
Abstract
The cerebral oxygen cascade includes three key stages: (a) convective oxygen delivery representing the bulk flow of oxygen to the cerebral vascular bed; (b) diffusion of oxygen from the blood into brain tissue; and (c) cellular utilisation of oxygen for aerobic metabolism. All three stages may become dysfunctional after resuscitation from cardiac arrest and contribute to hypoxic–ischaemic brain injury (HIBI). Improving convective cerebral oxygen delivery by optimising cerebral blood flow has been widely investigated as a strategy to mitigate HIBI. However, clinical trials aimed at optimising convective oxygen delivery have yielded neutral results. Advances in the understanding of HIBI pathophysiology suggest that impairments in the stages of the oxygen cascade pertaining to oxygen diffusion and cellular utilisation of oxygen should also be considered in identifying therapeutic strategies for the clinical management of HIBI patients. Culprit mechanisms for these impairments may include a widening of the diffusion barrier due to peri-vascular oedema and mitochondrial dysfunction. An integrated approach encompassing both intra-parenchymal and non-invasive neuromonitoring techniques may aid in detecting pathophysiologic changes in the oxygen cascade and enable patient-specific management aimed at reducing the severity of HIBI.
Similar content being viewed by others
The successful treatment of hypoxic–ischaemic brain injury will likely require a multi-pronged approach that aims to resolve dysfunction within each step of the oxygen cascade, including convective oxygen delivery, oxygen diffusion, and oxygen utilisation. Further, the timing of interventions after resuscitation from cardiac arrest and patient-specific pathophysiology must be considered in future studies. |
Introduction
In patients resuscitated from cardiac arrest, hypoxic–ischaemic brain injury (HIBI) is the primary cause of mortality [1, 2] and is associated with significant disability in survivors [1]. The pathophysiology of HIBI includes three phases: (1) global brain ischaemia occurring in the interval between circulatory arrest and the start of cardiopulmonary resuscitation (CPR) (no-flow); (2) global brain hypoperfusion occurring during CPR (low-flow); and (3) brain reperfusion after the return of spontaneous circulation (ROSC) [2]. A significant degree of HIBI occurs as a secondary injury after ROSC and part of this injury is associated with brain tissue hypoxia [3].
Observational studies have demonstrated a relationship between reductions in cerebral oxygen delivery (CDO2) due to arterial hypotension [4], anaemia [5], and hypocapnia [6] with adverse neurologic outcome following resuscitation. As such, significant focus has been placed on the post-resuscitation optimisation of CDO2 [2], although the mechanisms by which brain tissue hypoxia may persist after ROSC appear to be more complex [3].
The oxygen cascade encompasses oxygen transport from the atmosphere to mitochondria (Fig. 1). It requires integrating cardiorespiratory, microcirculatory, and cellular systems, and involves three key stages: (1) convective oxygen delivery, (2) diffusion of oxygen, and (3) cellular utilisation of oxygen. The lack of improved neurologic outcomes in HIBI trials attempting to optimise post-resuscitation CDO2 [7,8,9,10,11,12,13,14,15] (Table 1) may be explained by a disproportionate focus on solely optimising convective CDO2 without consideration of abnormalities in oxygen diffusion or utilisation. Further, integrating contemporary advances in our understanding of cerebrovascular physiology in health may provide improved contextualisation of the abnormalities seen in HIBI pathophysiology and help inform future clinical trial design. As such, we provide a review with three aims: (1) to review the cerebrovascular pathophysiology in humans with HIBI after cardiac arrest placed within the context of each stage of the oxygen cascade; (2) to review the utility of neuromonitoring techniques which assess the stages of the oxygen cascade; and (3) to highlight the clinical implications of dysfunction of the oxygen cascade for clinical management of HIBI patients and future research.

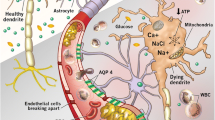
The oxygen cascade and oxygen delivery to the brain. A The oxygen cascade is a multi-step process involving the movement of oxygen from the atmosphere to the mitochondria. Oxygen transport depends upon both convective and diffusive oxygen delivery along the oxygen cascade and subsequent utilisation by the mitochondria. B Air is drawn into the lungs, where the partial pressure of inspired oxygen (PIO2) is ~ 150 mmHg. Subsequent mixing with residual volume renders an alveolar partial pressure of oxygen (PAO2) of ~ 103 mmHg, where at the alveolar–capillary junction oxygen then diffuses from the alveoli to the blood whilst carbon dioxide diffuses from the blood to the alveoli. This diffusion process is associated with a slight reduction in the partial pressure of arterial oxygen (PaO2) to approximately 98 mmHg. Thereafter, blood is pumped to the body by the heart. Importantly, cardiovascular (e.g., MAP), respiratory (e.g., PaO2/PaCO2), humoral (e.g., haemoglobin concentration, [Hb]), and microcirculatory (e.g., cerebrovascular resistance) factors influence CBF, which is determined by the integration of these physiologic factors and more [16]. Panel C depicts the neurovascular unit which is the anatomical and functional integration of cerebral microvasculature, peri-vascular glial cells and neurons, which ultimately maintains homeostasis in the brain parenchyma. D Convective oxygen delivery, denoted as (1), is determined by arterial oxygen content (CaO2) and cerebral blood flow (CBF). (2) Following oxygen delivery to the cerebral capillary network, where the partial pressure of capillary oxygen (PCO2) approximates 45 mmHg, oxygen diffusion from the cerebral vasculature to the cerebral parenchyma occurs. This diffusion process is determined by factors including the surface area for diffusion (A), the thickness of the diffusion barrier (T), and the pressure gradient for diffusion (ΔPO2), and results in a brain tissue partial pressure of oxygen (PbtO2) that is typically greater than 20 mmHg. Oxygen must then traverse the cytoplasm to reach the mitochondria, where the partial pressure of mitochondrial oxygen (PMITOO2) is 2–3 mmHg (estimation based upon measures of myoglobin saturation) [17]. (3) Finally, energetic homeostasis requires successfully utilising oxygen through aerobic mitochondrial respiration and generating adenosine triphosphate (ATP)
Stage 1: convective oxygen delivery
Convective CDO2 encompasses the circulatory system's delivery of oxygen from the pulmonary vasculature to the brain (Fig. 1). Convective CDO2 is the product of cerebral blood flow (CBF) and arterial oxygen content (CaO2), with the latter determined by arterial oxygen saturation (SaO2), haemoglobin (Hb) concentration, and, to a lesser extent, the partial pressure of arterial oxygen (PaO2) (Fig. 2). The physiologic components of convective CDO2 are summarised by Eq. 1

Regulation of cerebral blood flow and convective oxygen delivery. Panel A depicts the relationship between cerebral blood flow (CBF) and mean arterial blood pressure (MAP), the partial pressure of arterial carbon dioxide (PaCO2), and the partial pressure of oxygen (PaO2). During changes in blood pressure, the brain is more effective at combating increases as opposed to decreases in MAP. CBF changes linearly and proportionally to changes in PaCO2 until extreme levels of hypocapnia or hypercapnia. Decreases in PaO2 lead to a curvilinear increase in CBF in conjunction with the curvilinear nature of the oxyhaemoglobin dissociation curve. Panel B depicts the influence of PaO2 and haemoglobin concentration [Hb] on arterial oxygen content (CaO2). Separate lines for [Hb] concentrations are depicted for a haemoglobin concentration of 15 g/dL as well as two Hb thresholds that have been studied as transfusion thresholds in other patient groups in the intensive care unit. The minimal increase in CaO2 that results from supplemental oxygen leading to a PaO2 of up to 300 mmHg is also depicted. Panel C depicts a graphical overview of how increases and decreases in each of the factors depicted in panels A and B influence the overall convective cerebral delivery of oxygen (CDO2)
CBF is inversely proportional to cerebrovascular resistance (CVR) and proportional to cerebral perfusion pressure (CPP), which is the difference between the mean arterial pressure (MAP) and intracranial pressure (ICP).
