
Abstract
Many drugs have the potential to cause drug-induced liver injury (DILI); however, underlying mechanisms are diverse. The concept of adverse outcome pathways (AOPs) has become instrumental for risk assessment of drug class effects. We report AOPs specific for immune-mediated and drug hypersensitivity/allergic hepatitis by considering genomic, histo- and clinical pathology data of mice and dogs treated with diclofenac. The findings are relevant for other NSAIDs and drugs undergoing iminoquinone and quinone reactive metabolite formation. We define reactive metabolites catalyzed by CYP monooxygenase and myeloperoxidases of neutrophils and Kupffer cells as well as acyl glucuronides produced by uridine diphosphoglucuronosyl transferase as molecular initiating events (MIE). The reactive metabolites bind to proteins and act as neo-antigen and involve antigen-presenting cells to elicit B- and T-cell responses. Given the diverse immune systems between mice and dogs, six different key events (KEs) at the cellular and up to four KEs at the organ level are defined with mechanistic plausibility for the onset and progression of liver inflammation. With mice, cellular stress response, interferon gamma-, adipocytokine- and chemokine signaling provided a rationale for the AOP of immune-mediated hepatitis. With dogs, an erroneous programming of the innate and adaptive immune response resulted in mast cell activation; their infiltration into liver parenchyma and the shift to M2-polarized Kupffer cells signify allergic hepatitis and the occurrence of granulomas of the liver. Taken together, diclofenac induces divergent immune responses among two important preclinical animal species, and the injury pattern seen among clinical cases confirms the relevance of the developed AOP for immune-mediated hepatitis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The liver is a primary target organ of toxicity, and drug-induced liver injury (DILI) is a major concern that results in box warnings and even withdrawals of drugs from the market. It is a leading cause for acute liver failure (Przybylak and Cronin 2012). Importantly, with the advent of omics platforms and high-throughput technologies a plethora of diverse data have become available to significantly advance the field of toxicological sciences. Given that “omics and NGS” platform technologies enable the capturing of whole genome/proteome information, the findings permit the construction of circuitries within a cell, tissue and organ in response to specific treatments across different species. Yet, turning data into knowledge remains a fundamental challenge.
The AOP concept represents a paradigm shift in regulatory toxicology and risk assessment (Vinken 2013); it is a conceptual framework that provides information concerning the causal relationship between an MIE and adverse outcome (AO) at different levels of biology, i.e., molecular, cellular/organelle, organ and whole organism (Ankley et al. 2010; Vinken et al. 2017). The construction of an AOP is based on mechanistic consideration and requires expert knowledge across different fields of biomedical science, i.e., molecular and cellular, development, frank organ toxicity/pathology and so forth.
AOPs also support the development of alternative testing strategies by endorsing the 3R principle in experimental works while the need for AOP network analysis arises from the complex biological processes underlying toxicological events. Ideally key event relationships across different species are defined with the weight of evidence being assessed by considering biological and mechanistic information. The AOP knowledgebase (AOP-KB, https://aopkb.oecd.org/) facilitates the search for mutual KEs and key event relationship (KER) components (Knapen et al. 2018; Pollesch et al. 2019), and to reflect the more complex processes in a toxicological insult AOP network analysis has been advocated. Such networks can be analyzed by linking the shared KE and KER components as a modular unit of the AOP (Knapen et al. 2018; Pollesch et al. 2019). Therefore, the sharing of modular units like KE and KER of individual AOPs is encouraged (Knapen et al. 2018), and these de facto AOP networks can cover lacking information. However, to explore the connectivity of shared modular units, the biological and mechanistic information needs to be considered in the context of taxonomy, life stage, sex and target organs.
In an effort to define an AOP for immune-mediated and allergic hepatitis, we queried the AOP knowledgebase for drug-induced hepatotoxicity. This revealed 8 AOPs linked to liver fibrosis, cholestasis and steatosis, and the associated MIE are defined by protein alkylation, LXR and inhibition of the bile salt export pump ABCB11. However, none of the AOPs are specific for immune-mediated and/or allergic hepatitis and the lack of mechanistic and clinical consideration poorly reflects the complexity of DILI. Notwithstanding, AOPs are living documents and will improve over time as updated information is effectively integrated to avoid oversimplifications. Moreover, continuous research in the underlying mechanism of DILI provides new insights which enable the construction of more detailed AOPs (Vinken 2015).
We report an AOP for immune-mediated and allergic hepatitis based on comprehensive data obtained from mouse and dog diclofenac repeated-dose studies and literature findings with clinical relevance. This non-steroidal anti-inflammatory drug (NSAID) exerts anti-inflammatory, analgesic and anti-pyretic effects through various mechanisms; however, its use can lead to adverse drug reactions including DILI (Banks et al. 1995). We entrained the AOP on our previous genomic studies as well as serum biochemistry, histopathology and immunohistochemistry and Western immunoblotting data (Lee et al. 2016; Selvaraj et al. 2017) and show that diclofenac reactive metabolism causes divergent immune responses among the two animal species commonly used in toxicity studies. We define iminoquinone and quinone reactive metabolites as MIE and confirm the relevance of these structural alerts for a larger group of drugs and chemicals undergoing iminoquinone and quinone reactive metabolite formation. Collectively, an AOP for hepatitis based on experimental and computational biology studies is presented.
Methods
Construction of AOP framework
Previously reported genomic data of diclofenac-induced liver injury in mice and dog models were interrogated to construct this AOP framework (male C57BL/6 mice with daily intraperitoneal injection of 30 mg/kg/day and 150 mg/kg/days for 14 days; male beagle dogs with daily oral dosing of 1 mg/kg/day and 3 mg/kg/day for 28 days) (Lee et al. 2016; Selvaraj et al. 2017). Given the complex inference resulting from on-target but exaggerated pharmacological responses and toxicity related to the physicochemical characteristics of diclofenac and its effects on cells, organelles, membranes and/or metabolic pathways, a combined approach was taken to define MIE. The subsequently performed computational analysis enabled the development of an AOP of immune-mediated hepatitis, and the concept is based on mechanistic plausibility. It recapitulates the pleiotropic effects induced by diclofenac treatment. Next to whole genome gene expression data, the weight of evidence includes histopathology, clinical chemistry and immunohistochemistry findings. Collectively, the strength, consistency and specificity of the AOPs are considered.
Results and discussions
Pharmacological mode of action of diclofenac
Figure 1 illustrates the pharmacological mode of action of diclofenac. It inhibits cyclooxygenase 1 and 2 which catalyze arachidonic acid metabolism (Gan 2010). Likewise, it suppresses the production of leukotrienes by inhibiting lipoxygenases and prostaglandin E2 and thromboxane A2 synthesis (Gan 2010). Its analgesic activity resides in an activation of the nitric oxide–cGMP nociceptive pathway as well as inhibition of N-methyl-d-aspartate (NMDA) receptor. Note, NMDA is a receptor for the neurotransmitter glutamate and diclofenac dampens NMDA-evoked nociceptor activity by modulating voltage-gated neuronal potassium channel activity. Likewise, activated NMDA receptors stimulate neuronal NO-synthase activity with increased production of NO. At the postsynaptic neuron NO activates the guanyl cyclase, and through retrograde diffusion NO reinforces the glutamatergic signaling in the presynaptic neuron thereby reinforcing nociception. NO may also induce an antinociceptive effect (Gan 2010). In addition, diclofenac suppresses activity of the neuropeptide substance P and is also a partial agonist of PPARɣ to augment lipogenesis (Gan 2010).

