020) Inhibitors of monoamine oxidase and acetylcholinesterase as a front runner in CNS drug discovery. Comb Chem High Throughput Screen 23(9):834–835. https://doi.org/10.2174/138620732309201127093123
Mathew B, Suresh J, Anbazhagan S, Mathew GE (2013) Pyrazoline: a promising scaffold for the inhibition of monoamine oxidase. Cent Nerv Syst Agents Med Chem 13(3):195–206. https://doi.org/10.2174/1871524914666140129122632
Mathew B, Mathew GE, Petzer JP, Petzer A (2017) Structural exploration of synthetic chromones as selective MAO-B inhibitors: a mini review. Comb Chem High Throughput Screen 20(6):522–532. https://doi.org/10.2174/1386207320666170227155517
Matos MJ, Viña D, Picciau C, Orallo F, Santana L, Uriarte E (2009) Synthesis and evaluation of 6-methyl-3-phenylcoumarins as strong and selective MAO-B inhibitors. Bioorg Med Chem Lett 19(17):5053–5055. https://doi.org/10.1016/j.bmcl.2009.07.039
Matos MJ, Viña D, Janeiro P, Borges F, Santana L, Uriarte E (2010) New halogenated 3-phenylcoumarins as strong and selective MAO-B inhibitors. Bioorg Med Chem Lett 20(17):5157–5160. https://doi.org/10.1016/j.bmcl.2010.07.013
Matos MJ, Terán C, Pérez-Castillo Y, Uriarte E, Santana L, Viña D (2011a) Synthesis and study of a series of 3-arylcoumarins as strong and selective monoamine oxidase B inhibitors. J Med Chem 54(20):7127–7137. https://doi.org/10.1021/jm200716y
Matos MJ, Vazquez-Rodriguez S, Uriarte E, Santana L, Viña D (2011b) MAO inhibitory activity modulation: 3-Phenylcoumarins versus 3-benzoylcoumarins. Bioorg Med Chem Lett 21(14):4224–4227. https://doi.org/10.1016/j.bmcl.2011.05.074
Matos MJ, Viña D, Vazquez-Rodriguez S, Uriarte E, Santana L (2012) Focusing on new monoamine oxidase inhibitors: differently substituted coumarins as an interesting scaffold. Curr Top Med Chem 12(20):2210–2239. https://doi.org/10.2174/156802612805220002
Matsukawa M, Hirai T, Karita S et al (2004) A screening system of prodrugs selective for MAO-A or MAO-B. Neurotoxicology 25(1–2):293–302. https://doi.org/10.1016/s0161-813x(03)00108-6
Matsumoto K, Okamoto K, Ashizawa N, Nishino T (2011) FYX-051: a novel and strong hybrid-type inhibitor of xanthine oxidoreductase. J Pharmacol Exp Ther 336(1):95–103. https://doi.org/10.1124/jpet.110.174540
Matsumoto K, Hasegawa T, Ohara K, Kamei T, Koyanagi J, Akimoto M (2021) Role of human flavin-containing monooxygenase (FMO) 5 in the metabolism of nabumetone: Baeyer-Villiger oxidation in the activation of the intermediate metabolite, 3-hydroxy nabumetone, to the active metabolite, 6-methoxy-2-naphthylacetic acid in vitro. Xenobiotica 51(2):155–166. https://doi.org/10.1080/00498254.2020.1843089
Mattsson C, Svensson P, Sonesson C (2014) A novel series of 6-substituted 3-(pyrrolidin-1-ylmethyl)chromen-2-ones as selective monoamine oxidase (MAO) A inhibitors. Eur J Med Chem 73:177–186. https://doi.org/10.1016/j.ejmech.2013.11.035
Matveychuk D, MacKenzie EM, Kumpula D et al (2021) Overview of the neuroprotective effects of the MAO-inhibiting antidepressant phenelzine. Cell Mol Neurobiol. https://doi.org/10.1007/s10571-021-01078-3
McCabe BJ (1986) Dietary tyramine and other pressor amines in MAOI regimens: a review. J Am Diet Assoc 86(8):1059–1064
McEnroe JD, Fleishaker JC (2005) Clinical pharmacokinetics of almotriptan, a serotonin 5-HT(1B/1D) receptor agonist for the treatment of migraine. Clin Pharmacokinet 44(3):237–246. https://doi.org/10.2165/00003088-200544030-00002
McManaman JL, Hanson L, Neville MC, Wright RM (2000) Lactogenic hormones regulate xanthine oxidoreductase and beta-casein levels in mammary epithelial cells by distinct mechanisms. Arch Biochem Biophys 373(2):318–327. https://doi.org/10.1006/abbi.1999.1573
McManus ME, Stupans I, Burgess W, Koenig JA, Hall PM, Birkett DJ (1987) Flavin-containing monooxygenase activity in human liver microsomes. Drug Metab Dispos 15(2):256–261
Megarity CF, Timson DJ (2019) Cancer-associated variants of human NQO1: impacts on inhibitor binding and cooperativity. Biosci Rep. https://doi.org/10.1042/bsr20191874
Meiser J, Weindl D, Hiller K (2013) Complexity of dopamine metabolism. Cell Commun Signal 11(1):34. https://doi.org/10.1186/1478-811x-11-34
Mellado M, González C, Mella J et al (2021) Combined 3D-QSAR and docking analysis for the design and synthesis of chalcones as strong and selective monoamine oxidase B inhibitors. Bioorg Chem 108:104689. https://doi.org/10.1016/j.bioorg.2021.104689
Meng J, Zhong D, Li L et al (2015) Metabolism of MRX-I, a novel antibacterial oxazolidinone, in humans: the oxidative ring opening of 2,3-dihydropyridin-4-one catalyzed by non-P450 enzymes. Drug Metab Dispos 43(5):646–659. https://doi.org/10.1124/dmd.114.061747
Miao Z, Kamel A, Prakash C (2005) Characterization of a novel metabolite intermediate of ziprasidone in hepatic cytosolic fractions of rat, dog, and human by ESI-MS/MS, hydrogen/deuterium exchange, and chemical derivatization. Drug Metab Dispos 33(7):879–883. https://doi.org/10.1124/dmd.105.004036
Mihaly GW, Ward SA, Edwards G, Orme ML, Breckenridge AM (1984) Pharmacokinetics of primaquine in man: Identification of the carboxylic acid derivative as a major plasma metabolite. Br J Clin Pharmacol 17(4):441–446. https://doi.org/10.1111/j.1365-2125.1984.tb02369.x
Milczek EM, Bonivento D, Binda C, Mattevi A, McDonald IA, Edmondson DE (2008) Structural and mechanistic studies of mofegiline inhibition of recombinant human monoamine oxidase B. J Med Chem 51(24):8019–8026. https://doi.org/10.1021/jm8011867
Miller MM, James RA, Richer JK, Gordon DF, Wood WM, Horwitz KB (1997) Progesterone regulated expression of flavin-containing monooxygenase 5 by the B-isoform of progesterone receptors: implications for tamoxifen carcinogenicity. J Clin Endocrinol Metab 82(9):2956–2961. https://doi.org/10.1210/jcem.82.9.4239
Miners JO, Birkett DJ (1996) The use of caffeine as a metabolic probe for human drug metabolizing enzymes. Gen Pharmacol 27(2):245–249. https://doi.org/10.1016/0306-3623(95)02014-4
Miners JO, Attwood J, Birkett DJ (1982) Theobromine metabolism in man. Drug Metab Dispos 10(6):672–675
Miranda GE, Sordo M, Salazar AM et al (2007) Determination of amphetamine, methamphetamine, and hydroxyamphetamine derivatives in urine by gas chromatography-mass spectrometry and its relation to CYP2D6 phenotype of drug users. J Anal Toxicol 31(1):31–36. https://doi.org/10.1093/jat/31.1.31
Mishima E, Anzai N, Miyazaki M, Abe T (2020) Uric acid elevation by favipiravir, an antiviral drug. Tohoku J Exp Med 251(2):87–90. https://doi.org/10.1620/tjem.251.87
Miura M, Ohkubo T (2004) In vitro metabolism of quazepam in human liver and intestine and assessment of drug interactions. Xenobiotica 34(11–12):1001–1011. https://doi.org/10.1080/02772240400015214
Montefiori M, Jørgensen FS, Olsen L (2017) Aldehyde oxidase: reaction mechanism and prediction of site of metabolism. ACS Omega 2(8):4237–4244. https://doi.org/10.1021/acsomega.7b00658
Moretti A, Caccia C, Martini A et al (1981) Effect of caroxazone, a new antidepressant drug, on monoamine oxidases in healthy volunteers. Br J Clin Pharmacol 11(5):511–515. https://doi.org/10.1111/j.1365-2125.1981.tb01158.x
Moriwaki Y, Yamamoto T, Suda M et al (1993) Purification and immunohistochemical tissue localization of human xanthine oxidase. Biochim Biophys Acta 1164(3):327–330. https://doi.org/10.1016/0167-4838(93)90266-t
Moriwaki Y, Yamamoto T, Yamaguchi K et al (1996) Immunohistochemical localization of xanthine oxidase in human tissues. Acta Histochem Cytochem 29(2):153–162. https://doi.org/10.1267/ahc.29.153
Moriwaki Y, Yamamoto T, Takahashi S, Tsutsumi Z, Hada T (2001) Widespread cellular distribution of aldehyde oxidase in human tissues found by immunohistochemistry staining. Histol Histopathol 16(3):745–753
Morrison RD, Blobaum AL, Byers FW et al (2012) The role of aldehyde oxidase and xanthine oxidase in the biotransformation of a novel negative allosteric modulator of metabotropic glutamate receptor subtype 5. Drug Metab Dispos 40(9):1834–1845. https://doi.org/10.1124/dmd.112.046136
Mostert S, Petzer A, Petzer JP (2015) Indanones as high-potency reversible inhibitors of monoamine oxidase. ChemMedChem 10(5):862–873. https://doi.org/10.1002/cmdc.201500059
Mota C, Esmaeeli M, Coelho C et al (2019) Human aldehyde oxidase (hAOX1): structure determination of the Moco-free form of the natural variant G1269R and biophysical studies of single nucleotide polymorphisms. FEBS Open Bio 9(5):925–934. https://doi.org/10.1002/2211-5463.12617
Mota C, Diniz A, Coelho C et al (2021) Interrogating the inhibition mechanisms of human aldehyde oxidase by X-ray crystallography and NMR spectroscopy: the raloxifene case. J Med Chem 64(17):13025–13037. https://doi.org/10.1021/acs.jmedchem.1c01125
Murase T, Nampei M, Oka M, Miyachi A, Nakamura T (2016a) A highly sensitive assay of human plasma xanthine oxidoreductase activity using stable isotope-labeled xanthine and LC/TQMS. J Chromatogr B Analyt Technol Biomed Life Sci 1039:51–58. https://doi.org/10.1016/j.jchromb.2016.10.033
Murase T, Oka M, Nampei M, Miyachi A, Nakamura T (2016b) A highly sensitive assay for xanthine oxidoreductase activity using a combination of [13C2,15N2]-xanthine and liquid chromatography/triple quadrupole mass spectrometry. J Labelled Comp Radiopharm 59(5):214–220. https://doi.org/10.1002/jlcr.3390
Murphy DL, Lipper S, Slater S, Shiling D (1979) Selectivity of clorgyline and pargyline as inhibitors of monoamine oxidases A and B in vivo in man. Psychopharmacology 62(2):129–132. https://doi.org/10.1007/bf00427125
Murphy DL, Tamarkin L, Sunderland T, Garrick NA, Cohen RM (1986) Human plasma melatonin is elevated during treatment with the monoamine oxidase inhibitors clorgyline and tranylcypromine but not deprenyl. Psychiatry Res 17(2):119–127. https://doi.org/10.1016/0165-1781(86)90067-3