Cerebral blood flow
Cerebrovascular resistance is increased following HIBI, which may be due to mechanisms encompassing cerebral endothelial dysfunction [18], pericyte constriction and death [19], oxidative stress, microvascular thrombosis in the setting of disseminated intravascular coagulopathy [20], and/or peri-vascular oedema resulting in microvascular collapse [21]. Worsening neurologic outcome with arterial hypotension [4] or sustained hypocapnia [6] suggests that reduced CBF during this period is injurious. Multiple physiologic mechanisms regulate CBF during physiologic perturbations [16, 22]. For the purposes of this review, clinically relevant CBF regulatory mechanisms include cerebral autoregulation [23] and cerebrovascular carbon dioxide reactivity [22, 24] (Fig. 2A).
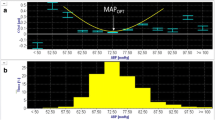
Cerebral autoregulation refers to intrinsic cerebral vasomotor responses that ‘buffer’ the influence of changes in MAP on CBF [23]. Pial arteriolar constriction and dilation in response to increases and decreases in MAP, respectively, were described as early as the 1930s [25, 26]. Thereafter, the notion that CBF is maintained constant between an MAP of 50–150 mmHg (i.e., Lassen’s curve) became very popular [27]. However, recent evidence [28,29,30,31] supports early critiques of Lassen’s curve [32] and it has been re-established that autoregulation only preserves CBF over a narrow plateau in health, which is not uniformly flat but often has a gradual upward slope [16, 30]. The magnitude by which MAP may be altered without a concomitant change in CBF ranges from approximately 10 to 20 mmHg (Fig. 2A) [30] and largely depends upon the rapidity of the change in MAP [33]. Importantly, the cerebral vasculature is more proficient at buffering increases rather than decreases in MAP in health, thereby rendering the brain vulnerable to ischaemia during hypotension [29, 33]. In health, the lower limit of autoregulation approximates 70 mmHg [16, 33, 34] and is highly variable; however, this limit is higher in HIBI patients [34,35,36,37], indicating a greater vulnerability for cerebral hypoperfusion. Using invasive neuromonitoring, the average lower limit of autoregulation has been observed to approximate 85 mmHg in HIBI patients, with significant inter-individual variability (range 60–100 mmHg) [38]. Clinically, this suggests that HIBI patients may experience cerebral hypoperfusion at standard MAP targets (i.e., > 65 mmHg) [39].
Authors have previously suggested that MAP augmentation may be an effective treatment strategy in the post-resuscitation setting [38, 40]. However, the neutral results of randomised control trials investigating MAP augmentation [7,8,9, 14], and lack of influence of MAP augmentation on neurologic outcome demonstrated in a recent meta-analysis [41], have led investigators to call into question the effectiveness of such an approach. Such discordance between the perceived importance of augmenting MAP and the lack of benefit for neurologic outcome demonstrated in clinical trials may be explained by dysfunction in the latter stages of the oxygen cascade (see Stage 2: oxygen diffusion & Stage 3: oxygen utilisation) and by individualised perfusion thresholds that may reflect patient-specific cerebrovascular physiology [42]. In addition to the potential influence of MAP augmentation on CDO2, MAP augmentation has been associated with improved renal function [8] and reduced myocardial injury [43]. Nevertheless, an ongoing multicenter international randomised control trial (STEPCARE: NCT05564754) will provide further insights into the impact of higher MAP targets on neurologic recovery in HIBI patients.
Another key regulator of CBF is PaCO2 [24]. In health, changes in PaCO2 cause directionally concordant changes in CBF. For every 1-mmHg change in PaCO2 above or below normal values, CBF increases by ~ 4–8% or decreases by ~ 1–4%, respectively (Fig. 2A) [44, 45]. Cerebrovascular CO2 reactivity regulates CBF throughout the cerebral vasculature [44, 46, 47], with the grey matter having a two- to three-fold higher cerebrovascular CO2 reactivity than the white matter [24, 48,49,50]. In healthy humans, normal cerebrovascular PaCO2 reactivity, as measured with transcranial Doppler ultrasound [i.e., Δ middle cerebral artery blood velocity (cm/s)/ΔPaCO2 (mmHg)], has been demonstrated to range from approximately 2.5–3.6 cm/s/mmHg [44, 51, 52]. However, in patients resuscitated from cardiac arrest, studies by Buunk et al. and [53] Bisschops et al. [54] reported values of 1.85 and 1.34 cm/s/mmHg PaCO2, respectively. These data suggest that cerebrovascular CO2 reactivity may be impaired in HIBI. Clinically, impaired CO2 reactivity may limit the ability of hypercapnia to improve convective CDO2. This may help explain why initial small clinical trials [10, 11] and the recently published TAME trial [55] did not demonstrate differences in neurologic outcome in HIBI patients with mild hypercapnia versus normocapnia. TAME randomised 1700 out-of-hospital cardiac arrest patients to mild hypercapnia (PaCO2 50–55 mmHg) vs normocapnia (PaCO2 35–45 mmHg) for 24 h post-ROSC. The primary outcome was favourable neurological outcome, defined as a Glasgow Outcome Scale-Extended ≥ 5. The trial did not demonstrate a difference in the favourable outcome rate of patients undergoing mild hypercapnia versus normocapnia (43.5% vs 44.6%, relative risk (RR) 0.98; 95% confidence interval (CI) 0.87–1.11; P = 0.76).
Arterial oxygen content
Maintenance and augmentation of arterial oxygen content have been extensively studied in critically ill patients in general, but only recently in HIBI [11,12,13, 56, 57]. Reductions in CaO2, secondary to anaemia or hypoxaemia (PaO2 < 60 mmHg) [22, 58] (Fig. 2B), increase CBF in health [22]. Specifically, a 1% reduction in CaO2 leads to a 2% increase in CBF [22]. The magnitude of this CBF response is sufficient to maintain convective CDO2 in health [22]. Whether HIBI alters this relationship is unknown. Hypoxaemia is associated with higher mortality in HIBI patients [59, 60]. However, the recent BOX trial comparing normal (e.g., PaO2 = 98–105 mmHg) versus restrictive arterial oxygen tension (e.g., PaO2 = 68–75 mmHg) did not demonstrate a difference in neurologic outcome [12], which may indicate that CDO2 is maintained in HIBI during modest reductions in PaO2 that remain above 60 mmHg.
Observational evidence suggests that severe hyperoxaemia (e.g., PaO2 > 300 mmHg) [59,60,61,62,63] is associated with worse outcomes after cardiac arrest, although specific harmful PaO2 thresholds are not well established [64]. A recent post hoc analysis of the TTM2 trial found that the best cut-off point associated with 6-month mortality for hyperoxaemia was 195 mmHg (RR 1.006, 95% CI 0.95–1.06) [60]. It has been suggested that hyperoxaemia may increase the production of reactive oxygen species worsening HIBI [65]. However, a post hoc analysis of COMACARE showed no difference in markers of cerebral lipid peroxidation between HIBI patients with a targeted PaO2 of 75–112 mmHg and 150–187 mmHg [66]. Further research is needed to elucidate the influence of moderate levels of oxygen supplementation on CDO2 in HIBI, and if individualised PaO2 goals are needed.
In addition to the influence of hyperoxaemia in the intensive care unit (ICU) setting, the influence of blood oxygen levels in the acute post-ROSC setting on convective CDO2 must be considered. The recent EXACT trial demonstrated that a modest reduction in blood oxygen levels (SpO2 97%) versus standard of care (SpO2 99%) did not significantly influence survival to hospital discharge (odds ratio [OR] 0.68 [95% CI 0.46–1.00]; P = 0.05) [13]. Further, there were no apparent differences in 12-month neurologic outcome in the patients that survived to hospital discharge or 12-month survival in all patients [13]. An important consideration is the extent to which PaO2 can improve the partial pressure of brain tissue oxygen (PbtO2) (see “Parenchymal brain tissue oxygenation”). Nonetheless, the findings of the EXACT trial do not support the use of lower oxygen targets in the pre-hospital phase following ROSC.