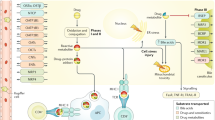
AOP of immune-mediated hepatitis in mice in response to diclofenac treatment. Diclofenac induced liver damage by the activation of complex immune-mediated inflammatory signaling cascades. This AOP illustrates the pharmacological mechanism of action of diclofenac and its adverse effects in liver. The MIE and KEs associated with immune-mediated hepatitis in mice are highlighted; the green boxes represent MIE, orange colored are KEs at the cellular level, and red-colored boxes define the KEs at the organ and organism level. The potential linkages among the KEs are based on experimental evidence and literature findings
AOP for immune-mediated hepatitis in mice
Diclofenac treatments caused an increase in serum biochemistry markers for liver injury (ALT and AST), and histopathology showed hepatomegaly, hepatocyte swelling, glycogen depletion, eosinophilic hepatocytes and cholesterolosis (Lee et al. 2016). Moreover, fatal acute liver failure was observed at a dose of 150 mg/kg. The genomic and biochemical validation study revealed immune-mediated responses to be considerably induced after diclofenac treatment (Lee et al. 2016).
MIE
Drug-induced hepatitis is a multistep process and frequently involves reactive metabolites which subsequently form drug–protein adducts. These are sensed by antigen-presenting cells to elicit innate and adaptive immune responses (Adams et al. 2010). Diclofenac is extensively metabolized by CYP monooxygenases and myeloperoxidases (MPO) of neutrophils and Kupffer cells into reactive metabolites notably quinoneimine intermediates (Table 1). The metabolism of diclofenac to reactive metabolites causes organ toxicity and this defines the MIE. A further example for a benzoquinone imine reactive metabolite causing liver injury is the metabolism of paracetamol to NAPQI. Correspondingly, NAPQI is the initial cause of hepatotoxicity and once again defines the MIE. Indeed, for a wide range of drugs quinoneimines are implicated in liver injury as summarized in supplementary Table S1.
Diclofenac caused significant regulation of CYP monooxygenases after single and repeated treatment of mice (Lee et al. 2016). Independent investigations also support the key role of the acyl glucuronide produced by uridine diphosphoglucuronosyl transferase as a molecular initiation event (Oda et al. 2017; Seitz and Boelsterli 1998), and among NSAIDs the salicyl acyl glucuronide derived from aspirin is a further example. Thus, reactive metabolites are formed by hepatocytes, neutrophils and Kupffer cells with benzoquinone imine intermediates and acyl glucuronides being particularly harmful. If not sufficiently detoxified the reactive metabolites damage organelles, proteins and membrane lipids and eventually trigger programmed cell death. In specific, the accumulation of the reactive metabolite triggers oxidative stress and mitochondrial permeability transitions, i.e., mitochondrial toxicity by inhibiting ATP synthesis that leads to hepatocellular damage (Syed et al. 2016). Next to direct effects the reactive metabolites can covalently bind to proteins to form adducts. These function as neoantigens and are sensed and phagocytozed by APCs. Through interaction with the major histocompatibility complex APCs elicit B and T cell responses (Aithal 2011; Boelsterli 2003). In addition, diclofenac acyl glucuronide inhibits the Mrp2 transport. This results in intrahepatic cholestasis and damage of the biliary epithelium (Boelsterli 2003; Lagas et al. 2010; Seitz and Boelsterli 1998). Similar to diclofenac the NSAIDs, lumiracoxib and indomethacin produce quinoneimine reactive intermediates, and next to ibuprofen and naproxen a wider range of carboxylic acid containing drugs are associated with allergic reactions (Stepan et al. 2011). The reactivity of acyl glucuronides derived from carboxylic acid containing drugs and the evidence for its toxicological concerns was recently summarized (Darnell et al. 2015; Van Vleet et al. 2017). Note, the covalent binding of acyl glucuronides to proteins constitutes a mechanism of toxicity, and the safety assessment of acyl glucuronides was the subject of a recent commentary with zomepirac being a prominent example for NSAID toxicity (Smith et al. 2018). Importantly, inhibition of MPO ameliorates adverse effects of MPO-derived oxidants (Malle et al. 2007) and MPO ko mice are an excellent system to study the importance of MPO in systemic inflammatory reactions. Alike, amelioration of diclofenac-induced toxicity was observed with cytochrome P450 reductase (CPR) null mice (Zhu and Zhang 2012), and multidrug resistance-associated protein 3 plays an important role in protection against acute toxicity of diclofenac acyl glucuronide as evidenced in Mrp3-null (KO) mice (Scialis et al. 2015). Moreover, NSAIDs are known to produce reactive oxygen species that result in cardiovascular disease (Ghosh et al. 2015), and a structural alert/reactive metabolite concept of 200 common drugs producing a wide range of different reactive metabolites was reported (Stepan et al. 2011). Collectively, the structural alerts quinoneimine and acyl glucuronides function as MIEs in immune-mediated and allergic hepatitis. Notwithstanding, defining an unequivocal MIE in the AOP framework can be complex as discussed in the seminal paper of Allen and coworkers (Allen et al. 2014).
KEs related to immune-mediated hepatitis
The biochemical and genomic data revealed diclofenac treatment to induce complex immune-mediated inflammatory signaling particularly from resident and migratory cells of the sinusoid and the space of Disse (Fig. 1 and Table 1). Within hepatocytes, the reactive metabolites elicit cellular stress responses including oxidative stress, mitochondrial dysfunction, apoptosis and ER stress/unfolded protein responses. In repeated-dose studies with mice, diclofenac treatment induced expression of the plasma membrane cysteine carrier (Slc3a1) to imply adaptive responses to oxidative stress. Furthermore, cysteine is an essential building block for the hepatic synthesis of reduced GSH and therefore of fundamental importance in alleviating oxidative stresses.
KE1: mitochondrial dysfunction
Diclofenac caused mitochondrial dysfunction through an inhibition of ATP synthesis (Boelsterli 2003; Kang et al. 2016; Syed et al. 2016). The significant repression of mitochondrial membrane transport proteins and key members of the oxidative phosphorylation pathway is testimony of an impaired mitochondrial respiration and ATP synthesis (Lee et al. 2016). Conversely, the plasma membrane Mg2+ transporter is strongly induced to increase intracellular Mg2+ concentration. Note increased Mg2+ uptake counteracts the detrimental effects of diclofenac treatment to alleviate mitochondrial stress and the opening of the Ca2+-dependent permeability transition pore to dampen apoptotic signaling.