Murphy DL, Karoum F, Pickar D et al (1998) Differential trace amine alterations in individuals receiving acetylenic inhibitors of MAO-A (clorgyline) or MAO-B (selegiline and pargyline). J Neural Transm Suppl 52:39–48. https://doi.org/10.1007/978-3-7091-6499-0_5
Murray M, Zhang WV, Edwards RJ (2018) Variation in the response of clozapine biotransformation pathways in human hepatic microsomes to CYP1A2- and CYP3A4-selective Inhibitors. Basic Clin Pharmacol Toxicol 122(4):388–395. https://doi.org/10.1111/bcpt.12933
Mushiroda T, Douya R, Takahara E, Nagata O (2000) The involvement of flavin-containing monooxygenase but not CYP3A4 in metabolism of itopride hydrochloride, a gastroprokinetic agent: comparison with cisapride and mosapride citrate. Drug Metab Dispos 28(10):1231–1237
Nagashima S, Shimizu M, Yano H et al (2009) Inter-individual variation in flavin-containing monooxygenase 3 in livers from Japanese: correlation with hepatic transcription factors. Drug Metab Pharmacokinet 24(3):218–225. https://doi.org/10.2133/dmpk.24.218
Nair NP, Ahmed SK, Kin NM (1993) Biochemistry and pharmacology of reversible inhibitors of MAO-A agents: focus on moclobemide. J Psychiatry Neurosci 18(5):214–225
Nakamura M, Yuichiro Y, Sass JO et al (2012) Identification of a xanthinuria type I case with mutations of xanthine dehydrogenase in an Afghan child. Clin Chim Acta 414:158–160. https://doi.org/10.1016/j.cca.2012.08.011
Nakamura A, Latif MA, Deck PA, Castagnoli N Jr, Tanko JM (2020) Evidence for a proton-coupled electron transfer mechanism in a biomimetic system for monoamine oxidase B catalysis. Chemistry 26(4):823–829. https://doi.org/10.1002/chem.201904634
Nandigama RK, Newton-Vinson P, Edmondson DE (2002) Phentermine inhibition of recombinant human liver monoamine oxidases A and B. Biochem Pharmacol 63(5):865–869. https://doi.org/10.1016/s0006-2952(02)00840-7
Nimkar SK, Anderson AH, Rimoldi JM et al (1996) Synthesis and monoamine oxidase B catalyzed oxidation of C-4 heteroaromatic substituted 1,2,3,6-tetrahydropyridine derivatives. Chem Res Toxicol 9(6):1013–1022. https://doi.org/10.1021/tx960063o
Nirogi R, Kandikere V, Palacharla RC et al (2014) Identification of a suitable and selective inhibitor towards aldehyde oxidase catalyzed reactions. Xenobiotica 44(3):197–204. https://doi.org/10.3109/00498254.2013.819594
Niwa T, Hiroi T, Tsuzuki D et al (2004) Effect of genetic polymorphism on the metabolism of endogenous neuroactive substances, progesterone and p-tyramine, catalyzed by CYP2D6. Brain Res Mol Brain Res 129(1–2):117–123. https://doi.org/10.1016/j.molbrainres.2004.06.030
Niwa T, Murayama N, Umeyama H, Shimizu M, Yamazaki H (2011) Human liver enzymes responsible for metabolic elimination of tyramine; a vasopressor agent from daily food. Drug Metab Lett 5(3):216–219. https://doi.org/10.2174/187231211796905026
Nolan KA, Dunstan MS, Caraher MC, Scott KA, Leys D, Stratford IJ (2012) In silico screening reveals structurally diverse, nanomolar inhibitors of NQO2 that are functionally active in cells and can modulate NF-κB signaling. Mol Cancer Ther 11(1):194–203. https://doi.org/10.1158/1535-7163.Mct-11-0543
Obach RS (2004) Strong inhibition of human liver aldehyde oxidase by raloxifene. Drug Metab Dispos 32(1):89–97. https://doi.org/10.1124/dmd.32.1.89
Obach RS, Dalvie DK (2006) Metabolism of nomifensine to a dihydroisoquinolinium ion metabolite by human myeloperoxidase, hemoglobin, monoamine oxidase A, and cytochrome P450 enzymes. Drug Metab Dispos 34(8):1310–1316. https://doi.org/10.1124/dmd.106.010173
Obach RS, Walsky RL (2005) Drugs that inhibit oxidation reactions catalyzed by aldehyde oxidase do not inhibit the reductive metabolism of ziprasidone to its major metabolite, S-methyldihydroziprasidone: an in vitro study. J Clin Psychopharmacol 25(6):605–608. https://doi.org/10.1097/01.jcp.0000186740.22395.50
Obach RS, Huynh P, Allen MC, Beedham C (2004) Human liver aldehyde oxidase: inhibition by 239 drugs. J Clin Pharmacol 44(1):7–19. https://doi.org/10.1177/0091270003260336
Obach RS, Prakash C, Kamel AM (2012) Reduction and methylation of ziprasidone by glutathione, aldehyde oxidase, and thiol S-methyltransferase in humans: an in vitro study. Xenobiotica 42(11):1049–1057. https://doi.org/10.3109/00498254.2012.683203
Oberley TD (2002) Oxidative damage and cancer. Am J Pathol 160(2):403–408. https://doi.org/10.1016/s0002-9440(10)64857-2
O'Carroll AM, Fowler CJ, Phillips JP, Tobbia I, Tipton KF (1983) The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn Schmiedebergs Arch Pharmacol 322(3):198–202. https://doi.org/10.1007/bf00500765
Ogiso T, Fukami T, Mishiro K, Konishi K, Jones JP, Nakajima M (2018) Substrate selectivity of human aldehyde oxidase 1 in reduction of nitroaromatic drugs. Arch Biochem Biophys 659:85–92. https://doi.org/10.1016/j.abb.2018.10.017
Oguchi K, Kobayashi S, Uesato T, Kamijo K (1981) Studies on b-phenylethylamine deamination by human placental monoamine oxidase. Jpn J Pharmacol 31(1):7–14. https://doi.org/10.1254/jjp.31.7
Oh JM, Jang HJ, Kim WJ et al (2020) Calycosin and 8-O-methylretusin isolated from Maackia amurensis as strong and selective reversible inhibitors of human monoamine oxidase-B. Int J Biol Macromol 151:441–448. https://doi.org/10.1016/j.ijbiomac.2020.02.144
Ohmi N, Yoshida H, Endo H, Hasegawa M, Akimoto M, Higuchi S (2003) S-Oxidation of S-methyl-esonarimod by flavin-containing monooxygenases in human liver microsomes. Xenobiotica 33(12):1221–1231. https://doi.org/10.1080/00498250310001624627
Ohmiya Y, Mehendale HM (1984) Species differences in pulmonary N-oxidation of chlorpromazine and imipramine. Pharmacology 28(5):289–295. https://doi.org/10.1159/000137976
Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T (2003) An extremely strong inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem 278(3):1848–1855. https://doi.org/10.1074/jbc.M208307200
Okamoto K, Eger BT, Nishino T, Pai EF, Nishino T (2008) Mechanism of inhibition of xanthine oxidoreductase by allopurinol: Crystal structure of reduced bovine milk xanthine oxidoreductase bound with oxipurinol. Nucleosides Nucleotides Nucleic Acids 27(6):888–893. https://doi.org/10.1080/15257770802146577
Okamoto K, Kusano T, Nishino T (2013) Chemical nature and reaction mechanisms of the molybdenum cofactor of xanthine oxidoreductase. Curr Pharm Des 19(14):2606–2614. https://doi.org/10.2174/1381612811319140010
Oliveira NG, Ramos DL, Dinis-Oliveira RJ (2021) Genetic toxicology and toxicokinetics of arecoline and related areca nut compounds: an updated review. Arch Toxicol 95(2):375–393. https://doi.org/10.1007/s00204-020-02926-9
Onderwater RC, Rettie AE, Commandeur JN, Vermeulen NP (2006) Bioactivation of N-substituted N′-(4-imidazole-ethyl)thioureas by human FMO1 and FMO3. Xenobiotica 36(7):645–657. https://doi.org/10.1080/00498250500354329
Ortiz de Montellano PR, Augusto O, Viola F, Kunze KL (1983) Carbon radicals in the metabolism of alkyl hydrazines. J Biol Chem 258(14):8623–8629
Overby LH, Buckpitt AR, Lawton MP, Atta-Asafo-Adjei E, Schulze J, Philpot RM (1995) Characterization of flavin-containing monooxygenase 5 (FMO5) cloned from human and guinea pig: evidence that the unique catalytic properties of FMO5 are not confined to the rabbit ortholog. Arch Biochem Biophys 317(1):275–284. https://doi.org/10.1006/abbi.1995.1163
Overby LH, Carver GC, Philpot RM (1997) Quantitation and kinetic properties of hepatic microsomal and recombinant flavin-containing monooxygenases 3 and 5 from humans. Chem-Biol Interact 106(1):29–45. https://doi.org/10.1016/s0009-2797(97)00055-0
Özdemir Z, Alagöz MA, Bahçecioğlu ÖF, Gök S (2021) Monoamine oxidase-B (MAO-B) inhibitors in the treatment of Alzheimer’s and Parkinson’s Disease. Curr Med Chem 28(29):6045–6065. https://doi.org/10.2174/0929867328666210203204710
Pacher P, Nivorozhkin A, Szabó C (2006) Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev 58(1):87–114. https://doi.org/10.1124/pr.58.1.6
Page S, Powell D, Benboubetra M et al (1998) Xanthine oxidoreductase in human mammary epithelial cells: activation in response to inflammatory cytokines. Biochim Biophys Acta 1381(2):191–202. https://doi.org/10.1016/s0304-4165(98)00028-2
Pang X, Tang C, Guo R, Chen X (2022) Non-cytochrome P450 enzymes involved in the oxidative metabolism of xenobiotics: Focus on the regulation of gene expression and enzyme activity. Pharmacol Ther 233:108020. https://doi.org/10.1016/j.pharmthera.2021.108020
Papandreou C, Moré M, Bellamine A (2020) Trimethylamine N-oxide in relation to cardiometabolic health-cause or effect? Nutrients 12(5). https://doi.org/10.3390/nu12051330
Paragas EM, Humphreys SC, Min J, Joswig-Jones CA, Jones JP (2017a) The two faces of aldehyde oxidase: oxidative and reductive transformations of 5-nitroquinoline. Biochem Pharmacol 145:210–217. https://doi.org/10.1016/j.bcp.2017.09.002
Paragas EM, Humphreys SC, Min J, Joswig-Jones CA, Leimkühler S, Jones JP (2017b) ecoAO: a simple system for the study of human aldehyde oxidases role in drug metabolism. ACS Omega 2(8):4820–4827. https://doi.org/10.1021/acsomega.7b01054
Parent MB, Master S, Kashlub S, Baker GB (2002) Effects of the antidepressant/antipanic drug phenelzine and its putative metabolite phenylethylidenehydrazine on extracellular gamma-aminobutyric acid levels in the striatum. Biochem Pharmacol 63(1):57–64. https://doi.org/10.1016/s0006-2952(01)00856-5
Park SB, Jacob P 3rd, Benowitz NL, Cashman JR (1993) Stereoselective metabolism of (S)-(−)-nicotine in humans: formation of trans-(S)-(−)-nicotine N-1′-oxide. Chem Res Toxicol 6(6):880–888. https://doi.org/10.1021/tx00036a019