Whilst the CBF response during PaO2-dependent reductions in CaO2 (i.e., hypoxaemia) is sufficient—to a certain extent—to maintain CDO2, the CBF response to acute anaemia (e.g., decreased CaO2 from acute haemorrhage) is insufficient to maintain CDO2 (Fig. 2C) [67]. Therefore, acute anaemia may contribute to brain tissue hypoxia in HIBI patients [5] and worsen neurologic outcome after cardiac arrest [5, 68,69,70]. In an observational study on 118 patients with HIBI, higher mean haemoglobin concentration in the first 48 h and 7 days following cardiac arrest was associated with lower adjusted odds of unfavourable neurologic outcome at hospital discharge (OR 0.69/10 unit decrease in Hb, 95% CI 0.54–0.88, P < 0.01) [68].
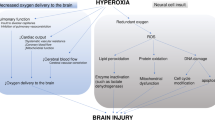
Clinical interventions aimed at optimising convective CDO2 continue to be a focus of active research in HIBI (Fig. 3). Conceptually, studies on the potential utility of MAP augmentation, mild hypercapnia, hyperoxaemia, and red blood cell transfusion are logical (Fig. 3A). However, their clinical efficacy has not been established (Table 1), suggesting that HIBI pathophysiology is more complex than dysregulation of only convective CDO2. Importantly, consideration of intracranial pressure and compliance in individual patients may be crucial in selecting the correct clinical interventions to augment convective CDO2. For example, in HIBI patients with elevated ICP, mild hypercapnia may lead to cerebral vasodilation, increased cerebrovascular blood volume [71], and intracranial hypertension [72, 73]. Similarly, such patients may also experience dangerous elevations in ICP with compensatory vasodilatory responses in the setting of severe hypoxaemia or reduced CPP. Thus, assessment of ICP (see “Neuromonitoring: intracranial pressure” below) is a key physiologic variable which must be accounted for in optimising convective CDO2 strategies after ROSC.

Therapies for hypoxic–ischemic brain injury in the context of the oxygen cascade. This figure illustrates the dysfunction of the oxygen cascade in hypoxic–ischaemic brain injury and the targeted therapies aimed at improving oxygen transport to the brain, brain oxygenation, and/or oxygen utilisation (mitochondrial function). A To therapeutically target convection oxygen delivery, MAP augmentation and hypercapnia aim to increase cerebral perfusion by increasing the hydraulic pressure head for flow and lower cerebral vascular resistance through CO2-mediated cerebral vasodilation, respectively. Conversely, hyperoxia and transfusion aim to increase oxygen content by increasing the pressure of dissolved O2 and haemoglobin concentration, respectively. B To therapeutically target diffusion limitations, hypertonic saline has been shown to reduce cerebral oedema as well as the oxygen gradient between cerebral venous blood and parenchyma, indicating improved oxygen diffusion. C Oxygen utilisation and mitochondrial dysfunction impairments have been well documented in HIBI and global ischemic brain disease models. Complex 1 generates excess ROS that leads to the dysfunction of key enzymes and metabolic processes within the TCA cycle. Further, increased calcium leads to mitochondrial efflux of cytochrome C and pro-apoptotic signalling. Experimental models have used dimethylmalonate to block excessive post-ischaemia oxidation of succinate, whilst MitoSNO and Rotenone have been used to selectively block the downstream ROS production by complex 1 following reverse electron transport. Cyclosporin A has been administered to inhibit the mitochondrial permeability transition pore and reduce cytochrome C's efflux and consequent apoptotic signalling. Finally, the co-factors thiamine and co-enzyme Q10 (Co-Q10) have been administered to restore metabolic function following ischaemia–reperfusion injury
Stage 2: oxygen diffusion
In a normal resting state, approximately 25% of oxygen carried to the brain diffuses into brain tissue. Consequently, normal cerebral venous haemoglobin oxygen saturation approximates 70–75% [74]. This relationship can be altered by reductions in CBF [45], when additional oxygen must be extracted from haemoglobin to maintain a constant cerebral metabolism. The diffusion of oxygen from the cerebral vasculature into brain tissue is governed by the biophysical principles of Fick’s law of diffusion, outlined in Eq. (2)
Specifically, the diffusion of oxygen is proportional to the surface area for diffusion (A), the diffusion coefficient (D), and the pressure gradient from the vasculature to tissue (ΔPO2). Most important to consider in the context of HIBI is that oxygen diffusion into the brain is inversely proportional to the thickness (T) of the diffusion barrier. However, it may be more appropriate to conceptualise this as the length of the diffusional path oxygen must take to successfully enter brain tissue. This barrier includes the cerebrovascular endothelium, vessel wall, and interstitial tissue, and the path length for diffusion may increase due to a multitude of factors including cerebral oedema. Further, regional microvascular shutdown due to microthrombosis or endothelial oedema may make areas of the brain dependent on oxygen that has to diffuse from distant capillaries that remain patent, with substantial increases in the path length for oxygen diffusion.
The concept of impaired oxygen diffusion from the blood into brain tissue (i.e., a diffusion limitation) as a pathophysiologic component of brain tissue hypoxia in acute brain injuries was first demonstrated by Menon et al. [21] in humans with traumatic brain injury. This study showed that the difference between the cerebral venous partial pressure of oxygen (PvO2) and brain tissue oxygen tension (PbtO2), termed the PvO2–PbtO2 gradient, was greater in patients with brain tissue hypoxia [21]. By acutely reducing CBF with a brief period of hypocapnia, the authors observed smaller increases in the cerebral oxygen extraction fraction in patients with brain tissue hypoxia compared to those with brain tissue normoxia, indicating the presence of impaired oxygen diffusion (7 ± 5% vs 16 ± 6%; P < 0.05) [21]. Similar observations have been made in humans with HIBI. Specifically, patients with post-resuscitation brain tissue hypoxia exhibit larger cerebral PvO2–PbtO2 gradients than those with brain tissue normoxia (39 mmHg [SD 11] vs 16 mmHg [SD 6]; P < 0.001) [75]. Moreover, increasing CPP was associated with a decrease in the PvO2–PbtO2 gradient in patients with brain tissue normoxia, whereby each 1 mmHg increase in CPP led to a 0.36 mmHg (95% CI 0.18–0.54, P < 0.001) decrease in the PvO2–PbtO2 gradient, indicating intact oxygen diffusion. Conversely, no relationship was observed between varying CPP and the PvO2–PbtO2 gradient in patients with brain tissue hypoxia (coefficient − 0.29, 95% CI − 0.17 to 0.11; P = 0.73), indicating a diffusion limitation [75].
There may be differential pathophysiologic phenotypes of HIBI, whereby some patients exhibit “perfusion-dependent” physiology (i.e., PbtO2 increases in response to augmented perfusion) [76]. In contrast, other patients exhibit “diffusion-limited” physiology (i.e., PbtO2 is unresponsive to augmented perfusion due to impaired oxygen diffusion) [76]. Clinically, HIBI patients exhibiting diffusion limitation would not exhibit increased brain tissue oxygenation with treatment approaches that aim to optimise convective CDO2 (e.g., MAP augmentation), rendering such interventions ineffective. Conversely, patients with intact diffusion of oxygen would likely exhibit improved brain tissue oxygen tension with convective CDO2 augmentation. Identifying these phenotypes in real time for bedside clinicians is a clear next step to facilitate individualised management paradigms in the post-resuscitation setting.
Research on interventions aimed at optimising oxygen diffusion is still in its preliminary phase. Diffusion limitation is associated with peri-vascular oedema on electron microscopy in humans [21, 77], which may be responsive to osmotherapy [78, 79]. Hypertonic saline reduces the PvO2–PbtO2 gradient and improves PbtO2 in HIBI patients with brain tissue hypoxia without significant changes in the other key physiologic variables, such as ICP, CPP, and MAP [3] (Fig. 3B). The decrease in the PvO2–PbtO2 gradient and concurrent improvement in PbtO2 provides preliminary evidence that osmotherapy may enhance oxygen diffusion into brain tissue by reducing the thickness of the diffusional barrier (Eq. 2). Although promising, considerable work remains to characterise this physiology further and evaluate its potential clinical efficacy.