KE2: apoptotic cell death
Reactive metabolites of diclofenac can directly or indirectly induce apoptotic cell death by activating several pro- and anti-apoptotic factors notably toll-like receptors, cytokine signaling inducible factors such as S100 calcium binding proteins and pro-inflammatory adipokines to augment ER stress-induced apoptosis (Lee et al. 2016; Sawa et al. 2009). Damaged hepatocytes send alarm signals like the damage-associated molecular patterns (DAMPs) that induce the immune and inflammatory response by activating immune cells. Although several DAMP molecules including S100 proteins were upregulated after repeated dosing of mice (supplementary Tables S2 and S3 for mice and dogs, respectively), the major components of the inflammasome are not regulated at the transcript level. Meanwhile, the danger hypothesis proposed that DAMPs can also be influenced by immune or inflammatory response. Our previous study demonstrated that diclofenac treatment induced the expression of inflammatory proteins which are released from macrophages/Kupffer cells. The subsequent inflammatory responses can reinforce the cellular damage of hepatocyte and in a vicious cycle strengthen inflammation (Lee et al. 2016).
KE3: ER stress/unfolded protein response
Genes involved in ER stress and unfolded protein response (UPR) were significantly regulated in the liver of diclofenac-treated mice. Independent studies evidenced diclofenac to trigger ER stress and UPR by PERK and ATF6 pathways as well as eIF2α phosphorylation (Foufelle and Fromenty 2016; Franceschelli et al. 2011; Fredriksson et al. 2014). However, the prolonged activation of PERK/eIF2α pathway induces apoptosis by activating the pro-apoptotic factor CHOP (Franceschelli et al. 2011; Fredriksson et al. 2014). In addition, ER stress can alter the lipid metabolism by UPR and leads to dyslipidemia (Basseri and Austin 2012). As reported by us, diclofenac caused hepatic cholesterolosis in mice with significant alteration in the transcription of genes coding for fatty acid and cholesterol metabolism (Lee et al. 2016). On the other hand, the repeated diclofenac treatment induced Cyp7a1 expression which catalyzes the hydroxylation of cholesterol into bile acids (Lee et al. 2016). Alike, the induction of the apical sodium–bile acid transporter (Slc10a2) evidences changes in the transcellular transport of bile acids across the biliary epithelium to support the enterohepatic cycling of bile acids (Lee et al. 2016). Moreover, the organic anion transporter Slc10a6 was induced and functions on taurolithocholic acid-3-sulfate (TCA-3S). Note, TCA-3S excretion into urine is about 90-fold higher in patients diagnosed with intrahepatic cholestasis of pregnancy thus highlighting its potential as a biomarker of hepatic cholestasis (Lee et al. 2016).
KE4: immune cell activation
Diclofenac adducts are sensed by APC and other phagocytic cells and trigger immune responses. The immune-mediated hepatitis is the result of complex interplay of innate and adaptive immune responses and involves the regulation of various cytokines/chemokines and their receptors (Lee et al. 2016). In specific, the released chemokines recruit the neutrophils, leukocytes and B lymphocytes to the sinusoidal space or to harmed hepatocytes, while cytokines endorse differentiation of myeloid and cytotoxic CD8+ T-cells (Saiman and Friedman 2012; Sawa et al. 2009). Increased expressions of interleukins modulate the activation and proliferation of T and/or NK cell responses (Hammerich and Tacke 2014; Zwirner and Domaica 2010) while members of the interleukin-1 superfamily stimulate the production of type 2 cytokines by T-helper cells (Miller 2011). Thus, diclofenac treatment resulted in an activation of several cytokines to affect T cell differentiation. Collectively, diclofenac stimulated an activation of diverse immune cells including monocytes, Kupffer cells and APC (Lee et al. 2016).
KE5: IFNγ signaling
Increased expression of IFNγ hallmarks innate and adaptive immune responses. IFNγ plays a pivotal role in host defense in response to infections and mediating the inflammation by producing the pro-inflammatory cytokines (Muhl and Pfeilschifter 2003). Genomic analysis revealed protein tyrosine phosphatase non-receptor type 2 (Ptpn2) to be significantly regulated in mice after diclofenac treatment and Ptpn2 plays a critical role in modulating IFNγ signaling (Lee et al. 2016; Scharl et al. 2010). Together, diclofenac treatment induced expression of inflammatory cytokines including IFNγ, interleukins and TNFα (Dutta et al. 2008; Yano et al. 2012).
KE6: inflammation
Our computational studies defined key master regulatory molecules and their associated networks. Based on independent RT-qPCR studies, induced expression of suppressor of cytokine signaling (Socs), leptin, growth hormone receptor (Ghr), and Ptpn proteins was confirmed and these function in IFNγ, Jak/Stat, pro- and anti-inflammatory signaling pathways. Stat3 is one of the major transcription factors activated by cytokines and growth factors to influence pro-inflammatory (Mapk, p38, Jnk, and IκB kinase) and anti-inflammatory signaling (Pi3k-Akt) events. Among individual animals marked induction of this protein was observed which controls transcription of Socs and inhibits Jak/Stat3 signaling (Kong et al. 2002). In addition, leptin and other adipokine signaling molecules play a major role in energy intake to influence monocyte and macrophage activity during inflammation (Fantuzzi and Faggioni 2000). Next to its role in the control of energy intake leptin and its receptor modulates Jak/Stat, Erk 1/2 and Pi3k signaling (Bjorbaek and Kahn 2004; Cottrell and Mercer 2012; Paz-Filho et al. 2012) and therefore plays a decisive role in inflammation. Similarly, the growth hormone receptor is influenced by multiple intracellular signaling cascades (Jak–Stat and chemokine signaling) and functions in liver regeneration. Diclofenac treatment reinforced Ghr degradation as evidenced by immunoblotting to suppress hepatic Ghr signaling (Takahashi 2017). Diclofenac also induced expression of the endothelial–leukocyte adhesion molecule selectin which recruits leukocytes to the inflammatory site and the computational analysis defined selectin as a master regulator (Lee et al. 2016; Ley 2003). Furthermore, the highly significant induction of lipocalin-2 (Lcn2) signifies sterile inflammation and neutralization of LCN2 controls neutrophilic inflammation as had been summarized (Moschen et al. 2017). Taken together, the IFNγ, Jak/Stat, adipocytokine and chemokine signaling pathways provide a rationale for the AOP of immune-mediated hepatitis observed in mice in response to diclofenac treatment.
AOP for diclofenac-induced immunoallergic hepatitis in dogs
To the best of our knowledge an animal model to investigate allergic hepatitis has not been developed so far. Our studies highlight the relevance of dogs as a surrogate for clinical immunoallergic DILI and its effects on the immune system. Further justification of the canine model resides in the similar expression of COX1/COX2 between dogs and humans (Kay-Mugford et al. 2000; Radi and Khan 2006; Radi 2009). To investigate diclofenac’s mechanism of immuno-allergic hepatitis, beagle dogs were given 1 or 3 mg/kg/day for 28 days. Histopathology revealed micro- and macrovesicular hepatic steatosis, glycogen depletion, apoptosis, acute lobular hepatitis, granulomas and mastocytosis. Biochemical and whole genome scans revealed that diclofenac induced hypersensitivity reactions. Key molecules related to oxidative stress, macrophage polarization, mast cell activation and complement cascade were regulated as the result of an erroneous programming of the innate and adaptive immune system to cause granulomatous hepatitis (Selvaraj et al. 2017). The AOP of immune-allergic hepatitis is depicted in Fig. 2 and Table 1.