Park CS, Kang JH, Chung WG et al (2002) Ethnic differences in allelic frequency of two flavin-containing monooxygenase 3 (FMO3) polymorphisms: linkage and effects on in vivo and in vitro FMO activities. Pharmacogenetics 12(1):77–80. https://doi.org/10.1097/00008571-200201000-00011
Parte P, Kupfer D (2005) Oxidation of tamoxifen by human flavin-containing monooxygenase (FMO) 1 and FMO3 to tamoxifen-N-oxide and its novel reduction back to tamoxifen by human cytochromes P450 and hemoglobin. Drug Metab Dispos 33(10):1446–1452. https://doi.org/10.1124/dmd.104.000802
Patil PO, Bari SB, Firke SD, Deshmukh PK, Donda ST, Patil DA (2013) A comprehensive review on synthesis and designing aspects of coumarin derivatives as monoamine oxidase inhibitors for depression and Alzheimer’s disease. Bioorg Med Chem 21(9):2434–2450. https://doi.org/10.1016/j.bmc.2013.02.017
Paudel P, Seong SH, Jung HA, Choi JS (2019) Rubrofusarin as a dual protein tyrosine phosphate 1B and human monoamine oxidase-A inhibitor: an in vitro and in silico study. ACS Omega 4(7):11621–11630. https://doi.org/10.1021/acsomega.9b01433
Peglow S, Toledo AH, Anaya-Prado R, Lopez-Neblina F, Toledo-Pereyra LH (2011) Allopurinol and xanthine oxidase inhibition in liver ischemia reperfusion. J Hepatobiliary Pancreat Sci 18(2):137–146. https://doi.org/10.1007/s00534-010-0328-7
Pelikant-Malecka I, Sielicka A, Kaniewska E, Smolenski RT, Slominska EM (2015) Endothelial toxicity of unusual nucleotide metabolites. Pharmacol Rep 67(4):818–822. https://doi.org/10.1016/j.pharep.2015.03.020
Perez-Paramo YX, Chen G, Ashmore JH et al (2019) Nicotine-N′-oxidation by flavin monooxygenase enzymes. Cancer Epidemiol Biomarkers Prev 28(2):311–320. https://doi.org/10.1158/1055-9965.Epi-18-0669
Peterson LA, Caldera PS, Trevor A, Chiba K, Castagnoli N Jr (1985) Studies on the 1-methyl-4-phenyl-2,3-dihydropyridinium species 2,3-MPDP+, the monoamine oxidase catalyzed oxidation product of the nigrostriatal toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). J Med Chem 28(10):1432–1436. https://doi.org/10.1021/jm00148a010
Pettit FH, Orme-Johnson W, Ziegler DM (1964) The requirement for flavin adenine dinucleotide by a liver microsmal oxygenase catalyzing the oxidation of alkylaryl amines. Biochem Biophys Res Commun 16(5):444–448. https://doi.org/10.1016/0006-291x(64)90373-0
Petzer JP, Petzer A (2015) Caffeine as a lead compound for the design of therapeutic agents for the treatment of Parkinson’s disease. Curr Med Chem 22(8):975–988. https://doi.org/10.2174/0929867322666141215160015
Petzer A, Pienaar A, Petzer JP (2013) The interactions of caffeine with monoamine oxidase. Life Sci 93(7):283–287. https://doi.org/10.1016/j.lfs.2013.06.020
Pey AL, Megarity CF, Timson DJ (2019) NAD(P)H quinone oxidoreductase (NQO1): an enzyme which needs just enough mobility, in just the right places. Biosci Rep. https://doi.org/10.1042/bsr20180459
Pfeffer KD, Huecksteadt TP, Hoidal JR (1994) Xanthine dehydrogenase and xanthine oxidase activity and gene expression in renal epithelial cells. Cytokine and steroid regulation. J Immunol 153(4):1789–1797
Phillips IR, Dolphin CT, Clair P et al (1995) The molecular biology of the flavin-containing monooxygenases of man. Chem-Biol Interact 96(1):17–32. https://doi.org/10.1016/0009-2797(94)03580-2
Phillips IR, Shephard EA (2017) Drug metabolism by flavin-containing monooxygenases of human and mouse. Expert Opin Drug Metab Toxicol 13(2):167–181. https://doi.org/10.1080/17425255.2017.1239718
Phillips IR, Shephard EA (2019) Endogenous roles of mammalian flavin-containing monooxygenases. Catalysts 9(12):1001
Phillips IR, Shephard EA (2020) Flavin-containing monooxygenase 3 (FMO3): genetic variants and their consequences for drug metabolism and disease. Xenobiotica 50(1):19–33. https://doi.org/10.1080/00498254.2019.1643515
Pichard-Garcia L, Weaver RJ, Eckett N et al (2004) The olivacine derivative 16020 (9-hydroxy-5,6-dimethyl-N-[2-(dimethylamino)ethyl)-6H-pyrido(4,3-b)-carbazole-1-carboxamide) induces CYP1A and its own metabolism in human hepatocytes in primary culture. Drug Metab Dispos 32(1):80–88. https://doi.org/10.1124/dmd.32.1.80
Pike MG, Martin YN, Mays DC, Benson LM, Naylor S, Lipsky JJ (1999) Roles of FMO and CYP450 in the metabolism in human liver microsomes of S-methyl-N, N-diethyldithiocarbamate, a disulfiram metabolite. Alcohol Clin Exp Res 23(7):1173–1179
Pike MG, Mays DC, Macomber DW, Lipsky JJ (2001) Metabolism of a disulfiram metabolite, S-methyl N, N-diethyldithiocarbamate, by flavin monooxygenase in human renal microsomes. Drug Metab Dispos 29(2):127–132
Polanski W, Reichmann H, Gille G (2011) Stimulation, protection and regeneration of dopaminergic neurons by 9-methyl-β-carboline: a new anti-Parkinson drug? Expert Rev Neurother 11(6):845–860. https://doi.org/10.1586/ern.11.1
Potega A, Dabrowska E, Niemira M et al (2011) The imidazoacridinone antitumor drug, C-1311, is metabolized by flavin monooxygenases but not by cytochrome P450s. Drug Metab Dispos 39(8):1423–1432. https://doi.org/10.1124/dmd.111.038984
Prakash C, Kamel A, Gummerus J, Wilner K (1997) Metabolism and excretion of a new antipsychotic drug, ziprasidone, in humans. Drug Metab Dispos 25(7):863–872
Prins LH, Petzer JP, Malan SF (2010) Inhibition of monoamine oxidase by indole and benzofuran derivatives. Eur J Med Chem 45(10):4458–4466. https://doi.org/10.1016/j.ejmech.2010.07.005
Prinsloo D, van Dyk S, Petzer A, Petzer JP (2019) Monoamine oxidase inhibition by kavalactones from kava (Piper methysticum). Planta Med 85(14–15):1136–1142. https://doi.org/10.1055/a-1008-9491
Pritsos CA (2000) Cellular distribution, metabolism and regulation of the xanthine oxidoreductase enzyme system. Chem-Biol Interact 129(1–2):195–208. https://doi.org/10.1016/s0009-2797(00)00203-9
Prueksaritanont T, Lu P, Gorham L, Sternfeld F, Vyas KP (2000) Interspecies comparison and role of human cytochrome P450 and flavin-containing monooxygenase in hepatic metabolism of L-775,606, a strong 5-HT(1D) receptor agonist. Xenobiotica 30(1):47–59. https://doi.org/10.1080/004982500237811
Pryde DC, Dalvie D, Hu Q, Jones P, Obach RS, Tran TD (2010) Aldehyde oxidase: an enzyme of emerging importance in drug discovery. J Med Chem 53(24):8441–8460. https://doi.org/10.1021/jm100888d
Qian L, Ortiz de Montellano PR (2006) Oxidative activation of thiacetazone by the Mycobacterium tuberculosis flavin monooxygenase EtaA and human FMO1 and FMO3. Chem Res Toxicol 19(3):443–449. https://doi.org/10.1021/tx050328b
Qiao Y, Maiti K, Sultana Z, Fu L, Smith R (2020) Inhibition of vertebrate aldehyde oxidase as a therapeutic treatment for cancer, obesity, aging and amyotrophic lateral sclerosis. Eur J Med Chem 187:111948. https://doi.org/10.1016/j.ejmech.2019.111948
Rae JM, Johnson MD, Lippman ME, Flockhart DA (2001) Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: studies with cDNA and oligonucleotide expression arrays. J Pharmacol Exp Ther 299(3):849–857
Ramanathan S, Jin F, Sharma S, Kearney BP (2016) Clinical pharmacokinetic and pharmacodynamic profile of idelalisib. Clin Pharmacokinet 55(1):33–45. https://doi.org/10.1007/s40262-015-0304-0
Ramírez J, Kim TW, Liu W et al (2014) A pharmacogenetic study of aldehyde oxidase I in patients treated with XK469. Pharmacogenet Genomics 24(2):129–132. https://doi.org/10.1097/fpc.0000000000000023
Ramsay RR (2012) Monoamine oxidases: the biochemistry of the proteins as targets in medicinal chemistry and drug discovery. Curr Top Med Chem 12(20):2189–2209. https://doi.org/10.2174/156802612805219978
Ramsay RR, Albreht A (2018) Kinetics, mechanism, and inhibition of monoamine oxidase. J Neural Transm (vienna) 125(11):1659–1683. https://doi.org/10.1007/s00702-018-1861-9
Ramsay RR, Hunter DJ (2003) Interactions of D-amphetamine with the active site of monoamine oxidase-A. Inflammopharmacology 11(2):127–133. https://doi.org/10.1163/156856003765764290
Rashid MH, Babu D, Siraki AG (2021) Interactions of the antioxidant enzymes NAD(P)H:quinone oxidoreductase 1 (NQO1) and NRH:quinone oxidoreductase 2 (NQO2) with pharmacological agents, endogenous biochemicals and environmental contaminants. Chem-Biol Interact 345:109574. https://doi.org/10.1016/j.cbi.2021.109574
Rashidi MR, Smith JA, Clarke SE, Beedham C (1997) In vitro oxidation of famciclovir and 6-deoxypenciclovir by aldehyde oxidase from human, guinea pig, rabbit, and rat liver. Drug Metab Dispos 25(7):805–813
Rasmussen BB, Brøsen K (1996) Determination of urinary metabolites of caffeine for the assessment of cytochrome P450 1A2, xanthine oxidase, and N-acetyltransferase activity in humans. Ther Drug Monit 18(3):254–262. https://doi.org/10.1097/00007691-199606000-00006
Rawden HC, Kokwaro GO, Ward SA, Edwards G (2000) Relative contribution of cytochromes P-450 and flavin-containing monoxygenases to the metabolism of albendazole by human liver microsomes. Br J Clin Pharmacol 49(4):313–322. https://doi.org/10.1046/j.1365-2125.2000.00170.x
Rees PJ, Selby P, Prentice HG, Whiteman PD, Grant DM (1986) A515U: a prodrug of acyclovir with increased oral bioavailability. J Antimicrob Chemother 18 Suppl B:215–222. https://doi.org/10.1093/jac/18.supplement_b.215
Rehuman NA, Oh JM, Nath LR et al (2021) Halogenated coumarin-chalcones as multifunctional monoamine oxidase-B and butyrylcholinesterase inhibitors. ACS Omega 6(42):28182–28193. https://doi.org/10.1021/acsomega.1c04252
Reid AA, Hill JL, Murphy DL (1988) Interactions of tricyclic antidepressant drugs with human and rat monoamine oxidase type B. Naunyn Schmiedebergs Arch Pharmacol 338(6):678–683. https://doi.org/10.1007/bf00165634