Stage 3: oxygen utilisation
At the cellular level, oxygen utilisation relies upon intact mitochondrial function and metabolic pathways. The main substrate used by the brain is glucose; however, alternative metabolic substrates such as lactate and ketones may be preferentially metabolised by the injured brain [80]. Clinical trial evidence has shown that intensive glycaemic control (4–6 mmol/L) in critically ill patients is associated with increased mortality [81], and can cause metabolic crisis in patients with acute brain injury [82]. As such, it is imperative to avoid hypoglycaemia (< 4 mmol/L) which may expose the injured brain to neuroglycopenia, and there is arguably a case for maintaining high normal blood sugar levels to optimise glucose delivery to the brain. The cerebral metabolic rate of oxygen (CMRO2) of the grey matter is higher than that of the white matter [83]. Neurons and glia are heavily distributed in grey matter and require adequate adenosine triphosphate to support the generation of action potentials, post-synaptic ion fluxes, and maintenance of resting potentials [84]. Conversely, the sub-cortical white matter, comprised largely of myelinated axons, requires less oxygen for basal metabolic consumption [85]. This heterogeneity in metabolism leads to regional differences in the requirements for sufficient CDO2 and vulnerability to ischaemia [86]. CMRO2 can be calculated as per the Fick principle (Eq. 3) [74]
Alterations to cerebral metabolic function occur following cerebral ischaemia [87] (Fig. 3C). A historical study demonstrated that CMRO2 decreased to approximately 50% of normal in humans resuscitated from cardiac arrest [88]. Interestingly, measures of the ratio of the cerebral venous-to-arterial differences of oxygen and carbon dioxide (i.e., Cv-aCO2/Cv-aO2), a global estimate of the balance between aerobic and anaerobic metabolism, indicate that low CBF may not necessarily lead to anaerobic metabolism in HIBI [89]. It remains unclear whether reductions in CMRO2 with HIBI are a regulated and adaptive response to maintain cerebral flow-metabolism coupling during the hypoperfusion that ensures following cardiac arrest However, the evidence to date indicates that a higher CMRO2 is associated with survival [90]. Important considerations for the interpretation of reduced cerebral metabolism include that: (1) global CMRO2 may not reflect regional physiologic differences of susceptible anatomic foci that are injured in HIBI, (2) reductions in metabolism could be the result of a down regulation of metabolism or irreversible cell death, and (3) sedative administration in the ICU setting will influence CMRO2 independent from HIBI-related pathophysiologic processes.
Mechanistic explanations for cerebral metabolic dysfunction finding in HIBI include an impairment in glycolysis [87] stemming from essential co-factor (e.g., thiamine) depletion and dysfunction of key enzymes (e.g., pyruvate dehydrogenase) [91, 92]. Further, animal models have demonstrated an accumulation of succinate during ischaemia followed by rapid oxidation of succinate and reactive oxygen species generation consequent to reverse electron transport at complex 1 [93]. Interestingly, plasma succinate levels are higher in HIBI that do not survive than in HIBI survivors [94]. This reactive oxygen species generation further exacerbates mitochondrial dysfunction whereby intracellular calcium accumulation induces cytochrome C release from mitochondria [95] and initiates apoptotic signalling following global ischaemia [96]. Importantly, reductions in antioxidant defences [97] and increased oxidative stress [66, 98] are associated with mitochondrial dysfunction in HIBI. The influence of anaesthetic administration on CMRO2 during intensive care management must also be considered [99]. For example, whilst propofol [100] and midazolam [101] reduce both CBF and CMRO2, and thus maintain coupling between blood flow and metabolism, other anaesthetic agents, such as volatile anaesthetics, may lead to uncoupling of CBF and CMRO2 [99].
To improve the balance between CDO2 and O2 utilisation, moderate therapeutic hypothermia has been ubiquitously employed in post-resuscitation care [102, 103]. Yet, supporting evidence has been conflicting and the most recent and comprehensive TTM2 trial showed no benefit of hypothermia in HIBI [104]. Although therapeutic hypothermia has demonstrated some potential benefit in patients presenting with non-shockable rhythms [105], the precise patient population who may benefit from therapeutic hypothermia has not been yet identified [106]. Given the considerable systemic side effects of sustained hypothermia, alternate treatments to optimise O2 utilisation and mitochondrial function are being investigated.
The administration of metabolic co-factors, antioxidants, and other treatments targeting mitochondrial function has gained interest as potential therapeutic strategies. For example, high-dose thiamine administration in a pre-clinical HIBI model reduced neurologic injury [92]; however, two recent phase-2 randomised control trials investigating the efficacy of high-dose thiamine administration (NCT03450707 and NCT02974257) were terminated early for futility. Administration of co-enzyme Q10, an essential co-factor in the electron transport chain, has demonstrated a potential benefit in patients undergoing therapeutic hypothermia (35 °C for 24 h) [107]. However, another recent randomised control trial showed no difference in cerebral metabolism, neurological biomarkers, or clinical outcomes compared to placebo despite increased plasma co-enzyme Q10 levels in the treatment group [108]. Regarding antioxidants, pre-clinical studies indicate that vitamin C reduces reactive oxygen species following cardiac arrest [18] and improves outcomes [18, 109]. A phase-2 randomised control trial (NCT03509662) investigating the efficacy of Vitamin C administration in HIBI patients is underway. Finally, cyclosporine [110,111,112], an inhibitor of the mitochondrial permeability transition pore, did not confer improved survival to hospital discharge when given at the onset of advanced cardiovascular life support [113].
Pre-clinical studies have illuminated additional intriguing therapeutic targets [114], but human studies have yet to show significant clinical benefits from mitochondrial-targeted therapies. For example, increasing S-nitrosylation of mitochondrial complexes/enzymes improves outcome following cardiac arrest [98], presumably by reducing reverse electron transport-mediated generation of reactive oxygen species by complex 1 [93, 115] and/or protecting mitochondrial enzymes from irreversible oxidation during ischaemia–reperfusion [116]. Further, the administration of antioxidants reduces cerebral lipid peroxidation [97] and may provide a modest benefit to cerebral perfusion and metabolism [117]. However, the applicability of these therapies in human patients remains to be determined. Given the complexity of the cellular pathophysiology in HIBI, the concept of rational polytherapy has emerged as a therapeutic strategy [114]. In this instance, simultaneous administration of multiple agents to reverse pathophysiologic processes at the cellular level has been advocated [114]. However, considerable work remains for translation of this approach into humans with HIBI.
Assessing the O2 cascade through neuromonitoring
Key considerations for the interpretation of neuromonitoring techniques with relevance to the oxygen cascade are presented below (Fig. 4). Currently, most published literature on HIBI patients includes neuromonitoring techniques focussing on convective CDO2 or oxygen diffusion. However, studies including techniques assessing oxygen utilisation are underway. A prospective interventional study examining surrogates of mitochondrial function (cerebral lactate/pyruvate ratio) using intra-parenchymal microdialysis (NCT05390060) may shed light on the utilisation stage of the oxygen cascade in HIBI.