AOP of immunoallergic hepatitis in dog in response to diclofenac. Diclofenac induced the key molecules related to the liver hypersensitivity reactions including oxidative stress, macrophage polarization, mastocytosis, complement activation and an erroneous programming of the innate and adaptive immune system that can cause granulomatous hepatitis. Putative MIE and KEs associated with immunoallergic hepatitis in dogs were highlighted; green boxes represent MIE, orange color is for KEs in the cellular level, and red-colored boxes define the KEs in organ and organism level. The potential linkages among the KEs are shown based on the experimental evidences and literature. The brown dotted line demonstrates the KEs occurred in the hepatocytes
MIE
With dogs the MIE is similar to the one described for mice, i.e., the formation of iminoquinone and acyl glucuronide reactive metabolites. However, the abundance of metabolite formation differs among species (Fujiwara et al. 2018; Hughes and Swamidass 2017; Pulli et al. 2013; Sasaki and Yokoi 2018; Smith et al. 2018).
KEs related to immunoallergic hepatitis
The genomic study revealed significant changes in the regulation of genes coding for immune, inflammation, apoptosis and oxidative stress responses to diclofenac treatment. In specific, the reduced expression of hepatic CYP monooxygenases and phase II enzymes is caused by inflammation and the immune response. Induced expression of acute-phase proteins (SAA), alpha-macroglobulin, fibrinogen, complement factors and cytokines evidences sterile inflammation. The significant up-regulation of mainly positive acute-phase proteins evidences inflammation in response to reactive metabolites and harmed hepatocytes; notwithstanding, hepatic SOD1 protein expression is decreased after diclofenac treatment as a result of oxidative stress.
KE1/KE2: mitochondrial dysfunction and apoptotic cell death
In addition, reactive metabolites of diclofenac cause cellular stress and increased the level of ROS which leads to mitochondrial damage and subsequent apoptosis in hepatocytes (Boelsterli 2003; Gomez-Lechon et al. 2003a). Consistent with the findings observed with mice, the genomic analysis of dog liver discovered genes related to oxidative stress, mitochondrial biogenesis and membrane transport and apoptosis as significantly regulated (Selvaraj et al. 2017).
KE3: microvesicular steatosis
Oxidative stress and impaired mitochondrial activity can result in drug-induced steatosis (Pessayre 2007; Sahini et al. 2014). The genes coding for lipogenesis, lipid transport, lipid droplet growth, ER stress and fatty acid oxidation were significantly regulated (Selvaraj et al. 2017) and histopathology evidenced hepatic steatosis in diclofenac-treated dogs. Note, drugs other than NSAIDs were reported to cause drug-induced steatosis in animal models and patients (Freneaux et al. 1990; Patel and Sanyal 2013; Sahini et al. 2014).
KE4: mast cell activation
Diclofenac caused mast cell activation and hepatic infiltration with strong induction of immunoglobulins, Fc-receptor signaling molecules and acute-phase proteins as well as the classical and alternative pathway components of the complement system to highlight allergic reactions. The marked mastocytosis hallmarks drug hypersensitivity. It is associated with granulocyte and mast cell degranulation and the release of pro-inflammatory mediators including histamines, prostaglandins, leukotrienes and other cytotoxic molecules (Selvaraj et al. 2017; Theoharides et al. 2012; Zhang et al. 2018) to aggravate the inflammatory responses and support migration of other inflammatory immune cells to sites of injury. Thus, the AOP highlights activation of the complement system as a mechanism of toxicity resulting in granulomatous hepatitis.
KE5: Kupffer cell activation and/polarization (M1/M2)
Diclofenac treatment caused an activation of Kupffer cells and strong expression of M1/M2 marker genes (Selvaraj et al. 2017). Activated Kupffer cells release a range of inflammatory mediators, growth factors and acute-phase proteins to perpetuate liver inflammation (Kolios et al. 2006; Roberts et al. 2007). Moreover, through complex cellular cross-talks macrophages influence the differentiation of Th-cell populations including Th17. Therefore, the induction of markers of M2-polarized Kupffer cells like IL10, IL4/IL13, the innate immune response (CD14), the scavenger receptors (CD163, MARCO and CXCL16), MHC class II molecules (CD74 and HLA-DRB1) as well as their sensor and effectors (complement genes C1QA-C, IGF) are suggestive for Th2 responses to alleviate the inflammatory reactions to diclofenac treatments (Selvaraj et al. 2017). Moreover, histopathology evidenced inflammatory infiltrates like immature/migrating macrophages, Kupffer cells, granulocytes and lymphocytes into interstitial and the sinusoidal space as well as harmed hepatic parenchyma (Selvaraj et al. 2017).
KE6: inflammation
Diclofenac treatment caused complex pro- and anti-inflammatory reactions in the liver (Chen et al. 2015; Njoku 2014; Yano et al. 2012). In specific, the pro-inflammatory chemokines and their receptors play a major role in cytotoxic T-cell activation and the trafficking of inflammatory immune cells (neutrophils, leukocytes, B-lymphocytes) to sites of injury (Karin 2010; Wong and Fish 2003). In addition, MAPK signaling molecules were induced in response to diclofenac treatment and their regulation can be triggered by cellular stress and pro-inflammatory cytokines to result in inflammation-mediated hepatotoxicity (Guegan et al. 2013; Kyriakis and Avruch 2012; Nakagawa and Maeda 2012; Nikolaou et al. 2013). Importantly, increased expression of TNF family members activates NF-κB, JNK, p38, and ERK1/ERK2 signaling and are involved in T-cell and T-helper (Th1, Th2 and Th17) cell-mediated responses and hepatic inflammation (Aiba and Nakamura 2013; Akiyama et al. 2012; Sakai et al. 2012; Zhang and Li 2012), whereas significant repression of the LY6 antigen indicates adaptive response to cytokine-induced inflammation (Begue et al. 2006). Collectively, the cytokines and chemokines bind and activate their corresponding receptors to promote immune-mediated inflammation of the liver.
The biological plausibility of KERs is summarized in supplementary Table S4 and Fig. 3 provides a simplified AOP for diclofenac-induced immune-mediated and allergic hepatitis.

A simplified AOP for immune-mediated and allergic hepatitis
Clinical relevance
A long-term prospective clinical trial involving 17,289 arthritis patients who were randomly assigned to diclofenac (150 mg daily) or etoricoxib (60 or 90 mg daily) evidenced diclofenac to cause common aminotransferase elevations (Laine et al. 2009). Alike, a systematic review of randomized clinical trial data of 3 NSAIDs revealed diclofenac to be top ranking for hepatotoxic events (Sriuttha et al. 2018). Earlier studies already reported cases of acute hepatitis induced by diclofenac (Helfgott et al. 1990; Iveson et al. 1990; Purcell et al. 1991; Sallie 1990), and liver biopsy findings defined a histological injury pattern of granulomatous hepatitis. Note, our studies with dogs also demonstrated granulomatous hepatitis as a key finding (see above KE4) and the granulomas are composed of inflammatory cells and histocytes (Ramachandran and Kakar 2009). Moreover, a current study compared 30 DILI cases among 8 NSAIDs and found diclofenac to be the most frequently implicated NSAID. The cases are characterized by hepatocellular injury, prolonged hospitalization and included a patient with fatal Stevens–Johnson syndrome (Schmeltzer et al. 2015). In their study 38% of diclofenac DILI cases presented fever, rash and eosinophilia, and the findings are consistent with the clinical features of drug hypersensitivity reactions (Schmeltzer et al. 2015). Altogether, the proposed AOP of immune-mediated and allergic hepatitis is relevant for human DILI induced by NSAIDs.