Reid JM, Walker DL, Miller JK et al (2004) The metabolism of pyrazoloacridine (NSC 366140) by cytochromes P450 and flavin monooxygenase in human liver microsomes. Clin Cancer Res 10(4):1471–1480. https://doi.org/10.1158/1078-0432.ccr-0557-03
Reiter S, Simmonds HA, Zöllner N, Braun SL, Knedel M (1990) Demonstration of a combined deficiency of xanthine oxidase and aldehyde oxidase in xanthinuric patients not forming oxipurinol. Clin Chim Acta 187(3):221–234. https://doi.org/10.1016/0009-8981(90)90107-4
Relling MV, Lin JS, Ayers GD, Evans WE (1992) Racial and gender differences in N-acetyltransferase, xanthine oxidase, and CYP1A2 activities. Clin Pharmacol Ther 52(6):643–658. https://doi.org/10.1038/clpt.1992.203
Rendić S (2002) Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev 34(1–2):83–448. https://doi.org/10.1081/dmr-120001392
Rendić S, Di Carlo FJ (1997) Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev 29(1–2):413–580. https://doi.org/10.3109/03602539709037591
Rendić S, Guengerich FP (2012) Contributions of human enzymes in carcinogen metabolism. Chem Res Toxicol 25(7):1316–1383. https://doi.org/10.1021/tx300132k
Rendić S, Guengerich FP (2015) Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem Res Toxicol 28(1):38–42. https://doi.org/10.1021/tx500444e
Rendić SP, Guengerich FP (2021) Human family 1–4 cytochrome P450 enzymes involved in the metabolic activation of xenobiotic and physiological chemicals: an update. Arch Toxicol 95(2):395–472. https://doi.org/10.1007/s00204-020-02971-4
Rendić S, Sunjić V, Toso R, Kajfez F, Ruf HH (1979) Interaction of cimetidine with liver microsomes. Xenobiotica 9(9):555–564. https://doi.org/10.3109/00498257909042321
Rendić S, Alebić-Kolbah T, Kajfez F, Ruf HH (1982) Interaction of ranitidine with liver microsomes. Xenobiotica 12(1):9–17. https://doi.org/10.3109/00498258209052450
Rendić S, Kajfez F, Ruf HH (1983) Characterization of cimetidine, ranitidine, and related structures’ interaction with cytochrome P-450. Drug Metab Dispos 11(2):137–142
Renwick AB, Ball SE, Tredger JM et al (2002) Inhibition of zaleplon metabolism by cimetidine in the human liver: in vitro studies with subcellular fractions and precision-cut liver slices. Xenobiotica 32(10):849–862. https://doi.org/10.1080/00498250210158221
Rettie AE, Lawton MP, Sadeque AJ, Meier GP, Philpot RM (1994) Prochiral sulfoxidation as a probe for multiple forms of the microsomal flavin-containing monooxygenase: studies with rabbit FMO1, FMO2, FMO3, and FMO5 expressed in Escherichia coli. Arch Biochem Biophys 311(2):369–377. https://doi.org/10.1006/abbi.1994.1250
Ricaurte GA, DeLanney LE, Irwin I, Witkin JM, Katz JL, Langston JW (1989) Evaluation of the neurotoxic potential of N, N-dimethylamphetamine: an illicit analog of methamphetamine. Brain Res 490(2):301–306. https://doi.org/10.1016/0006-8993(89)90247-3
Ring BJ, Catlow J, Lindsay TJ et al (1996) Identification of the human cytochromes P450 responsible for the in vitro formation of the major oxidative metabolites of the antipsychotic agent olanzapine. J Pharmacol Exp Ther 276(2):658–666
Ring BJ, Wrighton SA, Aldridge SL, Hansen K, Haehner B, Shipley LA (1999) Flavin-containing monooxygenase-mediated N-oxidation of the M(1)-muscarinic agonist xanomeline. Drug Metab Dispos 27(10):1099–1103
Ripp SL, Itagaki K, Philpot RM, Elfarra AA (1999a) Methionine S-oxidation in human and rabbit liver microsomes: evidence for a high-affinity methionine S-oxidase activity that is distinct from flavin-containing monooxygenase 3. Arch Biochem Biophys 367(2):322–332. https://doi.org/10.1006/abbi.1999.1247
Ripp SL, Itagaki K, Philpot RM, Elfarra AA (1999b) Species and sex differences in expression of flavin-containing monooxygenase form 3 in liver and kidney microsomes. Drug Metab Dispos 27(1):46–52
Rivera SP, Choi HH, Chapman B et al (2005) Identification of aldehyde oxidase 1 and aldehyde oxidase homologue 1 as dioxin-inducible genes. Toxicology 207(3):401–409. https://doi.org/10.1016/j.tox.2004.10.009
Rivett AJ, Eddy BJ, Roth JA (1982) Contribution of sulfate conjugation, deamination, and O-methylation to metabolism of dopamine and norepinephrine in human brain. J Neurochem 39(4):1009–1016. https://doi.org/10.1111/j.1471-4159.1982.tb11490.x
Rochat B, Amey M, Van Gelderen H, Testa B, Baumann P (1995) Determination of the enantiomers of citalopram, its demethylated and propionic acid metabolites in human plasma by chiral HPLC. Chirality 7(6):389–395. https://doi.org/10.1002/chir.530070602
Rochat B, Kosel M, Boss G, Testa B, Gillet M, Baumann P (1998) Stereoselective biotransformation of the selective serotonin reuptake inhibitor citalopram and its demethylated metabolites by monoamine oxidases in human liver. Biochem Pharmacol 56(1):15–23. https://doi.org/10.1016/s0006-2952(98)00008-2
Rodrigues AD (1994) Comparison of levels of aldehyde oxidase with cytochrome P450 activities in human liver in vitro. Biochem Pharmacol 48(1):197–200. https://doi.org/10.1016/0006-2952(94)90240-2
Rodriguez RJ, Miranda CL (2000) Isoform specificity of N-deacetyl ketoconazole by human and rabbit flavin-containing monooxygenases. Drug Metab Dispos 28(9):1083–1086
Rodrigues AD, Ferrero JL, Amann MT et al (1994) The in vitro hepatic metabolism of ABT-418, a cholinergic channel activator, in rats, dogs, cynomolgus monkeys, and humans. Drug Metab Dispos 22(5):788–798
Rodrigues AD, Kukulka MJ, Ferrero JL, Cashman JR (1995) In vitro hepatic metabolism of ABT-418 in chimpanzee (Pan troglodytes). A unique pattern of microsomal flavin-containing monooxygenase-dependent stereoselective N′-oxidation. Drug Metab Dispos 23(10):1143–1152
Rooseboom M, Commandeur JN, Floor GC, Rettie AE, Vermeulen NP (2001) Selenoxidation by flavin-containing monooxygenases as a novel pathway for b-elimination of selenocysteine Se-conjugates. Chem Res Toxicol 14(1):127–134. https://doi.org/10.1021/tx0001326
Rooseboom M, Commandeur JN, Vermeulen NP (2004) Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev 56(1):53–102. https://doi.org/10.1124/pr.56.1.3
Roy SK, Korzekwa KR, Gonzalez FJ, Moschel RC, Dolan ME (1995) Human liver oxidative metabolism of O6-benzylguanine. Biochem Pharmacol 50(9):1385–1389
Rumyantseva GV, Kennedy CH, Mason RP (1991) Trace transition metal-catalyzed reactions in the microsomal metabolism of alkyl hydrazines to carbon-centered free radicals. J Biol Chem 266(32):21422–21427
Rundles RW (1966) Effects of allopurinol on 6-mercaptopurine therapy in neoplastic diseases. Ann Rheum Dis 25(6 Suppl):655–656. https://doi.org/10.1136/ard.25.Suppl_6.655
Ryu SD, Yi HG, Cha YN et al (2004) Flavin-containing monooxygenase activity can be inhibited by nitric oxide-mediated S-nitrosylation. Life Sci 75(21):2559–2572. https://doi.org/10.1016/j.lfs.2004.05.018
Sağlık BN, Osmaniye D, Acar Çevik U et al (2020) Synthesis, in vitro enzyme activity and molecular docking studies of new benzylamine-sulfonamide derivatives as selective MAO-B inhibitors. J Enzyme Inhib Med Chem 35(1):1422–1432. https://doi.org/10.1080/14756366.2020.1784892
Sahi J, Khan KK, Black CB (2008) Aldehyde oxidase activity and inhibition in hepatocytes and cytosolic fractions from mouse, rat, monkey and human. Drug Metab Lett 2(3):176–183. https://doi.org/10.2174/187231208785425818
Saidemberg DM, Ferreira MA, Takahashi TN et al (2009) Monoamine oxidase inhibitory activities of indolylalkaloid toxins from the venom of the colonial spider Parawixia bistriata: functional characterization of PwTX-I. Toxicon 54(6):717–724. https://doi.org/10.1016/j.toxicon.2009.05.027
Salva M, Jansat JM, Martinez-Tobed A, Palacios JM (2003) Identification of the human liver enzymes involved in the metabolism of the antimigraine agent almotriptan. Drug Metab Dispos 31(4):404–411. https://doi.org/10.1124/dmd.31.4.404
Sanders SA, Eisenthal R, Harrison R (1997) NADH oxidase activity of human xanthine oxidoreductase–generation of superoxide anion. Eur J Biochem 245(3):541–548. https://doi.org/10.1111/j.1432-1033.1997.00541.x
Sanoh S, Nozaki K, Murai H, Terashita S, Teramura T, Ohta S (2012) Prediction of human metabolism of FK3453 by aldehyde oxidase using chimeric mice transplanted with human or rat hepatocytes. Drug Metab Dispos 40(1):76–82. https://doi.org/10.1124/dmd.111.041954
Santillo MF (2014) Inhibition of monoamine oxidase (MAO) by α-ethylphenethylamine and N, α-diethylphenethylamine, two compounds related to dietary supplements. Food Chem Toxicol 74:265–269. https://doi.org/10.1016/j.fct.2014.10.009
Sanz E, Quintana A, Battaglia V et al (2008) Anti-apoptotic effect of MAO-B inhibitor PF9601N [N-(2-propynyl)-2-(5-benzyloxy-indolyl) methylamine] is mediated by p53 pathway inhibition in MPP+-treated SH-SY5Y human dopaminergic cells. J Neurochem 105(6):2404–2417. https://doi.org/10.1111/j.1471-4159.2008.05326.x
Sawada H, Yokosawa H (1991) [Physiological roles of proteases in fertilization and development]. Tanpakushitsu Kakusan Koso 36(5):814–819 [Japanese]
Schlenk D, Cashman JR, Yeung C, Zhang X, Rettie AE (2002) Role of human flavin-containing monooxygenases in the sulfoxidation of [14C]aldicarb. Pesticide Biochem Physiol 73(2):67–73. https://doi.org/10.1016/S0048-3575(02)00013-5
Schneider J, Girreser U, Havemeyer A, Bittner F, Clement B (2018) Detoxification of trimethylamine N-oxide by the mitochondrial amidoxime reducing component mARC. Chem Res Toxicol 31(6):447–453. https://doi.org/10.1021/acs.chemrestox.7b00329
Schofield PC, Robertson IG, Paxton JW (2000) Inter-species variation in the metabolism and inhibition of N-[(2′-dimethylamino)ethyl]acridine-4-carboxamide (DACA) by aldehyde oxidase. Biochem Pharmacol 59(2):161–165. https://doi.org/10.1016/s0006-2952(99)00323-8