Measurement of cerebral oxygen delivery and oxygenation. Techniques employed to measure (or estimate) cerebral blood flow and oxygenation in HIBI patients are depicted, along with their pros and cons. The specific measurement principles of each technique should be considered when interpreting the influence of treatments aiming to improve cerebral oxygen delivery and/or cerebral oxygenation
Intracranial pressure
Coupled with measuring arterial blood pressure, ICP monitoring enables the continuous quantification of CPP, a key determinant of CBF and convective CDO2. Monitoring of ICP has traditionally been employed in traumatic brain injury management, but investigators have also started monitoring ICP in HIBI patients [3, 38, 75, 118]. There appears to be considerable patient heterogeneity regarding the burden of intracranial hypertension in HIBI, with the spectrum of disease severity encompassing normal ICP to fulminant cerebral oedema and brain death [119]. A recent prospective interventional study on a consecutive sample of HIBI patients demonstrated a mean ICP of 14 mmHg (SD 11) [119]. In this cohort, the percentage of time with ICP > 20 mmHg was 22% (range 0–100) during the monitoring period [119]. Importantly, these HIBI patients also exhibited limited compliance of the intracranial compartment presumably due to mild cerebral oedema. Therefore, HIBI patients with ‘normal’ ICP may remain at risk of developing intracranial hypertension if not managed with ICP lowering interventions [120]. Indeed, pre-clinical studies have shown that the administration of osmotherapy can attenuate cerebral oedema in HIBI [78, 79] and decrease brain injury biomarker release [121]. Monitoring ICP allows measurement of autoregulation indices, specifically the pressure reactivity index [38, 118] which may be used to estimate individualised optimal perfusion pressures. However, the clinical utility of these indices for patient management has yet to be determined [122]. Although ICP monitoring is an intriguing modality for use in HIBI, its invasive nature limits widespread implementation to guide HIBI management. An important limitation of the available literature describing ICP monitoring in HIBI is that it has largely been conducted in patients who present from non-cardiac causes of arrest (e.g., non-shockable rhythms) [118, 120]. In patients presenting with shockable rhythms, whose arrest is most often due to acute coronary occlusion, the consequent need for anti-platelet or anticoagulant therapy may preclude the implementation of invasive neuromonitoring. Considerable work remains to clarify the role of ICP monitoring in patients with HIBI and its indications and efficacy as part of critical care management after the return of spontaneous circulation.
Jugular venous bulb oximetry
Jugular venous bulb oximetry (SjvO2) measures the oxygen saturation of haemoglobin distal to the sigmoid sinus via an intravascular catheter placed retrograde in the dominant jugular vein. In a state of normal oxygen diffusion, SjvO2 can represent global cerebral haemodynamics by reflecting the overall balance between convective CDO2 and oxygen utilisation. However, when oxygen diffusion is abnormal in HIBI, increased SjvO2 may indicate underlying pathophysiologic processes, such as fulminant cerebral oedema, mitochondrial dysfunction [123], or widespread brain tissue death.
Increased SjvO2 is correlated to adverse neurologic outcome [124] and increased serum levels of neuron-specific enolase (NSE) [125], a biomarker of neuron cell body injury [126]. Richter et al. conducted a retrospective study of 40 out-of-hospital-cardiac-arrest patients in whom SjvO2 was intermittently sampled over 72 h after hospital admission [125]. They divided the participants into three study groups stratified by mean SjvO2 (Group 1: low SjvO2 < 55%; Group 2: SjvO2 55–75%; Group 3: SjvO2 > 75%). The authors found that 27/40 (68%) patients had mean SjvO2 > 75%, with the remaining exhibiting SjvO2 between 55 and 75% and none below 55% [125]. Further, they found that HIBI patients exhibiting SjvO2 55–75% had lower NSE levels compared to those with SjvO2 > 75% at 72 h (9 [interquartile range (IQR) 7–13] vs 46 [IQR 14–65] ng/mL; P < 0.01) [125]. Other studies integrating SjvO2 monitoring as part of a comprehensive neuromonitoring platform have found worse neurologic outcome in patients with elevated SjvO2 post-ROSC [75, 124].
Currently, the clinical utility of routine SjvO2 monitoring is unclear in HIBI. SjvO2 may provide insights into in vivo pathophysiology and help distinguish between HIBI patients with intact (low-normal SjvO2) or abnormal oxygen diffusion (high SjvO2). Whether or not increased SjvO2 in HIBI represents a sign of disease severity or could be a therapeutic goal remains to be seen and requires further study.
Parenchymal brain tissue oxygenation
The placement of a parenchymal brain tissue oxygen probe enables continuous assessment of brain tissue oxygen tension (i.e., PbtO2) within the sub-cortical white matter of the frontal lobe. A recent study by Sekhon et al. aimed to quantify the burden of brain tissue hypoxia in HIBI [38]. This prospective interventional study of invasive neuromonitoring found that patients spent ~ 40% (range 6–100%) of monitoring duration with a PbtO2 that is indicative of brain tissue hypoxia (less than 20 mmHg) [38]. Balu et al. demonstrated that a PbtO2 < 18 mmHg is associated with poor neurologic outcome in HIBI [118]. In a matched cohort study, Fergusson et al. stratified HIBI patients based on those who underwent management guided by PbtO2 (n = 21) versus standard of care (no PbtO2, n = 44) [119]. They observed that patients undergoing PbtO2 monitoring had a higher rate of favourable neurological outcome (cerebral performance category 1 or 2) than those without (44% vs 18%, P = 0.03). However, the small sample size and the post hoc design limit the strengths of this study [119].
Physiologically, PbtO2 reflects the balance between both convective CDO2 and O2 diffusion into the brain tissue and cerebral metabolism. A prospective study on HIBI patients undergoing PbtO2 monitoring demonstrated an association between increasing MAP and increased PbtO2 (R2 = 0.71, P < 0.001) [38]. However, the slope of the relationship between MAP and PbtO2 for each patient was heterogeneous [38]. This suggested diverse pathophysiologic HIBI phenotypes regarding coupling or uncoupling of convective CDO2 and O2 diffusion into the brain [75, 76] (see also the section “Stage 2: oxygen diffusion”). Therefore, the relationship between PbtO2 and other physiologic variables (e.g., MAP) provides insight into the functionality of oxygen diffusion into the brain.
Identifying patient-specific phenotypes with PbtO2 is clinically important, since patients exhibiting an uncoupling between convective CDO2 and PbtO2 would not likely benefit from MAP augmentation or other convective CDO2 focussed interventions [76]. Conversely, patients with intact O2 diffusion likely would benefit. As such, patient identification and selection stratified by physiologic phenotyping are key considerations for research using PbtO2 monitoring in HIBI. Future work in this area is needed to better understand the impact of post-resuscitation brain tissue hypoxia on neurologic outcome in HIBI, aid in determining methods to identify brain tissue hypoxia non-invasively and determine whether interventions that resolve brain tissue hypoxia are clinically efficacious.
An important limitation of PbtO2 monitoring that must be considered is the potential for confounding of the PbtO2 recording by dissolved oxygen within the cerebral vasculature. Whilst the catheter is thought to solely reflect tissue oxygen tension, it is likely unable to discriminate between the dissolved tension of oxygen within the brain parenchyma and within the microvasculature. Rosenthal et al. administered normobaric hyperoxia in humans undergoing multi-modal neuromonitoring following traumatic brain injury. The PaO2 increased from 127 (103–150) to 441 mmHg (363–518) and PbtO2 increased from 22.9 (17.2–28.6) to 77 mmHg (58.1–96). This increase in PbtO2 occurred despite a reduction in CBF from 23.9 (16.5–31.2) to 18.5 mL/100 g/min (12.2–24.8) [127]. That PbtO2 increased with normobaric hyperoxia despite a reduction in CBF and CDO2 likely indicates an independent effect of PaO2 on the recorded value of PbtO2. This notion has been supported by additional research [128].
Transcranial Doppler ultrasound
Transcranial Doppler ultrasound (TCD) may have a dual role in HIBI management and research: (1) measuring middle cerebral blood velocity to estimate CBF and (2) non-invasively estimating ICP. Thus, TCD provides estimates of physiologic variables that determine convective CDO2 (CBF and ICP). A linear relationship exists between CBF and flow velocities within the blood vessels insonated with TCD (e.g., middle cerebral artery), provided that the diameter of the vessel remains constant [129]. As such, TCD has been used as an indirect and non-invasive surrogate of CBF in HIBI [89, 90, 130,131,132,133]. Hoedemaekers et al. conducted a prospective observational study in 20 HIBI patients using TCD to estimate cerebral perfusion. The authors observed that the middle cerebral artery blood velocity of patients with HIBI (66 [59.5–73] years of age) was lower than healthy controls (28 ± 4.5 years of age) at 24 h (26 [18.6–40.4] vs. 59.1 cm/s [52.8–69], P < 0.001) but increased significantly at 72 h (63.9 cm/s [48.3–73.1]). Notwithstanding the potential influence of age on the lower CBF [134] observed in this study [89], these data suggest a dynamic nature of cerebral haemodynamics over time in HIBI. An ongoing clinical trial (NCT04000334) is assessing the feasibility of using TCD for goal-directed haemodynamic management in HIBI.