How can the AOP concept be translated into clinical and regulatory practice?
Biomarkers based on AOPs carry the potential to significantly improve an assessment of adverse drug reactions (ADRs). In specific, ADRs are assessed by expert opinion and the guidance given by the WHO and regulatory authorities. Furthermore, the magnitude and the incidence of ADRs are evaluated by postmarketing surveillance and the legally required period safety updated reports (PSUR). Although ADRs on single-drug treatment can be evaluated with certainty, the causality assessment of ADRs among comorbid patients which typically involves several drugs can be confounded by the complex drug properties–host factor interactions that need to be deciphered. Adding to complexity is the fact that scoring systems to evaluate organ specific toxicities are spares and with the exception of drug-induced liver injury, i.e., the RUCAM (Roussel Uclaf Causality Assessment Method) and the MELD (Model for End Stage Liver Disease) score in liver transplantation, there are no algorithms to assess more objectively organ-specific ADRs.
By probing for mechanistically plausible key events associated with drug injury, AOPs can help to define biomarkers to improve the causality assessment of ADRs. The development of AOPs for different drug classes and the clinical validation of AOP-defined biochemical markers will be a priority task.
Conclusion
Diclofenac treatment induced divergent immune responses among two important animal species commonly used in toxicity studies. The knowledge gain from these studies will be the base for the development of an integrated AOP for immune-mediated hepatitis.
Abbreviations
- ADR:
-
Adverse drug reaction
- ALT:
-
Alanine aminotransferase
- AOP:
-
Adverse outcome pathways
- APC:
-
Antigen-presenting cells
- AST:
-
Aspartate aminotransferase
- ATF6:
-
Activating transcription factor 6
- Atp5a1:
-
ATP synthase F1 subunit alpha
- Atp6v0d2:
-
ATPase H + transporting V0 subunit D2
- C1QA-C:
-
Complement C1q A chain
- CD14/68/74/163/205:
-
Cluster of differentiation 14/68/74/163/205
- CHOP:
-
C/EBP homologous protein
- COX 1/2:
-
Cyclooxygenase 1/2
- CXCL16:
-
C-X-C motif chemokine ligand 16
- CYP:
-
Cytochrome P450
- Cyp7a1:
-
Cytochrome P450 family 7 subfamily A member 1
- DAMPs:
-
Damage-associated molecular patterns
- DILI:
-
Drug-induced liver injury
- eIF2α:
-
Eukaryotic translation initiation factor 2A
- ER:
-
Endoplasmic reticulum
- Erk:
-
Extracellular signal-regulated kinase
- FasL:
-
Fas ligand
- Ghr:
-
Growth hormone receptor
- GSH:
-
Glutathione
- HIF1A:
-
Hypoxia inducible factor 1 subunit alpha
- HLA-DRB1:
-
Major histocompatibility complex, class II, DR beta 1
- IFNγ:
-
Interferon gamma
- IGF:
-
Insulin like growth factor 1
- IL4/10/13:
-
Interleukin 4/10/13
- Jak:
-
Janus kinase
- Jnk:
-
C-Jun N-terminal kinase
- KE:
-
Key event
- KER:
-
Key event relationship
- KLF6:
-
Kruppel like factor 6
- LBP:
-
Lipopolysaccharide binding protein
- LCN2:
-
Lipocalin 2
- LY6:
-
Lymphocyte antigen-6
- Mapk:
-
Mitogen-activated protein kinase 1
- MARCO:
-
Macrophage receptor with collagenous structure
- M-CSF:
-
Macrophage colony-stimulating factor
- MELD:
-
Model for end-stage liver disease
- MHC:
-
Major histocompatibility complex
- MIE:
-
Molecular initiating event
- MPO:
-
Myeloperoxidase
- MPTP:
-
Mitochondrial permeability transition pore
- Mrp2:
-
Multidrug resistance-associated protein 2
- Ndufb6:
-
NADH:Ubiquinone oxidoreductase subunit B6
- NGS:
-
Next-generation sequencing
- NK:
-
Natural killer
- NMDA:
-
N-Methyl-d-aspartate receptor
- NO:
-
Nitric oxide
- NSAID:
-
Non-steroidal anti-inflammatory drug
- PERK:
-
Protein kinase RNA-like ER kinase
- PI3K:
-
Phosphoinositide 3-kinases
- PPARɣ:
-
Peroxisome proliferator-activated receptor gamma
- Ptpn2:
-
Protein tyrosine phosphatase non-receptor type 2
- PSUR:
-
Periodic safety update report
- ROS:
-
Reactive oxygen species
- RUCAM:
-
Roussel Uclaf Causality Assessment Method
- S100a8/9:
-
S100 Calcium binding protein A8/9
- SAA:
-
Serum amyloid A
- SERPING1:
-
Serpin family G member 1
- Slc10a2/6:
-
Solute carrier family 10 member 2/6
- Slc3a1:
-
Solute carrier family 3 member 1
- Socs:
-
Suppressor of cytokine signaling
- SOD1:
-
Superoxide dismutase 1
- Stat3:
-
Signal transducer and activator of transcription 3
- TCA-3S:
-
Taurolithocholic acid-3-sulfate
- TNFα:
-
Tumor necrosis factor alpha
- UGT2B7:
-
UDP glucuronosyltransferase family 2 member B7
- UPR:
-
Unfolded protein response
- VCAM-1:
-
Vascular cell adhesion molecule 1
- WBC:
-
White blood cells
References
Adams DH, Ju C, Ramaiah SK, Uetrecht J, Jaeschke H (2010) Mechanisms of immune-mediated liver injury. Toxicol Sci 115:307–321. https://doi.org/10.1093/toxsci/kfq009
Aiba Y, Nakamura M (2013) The role of TL1A and DR3 in autoimmune and inflammatory diseases. Mediat Inflamm 2013:258164. https://doi.org/10.1155/2013/258164
Aithal GP (2011) Hepatotoxicity related to antirheumatic drugs. Nat Rev Rheumatol 7:139–150. https://doi.org/10.1038/nrrheum.2010.214
Akiyama T, Shinzawa M, Akiyama N (2012) RANKL-RANK interaction in immune regulatory systems. World J Orthop 3:142–150. https://doi.org/10.5312/wjo.v3.i9.142
Allen TE, Goodman JM, Gutsell S, Russell PJ (2014) Defining molecular initiating events in the adverse outcome pathway framework for risk assessment. Chem Res Toxicol 27:2100–2112. https://doi.org/10.1021/tx500345j
Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, Mount DR, Nichols JW, Russom CL, Schmieder PK, Serrrano JA, Tietge JE, Villeneuve DL (2010) Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem 29:730–741. https://doi.org/10.1002/etc.34
Banks AT, Zimmerman HJ, Ishak KG, Harter JG (1995) Diclofenac-associated hepatotoxicity: analysis of 180 cases reported to the Food and Drug Administration as adverse reactions. Hepatology 22:820–827
Basseri S, Austin RC (2012) Endoplasmic reticulum stress and lipid metabolism: mechanisms and therapeutic potential. Biochem Res Int 2012:841362. https://doi.org/10.1155/2012/841362