Schulz-Utermoehl T, Spear M, Pollard CR et al (2010) In vitro hepatic metabolism of cediranib, a strong vascular endothelial growth factor tyrosine kinase inhibitor: interspecies comparison and human enzymology. Drug Metab Dispos 38(10):1688–1697. https://doi.org/10.1124/dmd.110.033159
Scrutton NS (2004) Chemical aspects of amine oxidation by flavoprotein enzymes. Nat Prod Rep 21(6):722–730. https://doi.org/10.1039/b306788m
Secci D, Carradori S, Bolasco A et al (2011) Synthesis and selective human monoamine oxidase inhibition of 3-carbonyl, 3-acyl, and 3-carboxyhydrazido coumarin derivatives. Eur J Med Chem 46(10):4846–4852. https://doi.org/10.1016/j.ejmech.2011.07.017
Serra S, Ferino G, Matos MJ et al (2012) Hydroxycoumarins as selective MAO-B inhibitors. Bioorg Med Chem Lett 22(1):258–261. https://doi.org/10.1016/j.bmcl.2011.11.020
Seto Y, Guengerich FP (1993) Partitioning between N-dealkylation and N-oxygenation in the oxidation of N,N-dialkylarylamines catalyzed by cytochrome P450 2B1. J Biol Chem 268(14):9986–9997
Shaffer CL, Gunduz M, Scialis RJ, Fang AF (2007) Metabolism and disposition of a selective alpha(7) nicotinic acetylcholine receptor agonist in humans. Drug Metab Dispos 35(7):1188–1195. https://doi.org/10.1124/dmd.106.014449
Shaik AN, LeDuc BW, Khan AA (2017) Characterization of 1-aminobenzotriazole and ketoconazole as novel inhibitors of monoamine oxidase (MAO): an in vitro investigation. Eur J Drug Metab Pharmacokinet 42(5):827–834. https://doi.org/10.1007/s13318-017-0401-6
Sharma R, Eng H, Walker GS et al (2011) Oxidative metabolism of a quinoxaline derivative by xanthine oxidase in rodent plasma. Chem Res Toxicol 24(12):2207–2216. https://doi.org/10.1021/tx200329k
Sharma R, Strelevitz TJ, Gao H, et al. (2012) Deuterium isotope effects on drug pharmacokinetics. I. System-dependent effects of specific deuteration with aldehyde oxidase cleared drugs. Drug Metab Dispos 40(3):625–634. https://doi.org/10.1124/dmd.111.042770
Shen HW, Jiang XL, Winter JC, Yu AM (2010a) Psychedelic 5-methoxy-N, N-dimethyltryptamine: metabolism, pharmacokinetics, drug interactions, and pharmacological actions. Curr Drug Metab 11(8):659–666. https://doi.org/10.2174/138920010794233495
Shen HW, Wu C, Jiang XL, Yu AM (2010b) Effects of monoamine oxidase inhibitor and cytochrome P450 2D6 status on 5-methoxy-N,N-dimethyltryptamine metabolism and pharmacokinetics. Biochem Pharmacol 80(1):122–128. https://doi.org/10.1016/j.bcp.2010.02.020
Shi X, Dick RA, Ford KA, Casida JE (2009) Enzymes and inhibitors in neonicotinoid insecticide metabolism. J Agric Food Chem 57(11):4861–4866. https://doi.org/10.1021/jf900250f
Shibutani Y, Ueo T, Yamamoto T, Takahashi S, Moriwaki Y, Higashino K (1999) A case of classical xanthinuria (type 1) with diabetes mellitus and Hashimoto’s thyroiditis. Clin Chim Acta 285(1–2):18318–18319. https://doi.org/10.1016/s0009-8981(99)00070-4
Shih VE, Abroms IF, Johnson JL, et al. (1977) Sulfite oxidase deficiency. Biochemical and clinical investigations of a hereditary metabolic disorder in sulfur metabolism. N Engl J Med 297(19):1022–1028. https://doi.org/10.1056/nejm197711102971902
Shih JC, Chen K, Ridd MJ (1999) Monoamine oxidase: from genes to behavior. Annu Rev Neurosci 22:197–217. https://doi.org/10.1146/annurev.neuro.22.1.197
Shih JC, Grimsby J, Chen K (1990) The expression of human MAO-A and B genes. J Neural Transm Suppl 32:41–47. https://doi.org/10.1007/978-3-7091-9113-2_4
Shih TY, Pai CY, Yang P, Chang WL, Wang NC, Hu OY (2013) A novel mechanism underlies the hepatotoxicity of pyrazinamide. Antimicrob Agents Chemother 57(4):1685–1690. https://doi.org/10.1128/aac.01866-12
Shilliday FB, Walker DP, Gu C et al (2010) Multiple species metabolism of PHA-568487, a selective alpha 7 nicotinic acetylcholine receptor agonist. Drug Metab Lett 4(3):162–172
Shimizu M, Yano H, Nagashima S et al (2007) Effect of genetic variants of the human flavin-containing monooxygenase 3 on N- and S-oxygenation activities. Drug Metab Dispos 35(3):328–330. https://doi.org/10.1124/dmd.106.013094
Shimizu M, Denton T, Kozono M, Cashman JR, Leeder JS, Yamazaki H (2011) Developmental variations in metabolic capacity of flavin-containing mono-oxygenase 3 in childhood. Br J Clin Pharmacol 71(4):585–591. https://doi.org/10.1111/j.1365-2125.2010.03876.x
Shimizu M, Kobayashi Y, Hayashi S, Aoki Y, Yamazaki H (2012) Variants in the flavin-containing monooxygenase 3 (FMO3) gene responsible for trimethylaminuria in a Japanese population. Mol Genet Metab 107(3):330–334. https://doi.org/10.1016/j.ymgme.2012.06.014
Shimizu M, Allerston CK, Shephard EA, Yamazaki H, Phillips IR (2014) Relationships between flavin-containing mono-oxygenase 3 (FMO3) genotype and trimethylaminuria phenotype in a Japanese population. Br J Clin Pharmacol 77(5):839–851. https://doi.org/10.1111/bcp.12240
Shimizu M, Shiraishi A, Sato A, Nagashima S, Yamazaki H (2015) Potential for drug interactions mediated by polymorphic flavin-containing monooxygenase 3 in human livers. Drug Metab Pharmacokinet 30(1):70–74. https://doi.org/10.1016/j.dmpk.2014.09.008
Shimizu M, Yoda H, Igarashi N, Makino M, Tokuyama E, Yamazaki H (2019) Novel variants and haplotypes of human flavin-containing monooxygenase 3 gene associated with Japanese subjects suffering from trimethylaminuria. Xenobiotica 49(10):1244–1250. https://doi.org/10.1080/00498254.2018.1539279
Shulman KI, Herrmann N, Walker SE (2013) Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs 27(10):789–797. https://doi.org/10.1007/s40263-013-0097-3
Siddens LK, Krueger SK, Henderson MC, Williams DE (2014) Mammalian flavin-containing monooxygenase (FMO) as a source of hydrogen peroxide. Biochem Pharmacol 89(1):141–147. https://doi.org/10.1016/j.bcp.2014.02.006
Siegel D, Gustafson DL, Dehn DL et al (2004) NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger. Mol Pharmacol 65(5):1238–1247. https://doi.org/10.1124/mol.65.5.1238
Singer TP, Salach JI, Crabtree D (1985) Reversible inhibition and mechanism-based irreversible inactivation of monoamine oxidases by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Biochem Biophys Res Commun 127(2):707–712. https://doi.org/10.1016/s0006-291x(85)80219-9
Singer TP, Salach JI, Castagnoli N Jr, Trevor A (1986) Interactions of the neurotoxic amine 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine with monoamine oxidases. Biochem J 235(3):785–789. https://doi.org/10.1042/bj2350785
Singer TP, Ramsay RR, McKeown K, Trevor A, Castagnoli NE Jr (1988) Mechanism of the neurotoxicity of 1-methyl-4-phenylpyridinium (MPP+), the toxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 49(1):17–23. https://doi.org/10.1016/0300-483x(88)90169-2
Singla RK, Scotti L, Dubey AK (2017) In silico studies revealed multiple neurological targets for the antidepressant molecule ursolic acid. Curr Neuropharmacol 15(8):1100–1106. https://doi.org/10.2174/1570159x14666161229115508
Smith PB, Crespi C (2002) Thiourea toxicity in mouse C3H/10T1/2 cells expressing human flavin-dependent monooxygenase 3. Biochem Pharmacol 63(11):1941–1948. https://doi.org/10.1016/s0006-2952(02)00978-4
Söderberg MM, Haslemo T, Molden E, Dahl ML (2013) Influence of FMO1 and 3 polymorphisms on serum olanzapine and its N-oxide metabolite in psychiatric patients. Pharmacogenomics J 13(6):544–550. https://doi.org/10.1038/tpj.2012.47
Sodhi JK, Wong S, Kirkpatrick DS et al (2015) A novel reaction mediated by human aldehyde oxidase: amide hydrolysis of GDC-0834. Drug Metab Dispos 43(6):908–915. https://doi.org/10.1124/dmd.114.061804
Sozio P, Cerasa LS, Abbadessa A, Di Stefano A (2012) Designing prodrugs for the treatment of Parkinson’s disease. Expert Opin Drug Discov 7(5):385–406. https://doi.org/10.1517/17460441.2012.677025
Spector T (1988) Oxypurinol as an inhibitor of xanthine oxidase-catalyzed production of superoxide radical. Biochem Pharmacol 37(2):349–352. https://doi.org/10.1016/0006-2952(88)90739-3
Spector T, Hall WW, Krenitsky TA (1986) Human and bovine xanthine oxidases. Inhibition studies with oxipurinol. Biochem Pharmacol 35(18):3109–3114. https://doi.org/10.1016/0006-2952(86)90394-1
Spector T, Hall WW, Porter DJ, Lambe CU, Nelson DJ, Krenitsky TA (1989) Inhibition of xanthine oxidase by 4-hydroxy-6-mercaptopyrazolo[3,4-d]pyrimidine. Biochem Pharmacol 38(23):4315–4320. https://doi.org/10.1016/0006-2952(89)90531-5
Stahl SM (1998) Basic psychopharmacology of antidepressants, part 1: antidepressants have seven distinct mechanisms of action. J Clin Psychiatry 59(Suppl 4):5–14
Steel D, Bovill EG, Golden E, Tindle BH (1988) Hereditary hemorrhagic telangiectasia. A family study. Am J Clin Pathol 90(3):274–278. https://doi.org/10.1093/ajcp/90.3.274
Sternieri E, Coccia CP, Pinetti D, Ferrari A (2006) Pharmacokinetics and interactions of headache medications, part I: introduction, pharmacokinetics, metabolism and acute treatments. Expert Opin Drug Metab Toxicol 2(6):961–979. https://doi.org/10.1517/17425255.2.6.961
Stirpe F, Della Corte E (1969) The regulation of rat liver xanthine oxidase. Conversion in vitro of the enzyme activity from dehydrogenase (type D) to oxidase (type O). J Biol Chem 244(14):3855–3863
Störmer E, Roots I, Brockmöller J (2000) Benzydamine N-oxidation as an index reaction reflecting FMO activity in human liver microsomes and impact of FMO3 polymorphisms on enzyme activity. Br J Clin Pharmacol 50(6):553–561. https://doi.org/10.1046/j.1365-2125.2000.00296.x
Strelevitz TJ, Orozco CC, Obach RS (2012) Hydralazine as a selective probe inactivator of aldehyde oxidase in human hepatocytes: estimation of the contribution of aldehyde oxidase to metabolic clearance. Drug Metab Dispos 40(7):1441–1448. https://doi.org/10.1124/dmd.112.045195