The second key role of TCD for neuromonitoring in HIBI is the non-invasive estimation of ICP [135]. An important TCD-derived variable for this is the pulsatility index (PI). The PI is calculated as the difference between the peak systolic and diastolic flow velocities, divided by the mean flow velocity and a PI above 1.2 suggests intracranial hypertension. A recent multicentre study in neurocritically ill patients evaluating the accuracy of ICP prediction based on diastolic flow velocity and mean arterial pressure demonstrated a good negative predictive value in ruling out intracranial hypertension [136]. A high PI and low diastolic blood velocity in patients with HIBI are associated with poor neurological outcome [133]. Cardim et al. conducted an agreement study between invasively monitored ICP and non-invasive surrogates, including TCD in HIBI patients [135]. The authors found a linear relationship between ICP measured with intra-parenchymal monitoring and non-invasive ICP (R = 0.3, P = 0.01) measured with TCD. The area under the receiver-operating characteristic (ROC) curve of TCD for predicting intracranial hypertension (ICP > 20 mmHg) was 0.91 (95% CI 0.83–1.00). Although useful and risk-free, the need for technical expertise, potential inter-observer error, and difficulties acquiring high-fidelity continuous recordings may limit the widespread use of TCD to guide management in HIBI. Further studies are needed to better establish the utility of TCD in HIBI patient management [130].
Near-infrared spectroscopy
Near-infrared spectroscopy (NIRS) monitors the regional saturation of oxygen (rSO2). Simply requiring the bilateral application of adhesive oximetry pads to a patient’s forehead, NIRS is non-invasive and does not require high technical expertise to use. The NIRS-dependent rSO2 value represents an estimate of the oxygen saturation of haemoglobin within the cerebrovascular compartment and assumes a 25:75 or 30:70 ratio of cerebral arteriole to venule blood volume in the interrogated region [137]. In other words, rSO2 should approximate the sum of 0.25*SaO2 and 0.75*SjvO2. The advantages of NIRS include its non-invasive application and its low-risk profile. NIRS can be implemented quickly in the post-ROSC setting compared to other neuromonitoring devices. However, technical and methodological limitations hinder the widespread use of NIRS for clinical decision-making. Specifically, contamination of the rSO2 signal by cutaneous blood, non-adherence of the monitoring pads to skin, and ambient light interference present challenges to the accuracy of NIRS [138]. Further, pathophysiologic considerations in HIBI, such as diffusion limitation, may also limit the accuracy of NIRS. In this instance, the uncoupling between CDO2 and brain tissue oxygenation precludes the normal assumption of NIRS that haemoglobin saturation within the cerebrovascular compartment is reflective of brain tissue oxygen tension [139]. For example, in patients with HIBI, NIRS does not change concordantly with PbtO2 during MAP augmentation [38], nor does it change concordantly with CBF in health and in patients with HIBI [140]. Further, poor agreement has been shown between NIRS-derived cerebral autoregulation indices compared to established indices generated with parenchymal neuromonitoring [140].
Clinical implications
The restoration of adequate CDO2 in HIBI has intuitive importance; however, key clinical considerations remain regarding the implementation of CDO2-based patient management strategies for therapeutic benefit. At present, clinical interventions that only target a single stage of the oxygen cascade are unlikely to provide therapeutic efficacy (Table 1). Therefore, combined approaches are likely required to simultaneously assess convective CDO2, diffusion of oxygen, and oxygen utilisation, to ensure these critical stages of the oxygen cascade function optimally. In this regard, a stepwise approach to patient management that applies multiple interventions for the purpose of targeting each stage of the oxygen cascade represents a promising path forward (Fig. 5). For example, optimising convective CDO2 (stage 1) with CBF augmenting interventions should be sought prior to or in parallel with improving oxygen diffusion (stage 2) and cellular oxygen utilisation (stage 3). Potential candidate interventions for each stage of the oxygen cascade are described in Fig. 5. Such an approach would necessitate multi-modal neuromonitoring to assess the function of each stage of the oxygen cascade and their response (or lack thereof) to intervention.

Integration of hypoxic–ischaemic brain injury pathophysiology, neuromonitoring, and possible management interventions. Primary injury is characterised by global cerebral ischaemia during the initial cardiac arrest. Thereafter, secondary injury mechanisms take place. The pathophysiologic changes occurring in the oxygen cascade pertaining to oxygen convection, diffusion, and utilisation by mitochondria are central to secondary injury in the post-ROSC setting. Neuromonitoring devices provide physiologic data pertaining to specific stages of the oxygen cascade. Interventions can be compartmentalised based on specific stages of the oxygen cascade at which their physiologic effect is exerted. CBF cerebral blood flow, Hb haemoglobin, HTS hypertonic saline, ICP intracranial pressure, L/P ratio lactate/pyruvate ratio, MAP mean arterial pressure, MCAv middle cerebral artery blood velocity, MMM multi-modal monitoring, NIRS near infrared spectroscopy, PaCO2 arterial carbon dioxide tension, PbtO2 brain tissue oxygen tension, rSO2 regional oxygen saturation, SjvO2 jugular venous bulb oximetry, TCD transcranial Doppler
The above approach may lay the foundation for ‘personalised’ post-resuscitative care but requires recognising differing pathophysiologic patient phenotypes to apply the appropriate interventions [76]. Unfortunately, no widely implementable technique is presently available to identify these phenotypes at the bedside to provide clinicians with immediately actionable real-time data. The development of non-invasive techniques to identify patient-specific pathophysiology is an essential avenue for future research. An impairment of oxygen diffusion may explain why not all HIBI patients benefit from augmented convective CDO2 [7, 8, 14, 55, 76]. Studies to date have only targeted convective CDO2 or implemented singular interventions [7,8,9,10,11,12,13,14, 56]. It is becoming increasingly clear that no single treatment can resolve HIBI, and bundle-based management interventions are likely needed [114]. Translational studies should focus on establishing the biological plausibility of oxygen cascade-based therapeutic strategies. Clinical trial design will likely require platform-based adaptive or factorial design methodologies to assess the clinical efficacy of combined interventions.
Finally, the timing of the restoration of oxygen delivery to the injured brain is likely key. The longer the delay between resuscitation and the implementation of interventions aimed at restoring CDO2, the less likely it is that the restoration of CDO2 will confer a clinical benefit. This is analogous to the stroke literature's well-defined “time is brain” concept. The specific timing of when and by what magnitude the efficacy of CDO2 restoration is diminished after the return of spontaneous circulation in HIBI is unknown but is clearly an important variable when designing future clinical trials.
Conclusions
The successful treatment of HIBI will likely require a multi-pronged approach. A greater understanding of the factors that lead to dysfunction within the oxygen cascade in HIBI is needed to develop strategies to optimise adequate CDO2 and cellular utilisation. Given the complexity of HIBI pathophysiology, it is likely that optimisation of cerebral oxygen cascade will need to be paired with other neuroprotective strategies to confer clinical efficacy for patients. Key variables such as the timing of implementation for clinical interventions after resuscitation from cardiac arrest and patient-specific pathophysiology must be considered in future studies to effectively determine the efficacy of CDO2 restoring interventions as part of HIBI resuscitation in the intensive care setting.