Begue B, Wajant H, Bambou JC, Dubuquoy L, Siegmund D, Beaulieu JF, Canioni D, Berrebi D, Brousse N, Desreumaux P, Schmitz J, Lentze MJ, Goulet O, Cerf-Bensussan N, Ruemmele FM (2006) Implication of TNF-related apoptosis-inducing ligand in inflammatory intestinal epithelial lesions. Gastroenterology 130:1962–1974
Bjorbaek C, Kahn BB (2004) Leptin signaling in the central nervous system and the periphery. Recent Prog Horm Res 59:305–331
Boelsterli UA (2003) Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol Appl Pharmacol 192:307–322
Boelsterli UA, Lim PL (2007) Mitochondrial abnormalities—a link to idiosyncratic drug hepatotoxicity? Toxicol Appl Pharmacol 220:92–107
Bort R, Mace K, Boobis A, Gomez-Lechon MJ, Pfeifer A, Castell J (1999) Hepatic metabolism of diclofenac: role of human CYP in the minor oxidative pathways. Biochem Pharmacol 58:787–796
Chen M, Suzuki A, Borlak J, Andrade RJ, Lucena MI (2015) Drug-induced liver injury: interactions between drug properties and host factors. J Hepatol 63:503–514. https://doi.org/10.1016/j.jhep.2015.04.016
Cottrell EC, Mercer JG (2012) Leptin receptors. Handb Exp Pharmacol 209:3–21. https://doi.org/10.1007/978-3-642-24716-3_1
Darnell M, Breitholtz K, Isin EM, Jurva U, Weidolf L (2015) Significantly different covalent binding of oxidative metabolites, acyl glucuronides, and S-Acyl CoA conjugates formed from xenobiotic carboxylic acids in human liver microsomes. Chem Res Toxicol 28:886–896. https://doi.org/10.1021/tx500514z
Deng X, Luyendyk JP, Ganey PE, Roth RA (2009) Inflammatory stress and idiosyncratic hepatotoxicity: hints from animal models. Pharmacol Rev 61:262–282. https://doi.org/10.1124/pr.109.001727
Denson LA, Menon RK, Shaufl A, Bajwa HS, Williams CR, Karpen SJ (2001) TNF-alpha downregulates murine hepatic growth hormone receptor expression by inhibiting Sp1 and Sp3 binding. J Clin Investig 107:1451–1458. https://doi.org/10.1172/JCI10994
Dutta NK, Mazumdar K, Seok SH, Park JH (2008) The anti-inflammatory drug Diclofenac retains anti-listerial activity in vivo. Lett Appl Microbiol 47:106–111. https://doi.org/10.1111/j.1472-765X.2008.02391.x
Fantuzzi G, Faggioni R (2000) Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol 68:437–446
Foufelle F, Fromenty B (2016) Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol Res Perspect 4:e00211. https://doi.org/10.1002/prp2.211
Franceschelli S, Moltedo O, Amodio G, Tajana G, Remondelli P (2011) In the Huh7 hepatoma cells diclofenac and indomethacin activate differently the unfolded protein response and induce ER stress apoptosis. Open Biochem J 5:45–51. https://doi.org/10.2174/1874091X01105010045
Fredriksson L, Wink S, Herpers B, Benedetti G, Hadi M, de Bont H, Groothuis G, Luijten M, Danen E, de Graauw M, Meerman J, van de Water B (2014) Drug-induced endoplasmic reticulum and oxidative stress responses independently sensitize toward TNFalpha-mediated hepatotoxicity. Toxicol Sci 140:144–159. https://doi.org/10.1093/toxsci/kfu072
Freneaux E, Fromenty B, Berson A, Labbe G, Degott C, Letteron P, Larrey D, Pessayre D (1990) Stereoselective and nonstereoselective effects of ibuprofen enantiomers on mitochondrial beta-oxidation of fatty acids. J Pharmacol Exp Ther 255:529–535
Fujiwara R, Yoda E, Tukey RH (2018) Species differences in drug glucuronidation: humanized UDP-glucuronosyltransferase 1 mice and their application for predicting drug glucuronidation and drug-induced toxicity in humans. Drug Metab Pharmacokinet 33:9–16
Gan TJ (2010) Diclofenac: an update on its mechanism of action and safety profile. Curr Med Res Opin 26:1715–1731. https://doi.org/10.1185/03007995.2010.486301
Ghosh R, Alajbegovic A, Gomes AV (2015) NSAIDs and cardiovascular diseases: role of reactive oxygen species. Oxid Med Cell Longev 2015:536962. https://doi.org/10.1155/2015/536962
Gomez-Lechon MJ, Ponsoda X, O'Connor E, Donato T, Castell JV, Jover R (2003a) Diclofenac induces apoptosis in hepatocytes by alteration of mitochondrial function and generation of ROS. Biochem Pharmacol 66:2155–2167
Gomez-Lechon MJ, Ponsoda X, O'Connor E, Donato T, Jover R, Castell JV (2003b) Diclofenac induces apoptosis in hepatocytes. Toxicol In Vitro 17:675–680
Guegan JP, Ezan F, Theret N, Langouet S, Baffet G (2013) MAPK signaling in cisplatin-induced death: predominant role of ERK1 over ERK2 in human hepatocellular carcinoma cells. Carcinogenesis 34:38–47. https://doi.org/10.1093/carcin/bgs317
Hammerich L, Tacke F (2014) Interleukins in chronic liver disease: lessons learned from experimental mouse models. Clin Exp Gastroenterol 7:297–306. https://doi.org/10.2147/CEG.S43737
Helfgott SM, Sandberg-Cook J, Zakim D, Nestler J (1990) Diclofenac-associated hepatotoxicity. JAMA 264:2660–2662
Hughes TB, Swamidass SJ (2017) Deep learning to predict the formation of quinone species in drug metabolism. Chem Res Toxicol 30:642–656. https://doi.org/10.1021/acs.chemrestox.6b00385
Iveson TJ, Ryley NG, Kelly PM, Trowell JM, McGee JO, Chapman RW (1990) Diclofenac associated hepatitis. J Hepatol 10:85–89
Kang SW, Haydar G, Taniane C, Farrell G, Arias IM, Lippincott-Schwartz J, Fu D (2016) AMPK activation prevents and reverses drug-induced mitochondrial and hepatocyte injury by promoting mitochondrial fusion and function. PLoS One 11:e0165638. https://doi.org/10.1371/journal.pone.0165638
Karin N (2010) The multiple faces of CXCL12 (SDF-1alpha) in the regulation of immunity during health and disease. J Leukoc Biol 88:463–473. https://doi.org/10.1189/jlb.0909602
Kay-Mugford P, Benn SJ, LaMarre J, Conlon P (2000) In vitro effects of nonsteroidal anti-inflammatory drugs on cyclooxygenase activity in dogs. Am J Vet Res 61:802–810. https://doi.org/10.2460/ajvr.2000.61.802
Knapen D, Angrish MM, Fortin MC, Katsiadaki I, Leonard M, Margiotta-Casaluci L, Munn S, O'Brien JM, Pollesch N, Smith LC, Zhang X, Villeneuve DL (2018) Adverse outcome pathway networks I: development and applications. Environ Toxicol Chem 37:1723–1733. https://doi.org/10.1002/etc.4125