Strolin Benedetti M, Tipton KF, Whomsley R (2007) Amine oxidases and monooxygenases in the in vivo metabolism of xenobiotic amines in humans: has the involvement of amine oxidases been neglected? Fundam Clin Pharmacol 21(5):467–480. https://doi.org/10.1111/j.1472-8206.2007.00498.x
Subash S, Gogtay NJ, Iyer KR, Gandhe P, Budania R, Thatte UM (2021) Evaluation of vanillin as a probe drug for aldehyde oxidase and phenotyping for its activity in a Western Indian Cohort. Indian J Pharmacol 53(3):213–220. https://doi.org/10.4103/ijp.IJP_463_18
Suchting R, Tirumalajaru V, Gareeb R et al (2021) Revisiting monoamine oxidase inhibitors for the treatment of depressive disorders: a systematic review and network meta-analysis. J Affect Disord 282:1153–1160. https://doi.org/10.1016/j.jad.2021.01.021
Sugihara K, Kitamura S, Tatsumi K, Asahara T, Dohi K (1997) Differences in aldehyde oxidase activity in cytosolic preparations of human and monkey liver. Biochem Mol Biol Int 41(6):1153–1160. https://doi.org/10.1080/15216549700202241
Sugihara K, Kitamura S, Yamada T et al (2001) Aryl hydrocarbon receptor (AhR)-mediated induction of xanthine oxidase/xanthine dehydrogenase activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem Biophys Res Commun 281(5):1093–1099. https://doi.org/10.1006/bbrc.2001.4464
Sullivan JL, Dackis C, Stanfield C (1977) In vivo inhibition of platelet MAO activity by tricyclic antidepressants. Am J Psychiatry 134(2):188–190. https://doi.org/10.1176/ajp.134.2.188
Sun H, Ehlhardt WJ, Kulanthaivel P, Lanza DL, Reilly CA, Yost GS (2007) Dehydrogenation of indoline by cytochrome P450 enzymes: a novel "aromatase" process. J Pharmacol Exp Ther 322(2):843–851 (0022–3565). https://doi.org/10.1124/jpet.107.121723
Sung JW, Yun HY, Park S et al (2020) Population pharmacokinetics of sulindac and genetic polymorphisms of FMO3 and AOX1 in women with preterm labor. Pharm Res 37(3):44. https://doi.org/10.1007/s11095-020-2765-6
Surapaneni S, Yerramilli U, Bai A et al (2021) Absorption, metabolism, and excretion, in vitro pharmacology, and clinical pharmacokinetics of ozanimod, a novel sphingosine 1-phosphate receptor modulator. Drug Metab Dispos 49(5):405–419. https://doi.org/10.1124/dmd.120.000220
Suzuki O, Katsumata Y, Oya M (1981) Oxidation of b-phenylethylamine by both types of monoamine oxidase: examination of enzymes in brain and liver mitochondria of eight species. J Neurochem 36(3):1298–1301. https://doi.org/10.1111/j.1471-4159.1981.tb01734.x
Szöko E, Tábi T, Borbás T, Dalmadi B, Tihanyi K, Magyar K (2004) Assessment of the N-oxidation of deprenyl, methamphetamine, and amphetamine enantiomers by chiral capillary electrophoresis: an in vitro metabolism study. Electrophoresis 25(16):2866–2875. https://doi.org/10.1002/elps.200406023
Szökő É, Tábi T, Riederer P, Vécsei L, Magyar K (2018) Pharmacological aspects of the neuroprotective effects of irreversible MAO-B inhibitors, selegiline and rasagiline, in Parkinson’s disease. J Neural Transm (vienna) 125(11):1735–1749. https://doi.org/10.1007/s00702-018-1853-9
Szutowicz A, Tomaszewicz M, Orsulak PJ (1989) Modification of substrate-inhibitor affinities of human platelet monoamine oxidase B in vitro. J Biol Chem 264(30):17660–17664
Takamidoh H, Naoi M, Nagatsu T (1987) Inhibition of type A monoamine oxidase by 1-methyl-4-phenylpyridine. Neurosci Lett 73(3):293–297. https://doi.org/10.1016/0304-3940(87)90261-8
Takeuchi K, Yokouchi C, Goto H, Umehara K, Yamada H, Ishii Y (2018) Alleviation of fatty liver in a rat model by enhancing N1-methylnicotinamide bioavailability through aldehyde oxidase inhibition. Biochem Biophys Res Commun 507(1–4):203–210. https://doi.org/10.1016/j.bbrc.2018.11.008
Tan S, Radi R, Gaudier F et al (1993) Physiologic levels of uric acid inhibit xanthine oxidase in human plasma. Pediatr Res 34(3):303–307. https://doi.org/10.1203/00006450-199309000-00013
Tan WK, Tan ARY, Sivanandam P et al (2020) In vitro inhibition of human aldehyde oxidase activity by clinically relevant concentrations of gefitinib and erlotinib: comparison with select metabolites, molecular docking analysis, and impact on hepatic metabolism of zaleplon and methotrexate. J Pharmacol Exp Ther 374(2):295–307. https://doi.org/10.1124/jpet.120.265249
Tan X, Cai D, Chen N et al (2021) Methamphetamine mediates apoptosis of vascular smooth muscle cells via the chop-related endoplasmic reticulum stress pathway. Toxicol Lett 350:98–110. https://doi.org/10.1016/j.toxlet.2021.06.019
Taniguchi-Takizawa T, Shimizu M, Kume T, Yamazaki H (2015) Benzydamine N-oxygenation as an index for flavin-containing monooxygenase activity and benzydamine N-demethylation by cytochrome P450 enzymes in liver microsomes from rats, dogs, monkeys, and humans. Drug Metab Pharmacokinet 30(1):64–69. https://doi.org/10.1016/j.dmpk.2014.09.006
Taniguchi-Takizawa T, Kato H, Shimizu M, Yamazaki H (2021) Predicted contributions of flavin-containing monooxygenases to the N-oxygenation of drug candidates based on their estimated base dissociation constants. Curr Drug Metab 22(3):208–214. https://doi.org/10.2174/1389200221666201207195758
Tanoue C, Sugihara K, Tayama Y et al (2017) Variability of zaleplon 5-oxidase activity in mice and humans, and inhibition by raloxifene. Drug Metab Lett 10(4):278–285. https://doi.org/10.2174/1872312810666161227145358
Tao G, Irie Y, Li DJ, Keung WM (2005) Eugenol and its structural analogs inhibit monoamine oxidase A and exhibit antidepressant-like activity. Bioorg Med Chem 13(15):4777–4788. https://doi.org/10.1016/j.bmc.2005.04.081
Tayama Y, Miyake K, Sugihara K et al (2007) Developmental changes of aldehyde oxidase activity in young Japanese children. Clin Pharmacol Ther 81(4):567–572. https://doi.org/10.1038/sj.clpt.6100078
Tayama Y, Sugihara K, Sanoh S et al (2011) Effect of tea beverages on aldehyde oxidase activity. Drug Metab Pharmacokinet 26(1):94–101. https://doi.org/10.2133/dmpk.dmpk-10-nt-078
Tayama Y, Sugihara K, Sanoh S, Miyake K, Kitamura S, Ohta S (2012) Developmental changes of aldehyde oxidase activity and protein expression in human liver cytosol. Drug Metab Pharmacokinet 27(5):543–547. https://doi.org/10.2133/dmpk.dmpk-11-nt-124
Teffera Y, Liu J, Krolikowski P, Zhao Z (2021) The role of aldehyde oxidase in the metabolic clearance of substituted benzothiazoles. Drug Metab Lett 14(2):126–136. https://doi.org/10.2174/1872312814666210405101419
Teitelbaum AM, Murphy SE, Akk G et al (2018) Nicotine dependence is associated with functional variation in FMO3, an enzyme that metabolizes nicotine in the brain. Pharmacogenomics J 18(1):136–143. https://doi.org/10.1038/tpj.2016.92
Terao M, Romao MJ, Leimkühler S et al (2016) Structure and function of mammalian aldehyde oxidases. Arch Toxicol 90(4):753–780. https://doi.org/10.1007/s00204-016-1683-1
Terao M, Garattini E, Romão MJ, Leimkühler S (2020) Evolution, expression, and substrate specificities of aldehyde oxidase enzymes in eukaryotes. J Biol Chem 295(16):5377–5389. https://doi.org/10.1074/jbc.REV119.007741
Teufel R, Stull F, Meehan MJ et al (2015) Biochemical establishment and characterization of EncM’s flavin-N5-oxide cofactor. J Am Chem Soc 137(25):8078–8085. https://doi.org/10.1021/jacs.5b03983
Theobald DS, Maurer HH (2007) Identification of monoamine oxidase and cytochrome P450 isoenzymes involved in the deamination of phenethylamine-derived designer drugs (2C-series). Biochem Pharmacol 73(2):287–297. https://doi.org/10.1016/j.bcp.2006.09.022
Tran JQ, Zhang P, Walker S et al (2020) Multiple-dose pharmacokinetics of ozanimod and its major active metabolites and the pharmacodynamic and pharmacokinetic interactions with pseudoephedrine, a sympathomimetic agent, in healthy subjects. Adv Ther 37(12):4944–4958. https://doi.org/10.1007/s12325-020-01500-0
Trevor AJ, Castagnoli N Jr, Caldera P, Ramsay RR, Singer TP (1987a) Bioactivation of MPTP: reactive metabolites and possible biochemical sequelae. Life Sci 40(8):713–719. https://doi.org/10.1016/0024-3205(87)90298-0
Trevor AJ, Singer TP, Ramsay RR, Castagnoli N Jr (1987b) Processing of MPTP by monoamine oxidases: implications for molecular toxicology. J Neural Transm Suppl 23:73–89. https://doi.org/10.1007/978-3-7091-8901-6_5
Trevor AJ, Castagnoli N, Singer TP (1988) The formation of reactive intermediates in the MAO-catalyzed oxidation of the nigrostriatal toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 49(2–3):513–519. https://doi.org/10.1016/0300-483x(88)90037-6
Truman P, Stanfill S, Heydari A, Silver E, Fowles J (2019) Monoamine oxidase inhibitory activity of flavoured e-cigarette liquids. Neurotoxicology 75:123–128. https://doi.org/10.1016/j.neuro.2019.09.010
Tugnait M, Hawes EM, McKay G, Rettie AE, Haining RL, Midha KK (1997) N-Oxygenation of clozapine by flavin-containing monooxygenase. Drug Metab Dispos 25(4):524–527
Tugnait M, Hawes EM, McKay G, Eichelbaum M, Midha KK (1999) Characterization of the human hepatic cytochromes P450 involved in the in vitro oxidation of clozapine. Chem-Biol Interact 118(2):171–189. https://doi.org/10.1016/s0009-2797(99)00006-x
Uebelhack R, Franke L, Schewe HJ (1998) Inhibition of platelet MAO-B by kava pyrone-enriched extract from Piper methysticum Forster (kava-kava). Pharmacopsychiatry 31(5):187–192. https://doi.org/10.1055/s-2007-979325
Uehara S, Uno Y, Inoue T et al (2015) Activation and deactivation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by cytochrome P450 enzymes and flavin-containing monooxygenases in common marmosets (Callithrix jacchus). Drug Metab Dispos 43(5):735–742. https://doi.org/10.1124/dmd.115.063594
Uehara S, Yoneda N, Higuchi Y, Yamazaki H, Suemizu H (2020) Human aldehyde oxidase 1-mediated carbazeran oxidation in chimeric TK-NOG mice transplanted with human hepatocytes. Drug Metab Dispos 48(7):580–586. https://doi.org/10.1124/dmd.120.091090