References
Secher N, Adelborg K, Szentkúti P et al (2022) Evaluation of neurologic and psychiatric outcomes after hospital discharge among adult survivors of cardiac arrest. JAMA Netw Open 5:e2213546. https://doi.org/10.1001/jamanetworkopen.2022.13546
Sandroni C, Cronberg T, Sekhon M (2021) Brain injury after cardiac arrest: pathophysiology, treatment, and prognosis. Intensive Care Med 47:1393–1414. https://doi.org/10.1007/s00134-021-06548-2
Hoiland RL, Ainslie PN, Wellington CL et al (2021) Brain hypoxia is associated with neuroglial injury in humans post-cardiac arrest. Circ Res 129:583–597. https://doi.org/10.1161/CIRCRESAHA.121.319157
Bhate TD, Mcdonald B, Sekhon MS, Griesdale DEG (2015) Association between blood pressure and outcomes in patients after cardiac arrest: a systematic review. Resuscitation 97:1–6. https://doi.org/10.1016/j.resuscitation.2015.08.023
Ameloot K, Genbrugge C, Meex I et al (2015) Low hemoglobin levels are associated with lower cerebral saturations and poor outcome after cardiac arrest. Resuscitation 96:280–286. https://doi.org/10.1016/j.resuscitation.2015.08.015
Schneider AG, Eastwood GM, Bellomo R et al (2013) Arterial carbon dioxide tension and outcome in patients admitted to the intensive care unit after cardiac arrest. Resuscitation 84:927–934. https://doi.org/10.1016/j.resuscitation.2013.02.014
Kjaergaard J, Møller JE, Schmidt H et al (2022) Blood-pressure targets in comatose survivors of cardiac arrest. N Engl J Med 387:1456–1466. https://doi.org/10.1056/nejmoa2208687
Ameloot K, De Deyne C, Eertmans W et al (2019) Early goal-directed haemodynamic optimization of cerebral oxygenation in comatose survivors after cardiac arrest: the neuroprotect post-cardiac arrest trial. Eur Heart J 40:1804–1814. https://doi.org/10.1093/eurheartj/ehz120
Grand J, Meyer AS, Kjaergaard J et al (2020) A randomised double-blind pilot trial comparing a mean arterial pressure target of 65 mm Hg versus 72 mm Hg after out-of-hospital cardiac arrest. Eur Heart J Acute Cardiovasc Care 9:S100–S109. https://doi.org/10.1177/2048872619900095
Eastwood GM, Schneider AG, Suzuki S et al (2016) Targeted therapeutic mild hypercapnia after cardiac arrest: a phase II multi-centre randomised controlled trial (the CCC trial). Resuscitation 104:83–90. https://doi.org/10.1016/j.resuscitation.2016.03.023
Jakkula P, Reinikainen M, Hästbacka J et al (2018) Targeting two different levels of both arterial carbon dioxide and arterial oxygen after cardiac arrest and resuscitation: a randomised pilot trial. Intensive Care Med 44:2112–2121. https://doi.org/10.1007/s00134-018-5453-9
Schmidt H, Kjaergaard J, Hassager C et al (2022) Oxygen targets in comatose survivors of cardiac arrest. N Engl J Med 387:1467–1476. https://doi.org/10.1056/nejmoa2208686
Bernard SA, Bray JE, Smith K et al (2022) Effect of lower vs higher oxygen saturation targets on survival to hospital discharge among patients resuscitated after out-of-hospital cardiac arrest: the EXACT randomized clinical trial. JAMA. https://doi.org/10.1001/jama.2022.17701
Jakkula P, Pettilä V, Skrifvars MB et al (2018) Targeting low-normal or high-normal mean arterial pressure after cardiac arrest and resuscitation: a randomised pilot trial. Intensive Care Med 44:2091–2101. https://doi.org/10.1007/s00134-018-5446-8
Young P, Mackle D, Bellomo R et al (2020) Conservative oxygen therapy for mechanically ventilated adults with suspected hypoxic ischaemic encephalopathy. Intensive Care Med 46:2411–2422. https://doi.org/10.1007/s00134-020-06196-y
Willie CK, Tzeng Y-C, Fisher JA, Ainslie PN (2014) Integrative regulation of human brain blood flow. J Physiol 592:841–859. https://doi.org/10.1113/jphysiol.2013.268953
Richardson RS, Noyszewski EA, Kendrick KF et al (1995) Myoglobin 02 desaturation during exercise. Evidence of limited 02 transport. J Clin Investig 96:1916–1926. https://doi.org/10.1172/JCI118237
Xiao Y, Su C, Zhang G et al (2022) Vitamin C improves the outcomes of cardiopulmonary resuscitation and alters shedding of Syndecan-1 and p38/MAPK phosphorylation in a rat model. J Am Heart Assoc. https://doi.org/10.1161/JAHA.121.023787
Hall CN, Reynell C, Gesslein B et al (2014) Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508:55–60. https://doi.org/10.1038/nature13165
Gando S, Wada T (2019) Disseminated intravascular coagulation in cardiac arrest and resuscitation. J Thromb Haemost 17:1205–1216. https://doi.org/10.1111/jth.14480
Menon DK, Coles JP, Gupta AK et al (2004) Diffusion limited oxygen delivery following head injury*. Crit Care Med 32:1384–1390. https://doi.org/10.1097/01.CCM.0000127777.16609.08
Hoiland RL, Bain AR, Rieger MG et al (2016) Hypoxemia, oxygen content, and the regulation of cerebral blood flow. Am J Physiol Regul Integr Comp Physiol 310:R398–R413. https://doi.org/10.1152/ajpregu.00270.2015
Claassen JAHR, Thijssen DHJ, Panerai RB, Faraci FM (2021) Regulation of cerebral blood flow in humans: physiology and clinical implications of autoregulation. Physiol Rev 101:1487–1559. https://doi.org/10.1152/physrev.00022.2020
Hoiland RL, Fisher JA, Ainslie PN (2019) Regulation of the cerebral circulation by arterial carbon dioxide. Compr Physiol. https://doi.org/10.1002/cphy.c180021
Fog M (1939) Cerebral circulation II: reaction of pial arteries to increase in blood pressure. Arch Neurol Psychiatry 41:260–268
Fog M (1937) Cerebral circulation: the reaction of the pial arteries to a fall in blood pressure. Arch Neurol Psychiatry 37:351–364
Lassen NA (1959) Cerebral blood flow and oxygen consumption in man. Physiol Rev 39:183–238
Lucas SJE, Tzeng YC, Galvin SD et al (2010) Influence of changes in blood pressure on cerebral perfusion and oxygenation. Hypertension 55:698–705. https://doi.org/10.1161/HYPERTENSIONAHA.109.146290
Numan T, Bain AR, Hoiland RL et al (2014) Static autoregulation in humans: a review and reanalysis. Med Eng Phys. https://doi.org/10.1016/j.medengphy.2014.08.001
Brassard P, Labrecque L, Smirl JD et al (2021) Losing the dogmatic view of cerebral autoregulation. Physiol Rep. https://doi.org/10.14814/phy2.14982
Zhang R, Zuckerman JH, Giller CA et al (1998) Transfer function analysis of dynamic cerebral autoregulation in humans. Am J Physiol Heart Circ Physiol 274:H233–H241. https://doi.org/10.1016/j.preghy.2015.09.001
Heistad DD, Kontos HA (2011) Cerebral circulation. Compr Physiol Suppl. https://doi.org/10.1002/cphy.cp020305
Tzeng Y, Willie CK, Atkinson G et al (2010) Cerebrovascular regulation during transient hypotension and hypertension in humans. Hypertension 56:268–273. https://doi.org/10.1161/HYPERTENSIONAHA.110.152066
Sundgreen C, Larsen FS, Herzog TM et al (2001) Autoregulation of cerebral blood flow in patients resuscitated from cardiac arrest. Stroke 32:128–132
Joshi B, Ono M, Brown C et al (2012) Predicting the limits of cerebral autoregulation during cardiopulmonary bypass. Anesth Analg 114:503–510. https://doi.org/10.1213/ANE.0b013e31823d292a
Drummond JC (2019) Blood pressure and the brain: how low can you go? Anesth Analg 128:759–771. https://doi.org/10.1213/ANE.0000000000004034
Stolze Larsen F, Skovgaard Olsen K, Adel Hansen B et al (1994) Transcranial Doppler is valid for determination of the lower limit of cerebral blood flow autoregulation. Stroke 25:1985–1988