Kolios G, Valatas V, Kouroumalis E (2006) Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol 12:7413–7420
Kong F, Guo X, Noel JG, Wells DA, Lovell GJ, Ogle CK (2002) Thermal injury-induced increases of hepatocyte SOCS3 lead to decreases in STAT3. Shock 18:374–379
Kretz-Rommel A, Boelsterli UA (1993) Diclofenac covalent protein binding is dependent on acyl glucuronide formation and is inversely related to P450-mediated acute cell injury in cultured rat hepatocytes. Toxicol Appl Pharmacol 120:155–161
Kyriakis JM, Avruch J (2012) Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev 92:689–737. https://doi.org/10.1152/physrev.00028.2011
Lagas JS, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH (2010) Hepatic clearance of reactive glucuronide metabolites of diclofenac in the mouse is dependent on multiple ATP-binding cassette efflux transporters. Mol Pharmacol 77:687–694. https://doi.org/10.1124/mol.109.062364
Laine L, Goldkind L, Curtis SP, Connors LG, Yanqiong Z, Cannon CP (2009) How common is diclofenac-associated liver injury? Analysis of 17,289 arthritis patients in a long-term prospective clinical trial. Am J Gastroenterol 104:356–362. https://doi.org/10.1038/ajg.2008.149
Lee EH, Oh JH, Selvaraj S, Park SM, Choi MS, Spanel R, Yoon S, Borlak J (2016) Immunogenomics reveal molecular circuits of diclofenac induced liver injury in mice. Oncotarget. https://doi.org/10.18632/oncotarget.7698
Ley K (2003) The role of selectins in inflammation and disease. Trends Mol Med 9:263–268
Malle E, Furtmuller PG, Sattler W, Obinger C (2007) Myeloperoxidase: a target for new drug development? Br J Pharmacol 152:838–854
Masubuchi Y, Nakayama S, Horie T (2002) Role of mitochondrial permeability transition in diclofenac-induced hepatocyte injury in rats. Hepatology 35:544–551
Miller AM (2011) Role of IL-33 in inflammation and disease. J Inflamm (Lond) 8:22. https://doi.org/10.1186/1476-9255-8-22
Moschen AR, Adolph TE, Gerner RR, Wieser V, Tilg H (2017) Lipocalin-2: a master mediator of intestinal and metabolic inflammation. Trends Endocrinol Metab 28:388–397
Muhl H, Pfeilschifter J (2003) Anti-inflammatory properties of pro-inflammatory interferon-gamma. Int Immunopharmacol 3:1247–1255
Naisbitt DJ, Sanderson LS, Meng X, Stachulski AV, Clarke SE, Park BK (2007) Investigation of the immunogenicity of diclofenac and diclofenac metabolites. Toxicol Lett 168:45–50
Nakagawa H, Maeda S (2012) Molecular mechanisms of liver injury and hepatocarcinogenesis: focusing on the role of stress-activated MAPK. Patholog Res Int 2012:172894. https://doi.org/10.1155/2012/172894
Nikolaou K, Sarris M, Talianidis I (2013) Molecular pathways: the complex roles of inflammation pathways in the development and treatment of liver cancer. Clin Cancer Res 19:2810–2816. https://doi.org/10.1158/1078-0432.CCR-12-1961
Njoku DB (2014) Drug-induced hepatotoxicity: metabolic, genetic and immunological basis. Int J Mol Sci 15:6990–7003. https://doi.org/10.3390/ijms15046990
Oda S, Shirai Y, Akai S, Nakajima A, Tsuneyama K, Yokoi T (2017) Toxicological role of an acyl glucuronide metabolite in diclofenac-induced acute liver injury in mice. J Appl Toxicol 37:545–553. https://doi.org/10.1002/jat.3388
Patel V, Sanyal AJ (2013) Drug-induced steatohepatitis. Clin Liver Dis 17(533–46):vii. https://doi.org/10.1016/j.cld.2013.07.012
Paz-Filho G, Mastronardi C, Franco CB, Wang KB, Wong ML, Licinio J (2012) Leptin: molecular mechanisms, systemic pro-inflammatory effects, and clinical implications. Arq Bras Endocrinol Metabol 56:597–607
Pessayre D (2007) Role of mitochondria in non-alcoholic fatty liver disease. J Gastroenterol Hepatol 22(Suppl 1):S20–S27
Pollesch NL, Villeneuve DL, O'Brien JM (2019) Extracting and benchmarking emerging adverse outcome pathway knowledge. Toxicol Sci 168:349–364. https://doi.org/10.1093/toxsci/kfz006
Przybylak KR, Cronin MT (2012) In silico models for drug-induced liver injury—current status. Expert Opin Drug Metab Toxicol 8:201–217. https://doi.org/10.1517/17425255.2012.648613
Pulli B, Ali M, Forghani R, Schob S, Hsieh KL, Wojtkiewicz G, Linnoila JJ, Chen JW (2013) Measuring myeloperoxidase activity in biological samples. PLoS One 8:e67976. https://doi.org/10.1371/journal.pone.0067976
Purcell P, Henry D, Melville G (1991) Diclofenac hepatitis. Gut 32:1381–1385. https://doi.org/10.1136/gut.32.11.1381
Radi ZA (2009) Pathophysiology of cyclooxygenase inhibition in animal models. Toxicol Pathol 37:34–46. https://doi.org/10.1177/0192623308329474
Radi ZA, Khan NK (2006) Effects of cyclooxygenase inhibition on the gastrointestinal tract. Exp Toxicol Pathol 58:163–173
Ramachandran R, Kakar S (2009) Histological patterns in drug-induced liver disease. J Clin Pathol 62:481–492. https://doi.org/10.1136/jcp.2008.058248
Ramm S, Mally A (2013) Role of drug-independent stress factors in liver injury associated with diclofenac intake. Toxicology 312:83–96. https://doi.org/10.1016/j.tox.2013.08.002
Ramm S, Morissey B, Hernandez B, Rooney C, Pennington SR, Mally A (2015) Application of a discovery to targeted LC-MS proteomics approach to identify deregulated proteins associated with idiosyncratic liver toxicity in a rat model of LPS/diclofenac co-administration. Toxicology 331:100–111. https://doi.org/10.1016/j.tox.2015.03.004
Roberts RA, Ganey PE, Ju C, Kamendulis LM, Rusyn I, Klaunig JE (2007) Role of the Kupffer cell in mediating hepatic toxicity and carcinogenesis. Toxicol Sci 96:2–15
Sahini N, Selvaraj S, Borlak J (2014) Whole genome transcript profiling of drug induced steatosis in rats reveals a gene signature predictive of outcome. PLoS One 9:e114085. https://doi.org/10.1371/journal.pone.0114085
Saiman Y, Friedman SL (2012) The role of chemokines in acute liver injury. Front Physiol 3:213. https://doi.org/10.3389/fphys.2012.00213
Sakai N, Van Sweringen HL, Schuster R, Blanchard J, Burns JM, Tevar AD, Edwards MJ, Lentsch AB (2012) Receptor activator of nuclear factor-kappaB ligand (RANKL) protects against hepatic ischemia/reperfusion injury in mice. Hepatology 55:888–897. https://doi.org/10.1002/hep.24756