Uehara S, Yoneda N, Higuchi Y, Yamazaki H, Suemizu H (2021) Methyl-hydroxylation and subsequent oxidation to produce carboxylic acid is the major metabolic pathway of tolbutamide in chimeric TK-NOG mice transplanted with human hepatocytes. Xenobiotica 51(5):582–589. https://doi.org/10.1080/00498254.2021.1875515
Unzeta M, Sanz E (2011) Novel MAO-B inhibitors: Potential therapeutic use of the selective MAO-B inhibitor PF9601N in Parkinson’s disease. Int Rev Neurobiol 100:217–236. https://doi.org/10.1016/b978-0-12-386467-3.00011-x
Urbain A, Marston A, Grilo LS, et al. (2008) Xanthones from Gentianella amarella ssp. acuta with acetylcholinesterase and monoamine oxidase inhibitory activities. J Nat Prod 71(5):895–897 doi:https://doi.org/10.1021/np070690l
Usmani KA, Karoly ED, Hodgson E, Rose RL (2004) In vitro sulfoxidation of thioether compounds by human cytochrome P450 and flavin-containing monooxygenase isoforms with particular reference to the CYP2C subfamily. Drug Metab Dispos 32(3):333–339. https://doi.org/10.1124/dmd/32.3.333(0090-9556)
Valerio LG Jr, Kepa JK, Pickwell GV, Quattrochi LC (2001) Induction of human NAD(P)H:quinone oxidoreductase (NQO1) gene expression by the flavonol quercetin. Toxicol Lett 119(1):49–57. https://doi.org/10.1016/s0378-4274(00)00302-7
van Diermen D, Marston A, Bravo J, Reist M, Carrupt PA, Hostettmann K (2009) Monoamine oxidase inhibition by Rhodiola rosea L. roots. J Ethnopharmacol 122(2):397–401. https://doi.org/10.1016/j.jep.2009.01.007
Van Haarst AD, Van Gerven JM, Cohen AF et al (1999) The effects of moclobemide on the pharmacokinetics of the 5-HT1B/1D agonist rizatriptan in healthy volunteers. Br J Clin Pharmacol 48(2):190–196. https://doi.org/10.1046/j.1365-2125.1999.00011.x
van Muiswinkel FL, Riemers FM, Peters GJ et al (2000) L-Dopa stimulates expression of the antioxidant enzyme NAD(P)H:quinone oxidoreductase (NQO) in cultured astroglial cells. Free Radic Biol Med 29(5):442–453. https://doi.org/10.1016/s0891-5849(00)00328-2
Van Scoik KG, Johnson CA, Porter WR (1985) The pharmacology and metabolism of the thiopurine drugs 6-mercaptopurine and azathioprine. Drug Metab Rev 16(1–2):157–174. https://doi.org/10.3109/03602538508991433
Veldman A, Santamaria-Araujo JA, Sollazzo S et al (2010) Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics 125(5):e1249-1254. https://doi.org/10.1542/peds.2009-2192
Vickers S, Schiller HJ, Hildreth JE, Bulkley GB (1998) Immunoaffinity localization of the enzyme xanthine oxidase on the outside surface of the endothelial cell plasma membrane. Surgery 124(3):551–560
Vickneson K, George J (2021) Xanthine oxidoreductase inhibitors. Handb Exp Pharmacol 264:205–228. https://doi.org/10.1007/164_2020_383
Vyas PM, Roychowdhury S, Koukouritaki SB et al (2006) Enzyme-mediated protein haptenation of dapsone and sulfamethoxazole in human keratinocytes: II. Expression and role of flavin-containing monooxygenases and peroxidases. J Pharmacol Exp Ther 319(1):497–505. https://doi.org/10.1124/jpet.106.105874
Wagmann L, Brandt SD, Kavanagh PV, Maurer HH, Meyer MR (2017) In vitro monoamine oxidase inhibition potential of α-methyltryptamine analog new psychoactive substances for assessing possible toxic risks. Toxicol Lett 272:84–93. https://doi.org/10.1016/j.toxlet.2017.03.007
Walsh C (1979) Enzymatic reaction mechanisms. W. H. Freeman Co., San Francisco
Walsh CT, Chen YCJ (1988) Enzymic Baeyer-Villiger oxidations by flavin-dependent monooxygenases. Angew Chem Int Ed 27(3):333–343. https://doi.org/10.1002/anie.198803331
Wang JS, Zhu HJ, Markowitz JS, Donovan JL, DeVane CL (2006) Evaluation of antipsychotic drugs as inhibitors of multidrug resistance transporter P-glycoprotein. Psychopharmacology 187(4):415–423. https://doi.org/10.1007/s00213-006-0437-9
Wang L, Christopher LJ, Cui D et al (2008) Identification of the human enzymes involved in the oxidative metabolism of dasatinib: an effective approach for determining metabolite formation kinetics. Drug Metab Dispos 36(9):1828–1839. https://doi.org/10.1124/dmd.107.020255
Wang X, Zhao J, Wen T, Liao X, Luo B (2021) Predictive value of FMO3 variants on plasma disposition and adverse reactions of oral voriconazole in febrile neutropenia. Pharmacology 106(3–4):202–210. https://doi.org/10.1159/000510327
Washio T, Arisawa H, Kohsaka K, Yasuda H (2001) Identification of human drug-metabolizing enzymes involved in the metabolism of SNI-2011. Biol Pharm Bull 24(11):1263–1266. https://doi.org/10.1248/bpb.24.1263
Watts RW, Watts JE, Seegmiller JE (1965) Xanthine oxidase activity in human tissues and its inhibition by allopurinol (4-hydroxypyrazolo[3,4-d] pyrimidine). J Lab Clin Med 66(4):688–697
Weidert ER, Schoenborn SO, Cantu-Medellin N, Choughule KV, Jones JP, Kelley EE (2014) Inhibition of xanthine oxidase by the aldehyde oxidase inhibitor raloxifene: implications for identifying molybdopterin nitrite reductases. Nitric Oxide 37:41–45. https://doi.org/10.1016/j.niox.2013.12.010
Weigert J, Neumeier M, Bauer S et al (2008) Small-interference RNA-mediated knock-down of aldehyde oxidase 1 in 3T3-L1 cells impairs adipogenesis and adiponectin release. FEBS Lett 582(19):2965–2972. https://doi.org/10.1016/j.febslet.2008.07.034
Weinreb O, Amit T, Bar-Am O, Youdim MB (2012) Ladostigil: A novel multimodal neuroprotective drug with cholinesterase and brain-selective monoamine oxidase inhibitory activities for Alzheimer’s disease treatment. Curr Drug Targets 13(4):483–494. https://doi.org/10.2174/138945012799499794
Weinstock M, Luques L, Bejar C, Shoham S (2006) Ladostigil, a novel multifunctional drug for the treatment of dementia co-morbid with depression. J Neural Transm Suppl(70):443–446. https://doi.org/10.1007/978-3-211-45295-0_67
Wellaway CR, Baldwin IR, Bamborough P et al (2022) Investigation of Janus kinase (JAK) inhibitors for lung delivery and the importance of aldehyde oxidase metabolism. J Med Chem 65(1):633–664. https://doi.org/10.1021/acs.jmedchem.1c01765
Whitehouse LW, Lodge BA, By AW, Thomas BH (1987) Metabolic disposition of pyrazinamide in the rat: identification of a novel in vivo metabolite common to both rat and human. Biopharm Drug Dispos 8(4):307–318. https://doi.org/10.1002/bdd.2510080402
Wild MJ, McKillop D, Butters CJ (1999) Determination of the human cytochrome P450 isoforms involved in the metabolism of zolmitriptan. Xenobiotica 29(8):847–857. https://doi.org/10.1080/004982599238290
Wilkinson DJ, Southall RL, Li M et al (2017) Minipig and human metabolism of aldehyde oxidase substrates: in vitro-in vivo comparisons. AAPS J 19(4):1163–1174. https://doi.org/10.1208/s12248-017-0087-3
Winter HR, Wang Y, Unadkat JD (2000) CYP2C8/9 mediate dapsone N-hydroxylation at clinical concentrations of dapsone. Drug Metab Dispos 28(8):865–868
Workman P (1994) Enzyme-directed bioreductive drug development revisited: a commentary on recent progress and future prospects with emphasis on quinone anticancer agents and quinone metabolizing enzymes, particularly DT-diaphorase. Oncol Res 6(10–11):461–475
Wu JB, Shih JC (2011) Valproic acid induces monoamine oxidase A via Akt/forkhead box O1 activation. Mol Pharmacol 80(4):714–723. https://doi.org/10.1124/mol.111.072744
Wu Z, Lee D, Joo J et al (2013) CYP2J2 and CYP2C19 are the major enzymes responsible for metabolism of albendazole and fenbendazole in human liver microsomes and recombinant P450 assay systems. Antimicrob Agents Chemother 57(11):5448–5456. https://doi.org/10.1128/aac.00843-13
Xie G, Wong CC, Cheng KW, Huang L, Constantinides PP, Rigas B (2012) Regioselective oxidation of phospho-NSAIDs by human cytochrome P450 and flavin monooxygenase isoforms: implications for their pharmacokinetic properties and safety. Br J Pharmacol 167(1):222–232. https://doi.org/10.1111/j.1476-5381.2012.01982.x
Xie J, Saburulla NF, Chen S et al (2019) Evaluation of carbazeran 4-oxidation and O6-benzylguanine 8-oxidation as catalytic markers of human aldehyde oxidase: impact of cytosolic contamination of liver microsomes. Drug Metab Dispos 47(1):26–37. https://doi.org/10.1124/dmd.118.082099
Xu P, LaVallee P, Hoidal JR (2000) Repressed expression of the human xanthine oxidoreductase gene. E-box and TATA-like elements restrict ground state transcriptional activity. J Biol Chem 275(8):5918–5926. https://doi.org/10.1074/jbc.275.8.5918
Yamada M, Yasuhara H (2004) Clinical pharmacology of MAO inhibitors: safety and future. Neurotoxicology 25(1–2):215–221. https://doi.org/10.1016/s0161-813x(03)00097-4
Yamada T, Mino Y, Naito T, Kawakami J (2019) Impact of flavin-containing monooxygenase 3 and CYP2C19 genotypes on plasma disposition and adverse effects of voriconazole administered orally in immunocompromised patients. J Infect Chemother 25(12):1019–1025. https://doi.org/10.1016/j.jiac.2019.05.032
Yamaguchi Y, Matsumura T, Ichida K, Okamoto K, Nishino T (2007) Human xanthine oxidase changes its substrate specificity to aldehyde oxidase type upon mutation of amino acid residues in the active site: roles of active site residues in binding and activation of purine substrate. J Biochem 141(4):513–524. https://doi.org/10.1093/jb/mvm053
Yamamoto T, Moriwaki Y, Takahashi S, Hada T, Higashino K (1987) In vitro conversion of pyrazinamide into 5-hydroxypyrazinamide and that of pyrazinoic acid into 5-hydroxypyrazinoic acid by xanthine oxidase from human liver. Biochem Pharmacol 36(19):3317–3318. https://doi.org/10.1016/0006-2952(87)90654-x
Yamamoto T, Moriwaki Y, Takahashi S et al (1996) Determination of human plasma xanthine oxidase activity by high-performance liquid chromatography. J Chromatogr B Biomed Appl 681(2):395–400. https://doi.org/10.1016/0378-4347(96)00071-0
Yamamoto BK, Moszczynska A, Gudelsky GA (2010) Amphetamine toxicities: classical and emerging mechanisms. Ann N Y Acad Sci 1187:101–121. https://doi.org/10.1111/j.1749-6632.2009.05141.x
Yamazaki M, Shimizu M, Uno Y, Yamazaki H (2014) Drug oxygenation activities mediated by liver microsomal flavin-containing monooxygenases 1 and 3 in humans, monkeys, rats, and minipigs. Biochem Pharmacol 90(2):159–165. https://doi.org/10.1016/j.bcp.2014.04.019
Yamazaki-Nishioka M, Shimizu M, Suemizu H, Nishiwaki M, Mitsui M, Yamazaki H (2018) Human plasma metabolic profiles of benzydamine, a flavin-containing monooxygenase probe substrate, simulated with pharmacokinetic data from control and humanized-liver mice. Xenobiotica 48(2):117–123. https://doi.org/10.1080/00498254.2017.1288280
Yang X, Johnson N, Di L (2019a) Evaluation of cytochrome P450 selectivity for hydralazine as an aldehyde oxidase inhibitor for reaction phenotyping. J Pharm Sci 108(4):1627–1630. https://doi.org/10.1016/j.xphs.2018.11.007
Yang Z, Li W, Chen H et al (2019b) Inhibitor structure-guided design and synthesis of near-infrared fluorescent probes for monoamine oxidase A (MAO-A) and its application in living cells and in vivo. Chem Commun (cambridge) 55(17):2477–2480. https://doi.org/10.1039/c8cc10084e
Yanni SB, Annaert PP, Augustijns P et al (2008) Role of flavin-containing monooxygenase in oxidative metabolism of voriconazole by human liver microsomes. Drug Metab Dispos 36(6):1119–1125. https://doi.org/10.1124/dmd.107.019646
Yanni SB, Annaert PP, Augustijns P, Ibrahim JG, Benjamin DK Jr, Thakker DR (2010) In vitro hepatic metabolism explains higher clearance of voriconazole in children versus adults: role of CYP2C19 and flavin-containing monooxygenase 3. Drug Metab Dispos 38(1):25–31. https://doi.org/10.1124/dmd.109.029769
Ye S, Yoshida S, Fröhlich R, Haufe G, Kirk KL (2005) Fluorinated phenylcyclopropylamines. Part 4: effects of aryl substituents and stereochemistry on the inhibition of monoamine oxidases by 1-aryl-2-fluoro-cyclopropylamines. Bioorg Med Chem 13(7):2489–2499. https://doi.org/10.1016/j.bmc.2005.01.043
Yeniceli D, Deng X, Adams E, Dogrukol-Ak D, Van Schepdael A (2013) Development of a CD-MEKC method for investigating the metabolism of tamoxifen by flavin-containing monooxygenases and the inhibitory effects of methimazole, nicotine and DMXAA. Electrophoresis 34(3):463–470. https://doi.org/10.1002/elps.201200356
Yeung CK, Lang DH, Thummel KE, Rettie AE (2000) Immunoquantitation of FMO1 in human liver, kidney, and intestine. Drug Metab Dispos 28(9):1107–1111
Yeung CK, Rettie AE (2006) Benzydamine N-oxygenation as a measure of flavin-containing monooxygenase activity. Methods Mol Biol 320:157–162. https://doi.org/10.1385/1-59259-998-2:157
Yeung CK, Adman ET, Rettie AE (2007) Functional characterization of genetic variants of human FMO3 associated with trimethylaminuria. Arch Biochem Biophys 464(2):251–259. https://doi.org/10.1016/j.abb.2007.04.014
Youdim MB (1975) Monoamine oxidase, its inhibition. Mod Probl Pharmacopsychiatry 10:65–88
Youdim MB, Bakhle YS (2006) Monoamine oxidase: isoforms and inhibitors in Parkinson's disease and depressive illness. Br J Pharmacol 147(Suppl 1):S287–S296. https://doi.org/10.1038/sj.bjp.0706464
Youdim MB, Weinstock M (2004) Therapeutic applications of selective and non-selective inhibitors of monoamine oxidase A and B that do not cause significant tyramine potentiation. Neurotoxicology 25(1–2):243–250. https://doi.org/10.1016/s0161-813x(03)00103-7
Yu PH (1986) Inhibition of monoamine oxidase activity by phenylpropanolamine, an anorectic agent. Res Commun Chem Pathol Pharmacol 51(2):163–171
Yu AM, Granvil CP, Haining RL et al (2003) The relative contribution of monoamine oxidase and cytochrome P450 isozymes to the metabolic deamination of the trace amine tryptamine. J Pharmacol Exp Ther 304(2):539–546. https://doi.org/10.1124/jpet.102.043786
Zapata-Torres G, Fierro A, Barriga-González G, Salgado JC, Celis-Barros C (2015) Revealing monoamine oxidase B catalytic mechanisms by means of the quantum chemical cluster approach. J Chem Inf Model 55(7):1349–1360. https://doi.org/10.1021/acs.jcim.5b00140
Zarmouh NO, Mazzio EA, Elshami FM, Messeha SS, Eyunni SV, Soliman KF (2015) Evaluation of the inhibitory effects of bavachinin and bavachin on human monoamine oxidases A and B. Evid Based Complement Alternat Med 2015:852194. https://doi.org/10.1155/2015/852194
Zarmouh NO, Messeha SS, Elshami FM, Soliman KF (2016) Natural products screening for the identification of selective monoamine oxidase-B inhibitors. Eur J Med Plants 15(1). https://doi.org/10.9734/ejmp/2016/26453
Zarmouh NO, Eyunni SK, Soliman KF (2017) The benzopyrone biochanin-A as a reversible, competitive, and selective monoamine oxidase B inhibitor. BMC Complement Altern Med 17(1):34. https://doi.org/10.1186/s12906-016-1525-y
Zetterberg C, Maltais F, Laitinen L et al (2016) VX-509 (decernotinib)-mediated CYP3A time-dependent inhibition: an aldehyde oxidase metabolite as a perpetrator of drug–drug interactions. Drug Metab Dispos 44(8):1286–1295. https://doi.org/10.1124/dmd.116.071100
Zhang X, Liu HH, Weller P et al (2011) In silico and in vitro pharmacogenetics: aldehyde oxidase rapidly metabolizes a p38 kinase inhibitor. Pharmacogenomics J 11(1):15–24. https://doi.org/10.1038/tpj.2010.8
Zhang JW, Xiao W, Gao ZT, Yu ZT, Zhang JYJ (2018) Metabolism of c-Met kinase inhibitors containing quinoline by aldehyde oxidase, electron donating, and steric hindrance effect. Drug Metab Dispos 46(12):1847–1855. https://doi.org/10.1124/dmd.118.081919
Zhang Y, Wang Q, Liu R et al (2019a) Rapid screening and identification of monoamine oxidase-A inhibitors from Corydalis rhizome using enzyme-immobilized magnetic beads based method. J Chromatogr A 1592:1–8. https://doi.org/10.1016/j.chroma.2019.01.062
Zhang Z, Hamada H, Gerk PM (2019b) Selectivity of dietary phenolics for inhibition of human monoamine oxidases A and B. Biomed Res Int 2019:8361858. https://doi.org/10.1155/2019/8361858
Zhao X, Lu J, Chen X et al (2021) Methamphetamine exposure induces neuronal programmed necrosis by activating the receptor-interacting protein kinase 3-related signalling pathway. FASEB J 35(5):e21561. https://doi.org/10.1096/fj.202100188R
Zheng J, Xin Y, Zhang J et al (2018) Pharmacokinetics and disposition of momelotinib revealed a disproportionate human metabolite-resolution for clinical development. Drug Metab Dispos 46(3):237–247. https://doi.org/10.1124/dmd.117.078899
Zhong G, Seaman CJ, Paragas EM et al (2021) Aldehyde oxidase contributes to all-trans-retinoic acid biosynthesis in human liver. Drug Metab Dispos 49(3):202–211. https://doi.org/10.1124/dmd.120.000296
Zhou S, Kestell P, Paxton JW (2002) 6-Methylhydroxylation of the anti-cancer agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA) by flavin-containing monooxygenase 3. Eur J Drug Metab Pharmacokinet 27(3):179–183. https://doi.org/10.1007/bf03190455
Zhou W, Humphries H, Neuhoff S et al (2017) Development of a physiologically based pharmacokinetic model to predict the effects of flavin-containing monooxygenase 3 (FMO3) polymorphisms on itopride exposure. Biopharm Drug Dispos 38(6):389–393. https://doi.org/10.1002/bdd.2074
Zhou S, Chen G, Huang G (2018) Design, synthesis and biological evaluation of lazabemide derivatives as inhibitors of monoamine oxidase. Bioorg Med Chem 26(17):4863–4870. https://doi.org/10.1016/j.bmc.2018.08.024
Zhou L, Pang XY, Hou XY, Liu L, Guo ZT, Chen XY (2020) Nimesulide increases the aldehyde oxidase activity of humans and rats. Acta Pharmacol Sin 41(6):843–851. https://doi.org/10.1038/s41401-019-0336-3
Zhu W, Buffa JA, Wang Z et al (2018) Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J Thromb Haemost 16(9):1857–1872. https://doi.org/10.1111/jth.14234
Ziegler DM (1988) Flavin-containing monooxygenases: catalytic mechanism and substrate specificities. Drug Metab Rev 19(1):1–32. https://doi.org/10.3109/03602538809049617
Ziegler DM (2002) An overview of the mechanism, substrate specificities, and structure of FMOs. Drug Metab Rev 34(3):503–511. https://doi.org/10.1081/dmr-120005650
Ziegler DM, Pettit FH (1966) Microsomal oxidases. I. The isolation and dialkylarylamine oxygenase activity of pork liver microsomes. Biochemistry 5(9):2932–2938. https://doi.org/10.1021/bi00873a024
Zientek MA, Youdim K (2015) Reaction phenotyping: advances in the experimental strategies used to characterize the contribution of drug-metabolizing enzymes. Drug Metab Dispos 43(1):163–181. https://doi.org/10.1124/dmd.114.058750
Zientek M, Jiang Y, Youdim K, Obach RS (2010) In vitro-in vivo correlation for intrinsic clearance for drugs metabolized by human aldehyde oxidase. Drug Metab Dispos 38(8):1322–1327. https://doi.org/10.1124/dmd.110.033555
Zimm S, Collins JM, O’Neill D, Chabner BA, Poplack DG (1983) Inhibition of first-pass metabolism in cancer chemotherapy: interaction of 6-mercaptopurine and allopurinol. Clin Pharmacol Ther 34(6):810–817. https://doi.org/10.1038/clpt.1983.254
Acknowledgements
We thank K. Trisler for assistance in the preparation of the manuscript.
Funding
F.P.G. acknowledges current support from the United States National Institutes of Health Grant R01 GM118122. This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
All authors participated in the writing of this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest, financial or otherwise.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rendić, S.P., Crouch, R.D. & Guengerich, F.P. Roles of selected non-P450 human oxidoreductase enzymes in protective and toxic effects of chemicals: review and compilation of reactions. Arch Toxicol 96, 2145–2246 (2022). https://doi.org/10.1007/s00204-022-03304-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-022-03304-3