Sallie R (1990) Diclofenac hepatitis. J Hepatol 11:281
Sasaki E, Yokoi T (2018) Role of cytochrome P450-mediated metabolism and involvement of reactive metabolite formations on antiepileptic drug-induced liver injuries. J Toxicol Sci 43:75–87. https://doi.org/10.2131/jts.43.75
Sawa Y, Arima Y, Ogura H, Kitabayashi C, Jiang JJ, Fukushima T, Kamimura D, Hirano T, Murakami M (2009) Hepatic interleukin-7 expression regulates T cell responses. Immunity 30:447–457. https://doi.org/10.1016/j.immuni.2009.01.007
Scharl M, Hruz P, McCole DF (2010) Protein tyrosine phosphatase non-receptor type 2 regulates IFN-gamma-induced cytokine signaling in THP-1 monocytes. Inflamm Bowel Dis 16:2055–2064. https://doi.org/10.1002/ibd.21325
Schmeltzer PA, Kosinski AS, Kleiner DE, Hoofnagle JH, Stolz A, Fontana RJ, Russo MW, Drug-Induced Liver Injury Network (DILIN) (2015) Liver injury from nonsteroidal anti-inflammatory drugs in the United States. Liver Int. https://doi.org/10.1111/liv.13032
Scialis RJ, Csanaky IL, Goedken MJ, Manautou JE (2015) Multidrug resistance-associated protein 3 plays an important role in protection against acute toxicity of diclofenac. Drug Metab Dispos 43:944–950. https://doi.org/10.1124/dmd.114.061705
Seitz S, Boelsterli UA (1998) Diclofenac acyl glucuronide, a major biliary metabolite, is directly involved in small intestinal injury in rats. Gastroenterology 115:1476–1482
Selvaraj S, Oh JH, Spanel R, Langer F, Han HY, Lee EH, Yoon S, Borlak J (2017) The pathogenesis of diclofenac induced immunoallergic hepatitis in a canine model of liver injury. Oncotarget 8:107763–107824. https://doi.org/10.18632/oncotarget.21201
Smith DA, Hammond T, Baillie TA (2018) Safety assessment of Acyl glucuronides-A simplified paradigm. Drug Metab Dispos 46:908–912. https://doi.org/10.1124/dmd.118.080515
Sriuttha P, Sirichanchuen B, Permsuwan U (2018) Hepatotoxicity of nonsteroidal anti-inflammatory drugs: a systematic review of randomized controlled trials. Int J Hepatol 2018:5253623. https://doi.org/10.1155/2018/5253623
Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD (2011) Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem Res Toxicol 24:1345–1410. https://doi.org/10.1021/tx200168d
Syed M, Skonberg C, Hansen SH (2016) Mitochondrial toxicity of diclofenac and its metabolites via inhibition of oxidative phosphorylation (ATP synthesis) in rat liver mitochondria: Possible role in drug induced liver injury (DILI). Toxicol In Vitro 31:93–102. https://doi.org/10.1016/j.tiv.2015.11.020
Takahashi Y (2017) The role of growth hormone and insulin-like growth factor-I in the liver. Int J Mol Sci. https://doi.org/10.3390/ijms18071447
Takayama F, Egashira T, Yamanaka Y (1994) Effect of diclofenac, a non-steroidal anti-inflammatory drug, on lipid peroxidation caused by ischemia–reperfusion in rat liver. Jpn J Pharmacol 64:71–78
Tang W (2003) The metabolism of diclofenac–enzymology and toxicology perspectives. Curr Drug Metab 4:319–329
Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, Asadi S, Vasiadi M, Weng Z, Miniati A, Kalogeromitros D (2012) Mast cells and inflammation. Biochim Biophys Acta 1822:21–33. https://doi.org/10.1016/j.bbadis.2010.12.014
Van Vleet TR, Liu H, Lee A, Blomme EAG (2017) Acyl glucuronide metabolites: implications for drug safety assessment. Toxicol Lett 272:1–7
Vinken M (2013) The adverse outcome pathway concept: a pragmatic tool in toxicology. Toxicology 312:158–165. https://doi.org/10.1016/j.tox.2013.08.011
Vinken M (2015) Adverse outcome pathways and drug-induced liver injury testing. Chem Res Toxicol 28:1391–1397. https://doi.org/10.1021/acs.chemrestox.5b00208
Vinken M, Knapen D, Vergauwen L, Hengstler JG, Angrish M, Whelan M (2017) Adverse outcome pathways: a concise introduction for toxicologists. Arch Toxicol 91:3697–3707. https://doi.org/10.1007/s00204-017-2020-z
Wong MM, Fish EN (2003) Chemokines: attractive mediators of the immune response. Semin Immunol 15:5–14
Yano A, Higuchi S, Tsuneyama K, Fukami T, Nakajima M, Yokoi T (2012) Involvement of immune-related factors in diclofenac-induced acute liver injury in mice. Toxicology 293:107–114. https://doi.org/10.1016/j.tox.2012.01.008
Zhang Z, Li LY (2012) TNFSF15 modulates neovascularization and inflammation. Cancer Microenviron 5:237–247. https://doi.org/10.1007/s12307-012-0117-8
Zhang B, Li Q, Shi C, Zhang X (2018) Drug-induced pseudoallergy: a review of the causes and mechanisms. Pharmacology 101:104–110. https://doi.org/10.1159/000479878
Zhu Y, Zhang QY (2012) Role of intestinal cytochrome p450 enzymes in diclofenac-induced toxicity in the small intestine. J Pharmacol Exp Ther 343:362–370. https://doi.org/10.1124/jpet.112.198077
Zwirner NW, Domaica CI (2010) Cytokine regulation of natural killer cell effector functions. BioFactors 36:274–288. https://doi.org/10.1002/biof.107
Acknowledgements
Open Access funding provided by Projekt DEAL.
Funding
The financial support from The Virtual Liver Network (Grant 031 6154) of the German Federal Ministry of Education and Research (BMBF) to JB is gratefully acknowledged. The cooperation between the Division of Toxicology, Korea Institute of Toxicology and the Institute for Pharmaco- and Toxicogenomics of Hannover Medical School is supported by a mobility program of the BMBF (Grant 01DR14013 to JB). Furthermore, the work was supported by a grant (NRF-2016M3A9C4953144, NRF-2014M3A7B6020163) from the Ministry of Science, ICT, and Future Planning and a general research grant from the Korea Institute of Toxicology to JHO. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Selvaraj, S., Oh, JH. & Borlak, J. An adverse outcome pathway for immune-mediated and allergic hepatitis: a case study with the NSAID diclofenac. Arch Toxicol 94, 2733–2748 (2020). https://doi.org/10.1007/s00204-020-02767-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-020-02767-6