Sekhon MS, Gooderham P, Menon DK et al (2019) The burden of brain hypoxia and optimal mean arterial pressure in patients with hypoxic ischemic brain injury after cardiac arrest. Crit Care Med 47:960–969
Nolan JP, Sandroni C, Böttiger BW et al (2021) European Resuscitation Council and European Society of Intensive Care Medicine guidelines 2021: post-resuscitation care. Intensive Care Med 47:369–421. https://doi.org/10.1007/s00134-021-06368-4
Rikhraj KJK, Wood MD, Hoiland RL et al (2020) Determining optimal mean arterial pressure after cardiac arrest: a systematic review. Neurocrit Care. https://doi.org/10.1007/s12028-020-01027-w
Rikhraj KJK, Ronsley C, Sekhon MS et al (2023) High-normal versus low-normal mean arterial pressure thresholds in critically ill patients: a systematic review and meta-analysis of randomized trials. Can J Anaesth. https://doi.org/10.1007/s12630-023-02494-3
Sekhon MS, Griesdale DE (2017) Individualized perfusion targets in hypoxic ischemic brain injury after cardiac arrest. Crit Care 21:1–5. https://doi.org/10.1186/s13054-017-1832-9
Ameloot K, Jakkula P, Hästbacka J et al (2020) Optimum blood pressure in patients with shock after acute myocardial infarction and cardiac arrest. J Am Coll Cardiol 76:812–824. https://doi.org/10.1016/j.jacc.2020.06.043
Willie CK, Macleod DB, Shaw AD et al (2012) Regional brain blood flow in man during acute changes in arterial blood gases. J Physiol 59014:3261–3275. https://doi.org/10.1113/jphysiol.2012.228551
Kety SS, Schmidt CF (1948) The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Investig 27:484–492
Coverdale NS, Gati JS, Opalevych O, Perrotta A, Shoemaker JK (2014) Cerebral blood flow velocity underestimates cerebral blood flow during modest hypercapnia and hypocapnia. J Appl Physiol (1985) 2014;117:1090–1096
Wolff HG, Lennox WG. The effect on pial vessels of variations in the oxygen and carbon dioxide content of the blood. Arch Neurol Psychiatry 1930;23:1097–1120
Ramsay SC, Murphy K, Shea SA et al (1993) Changes in global cerebal blood flow in humans: effect on regional cerebral blood flow during a neural activation task. J Physiol 471:521–534
Bhogal AA, Philippens MEP, Siero JCW et al (2015) Examining the regional and cerebral depth-dependent BOLD cerebrovascular reactivity response at 7T. Neuroimage 114:239–248. https://doi.org/10.1016/j.neuroimage.2015.04.014
Thomas BP, Liu P, Park DC et al (2014) Cerebrovascular reactivity in the brain white matter: magnitude, temporal characteristics, and age effects. J Cereb Blood Flow Metab 34:242–247. https://doi.org/10.1038/jcbfm.2013.194
Hoiland RL, Caldwell HG, Carr JMJR et al (2021) Nitric oxide contributes to cerebrovascular shear-mediated dilation but not steady-state cerebrovascular reactivity to carbon dioxide. J Physiol 600:1385–1403. https://doi.org/10.1113/JP282427
Carr JMJR, Ainslie PN, MacLeod DB et al (2023) Cerebral O2 and CO2 transport in isovolumic haemodilution: compensation of cerebral delivery of O2 and maintenance of cerebrovascular reactivity to CO2. J Cereb Blood Flow Metab 43:99–114. https://doi.org/10.1177/0271678X221119442
Buunk G, van der Hoeven JG, Meinders AE (1997) Cerebrovascular reactivity in comatose patients resuscitated from a cardiac arrest. Stroke 28:1569–1573. https://doi.org/10.1161/01.STR.28.8.1569
Bisschops LLA, Hoedemaekers CWE, Simons KS, van der Hoeven JG (2010) Preserved metabolic coupling and cerebrovascular reactivity during mild hypothermia after cardiac arrest. Crit Care Med 38:1542–1547. https://doi.org/10.1097/CCM.0b013e3181e2cc1e
Eastwood G, Nichol AD, Hodgson C et al (2023) Mild hypercapnia or normocapnia after out-of-hospital cardiac arrest. N Engl J Med. https://doi.org/10.1056/NEJMoa2214552
Young PJ, Bailey M, Bellomo R et al (2020) Conservative or liberal oxygen therapy in adults after cardiac arrest: an individual-level patient data meta-analysis of randomised controlled trials. Resuscitation 157:15–22. https://doi.org/10.1016/j.resuscitation.2020.09.036
Young P, Bailey M, Bellomo R et al (2014) HyperOxic Therapy OR NormOxic Therapy after out-of-hospital cardiac arrest (HOT OR NOT): a randomised controlled feasibility trial. Resuscitation 85:1686–1691. https://doi.org/10.1016/j.resuscitation.2014.09.011
Brown M, Wade J, Marshall J (1985) Fundamental importance of arterial oxygen content in the regulation of cerebral blood flow in man. Brain 108:81–93
Kilgannon JH, Jones AE, Shapiro NI et al (2010) Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. J Am Med Assoc 303:2165–2171
Robba C, Badenes R, Battaglini D et al (2022) Oxygen targets and 6-month outcome after out of hospital cardiac arrest: a pre-planned sub-analysis of the targeted hypothermia versus targeted normothermia after Out-of-Hospital Cardiac Arrest (TTM2) trial. Crit Care. https://doi.org/10.1186/s13054-022-04186-8
Elmer J, Scutella M, Pullalarevu R et al (2015) The association between hyperoxia and patient outcomes after cardiac arrest: analysis of a high-resolution database. Intensive Care Med 41:49–57. https://doi.org/10.1007/s00134-014-3555-6
Janz DR, Hollenbeck RD, Pollock JS et al (2012) Hyperoxia is associated with increased mortality in patients treated with mild therapeutic hypothermia after sudden cardiac arrest. Crit Care Med 40:3135–3139. https://doi.org/10.1097/CCM.0b013e3182656976
Pilcher J, Weatherall M, Shirtcliffe P et al (2012) The effect of hyperoxia following cardiac arrest—a systematic review and meta-analysis of animal trials. Resuscitation 83:417–422
Kilgannon JH, Jones AE, Parrillo JE et al (2011) Relationship between supranormal oxygen tension and outcome after resuscitation from cardiac arrest. Circulation 123:2717–2722. https://doi.org/10.1161/CIRCULATIONAHA.110.001016
Helmerhorst HJF, Schultz MJ, van der Voort PHJ et al (2015) Bench-to-bedside review: the effects of hyperoxia during critical illness. Crit Care 19
Humaloja J, Vento M, Kuligowski J et al (2021) High oxygen does not increase reperfusion injury assessed with lipid peroxidation biomarkers after cardiac arrest: a post hoc analysis of the COMACARE trial. J Clin Med. https://doi.org/10.3390/jcm10184226
Hoiland RL, MacLeod DB, Stacey BS et al (2023) Hemoglobin and cerebral hypoxic vasodilation in humans: evidence for nitric oxide-dependent and S-nitrosothiol mediated signal transduction. J Cereb Blood Flow Metab. https://doi.org/10.1177/0271678X231169579
Wormsbecker A, Sekhon MS, Griesdale DE et al (2017) The association between anemia and neurological outcome in hypoxic ischemic brain injury after cardiac arrest. Resuscitation. https://doi.org/10.1016/j.resuscitation.2016.12.010
Albaenia A, Eid SM, Akinyele B et al (2016) The association between post resuscitation hemoglobin level and survival with good neurological outcome following Out Of Hospital cardiac arrest. Resuscitation 99:7–12. https://doi.org/10.1016/j.resuscitation.2015.11.015
Johnson NJ, Rosselot B, Perman SM et al (2016) The association between hemoglobin concentration and neurologic outcome after cardiac arrest. J Crit Care 36:218–222. https://doi.org/10.1016/j.jcrc.2016.07.012
Ito H, Kanno I, Ibaraki M et al (2003) Changes in human cerebral blood flow and cerebral blood volume during hypercapnia and hypocapnia measured by positron emission tomography. J Cereb Blood Flow Metab 23:665–670. https://doi.org/10.1097/01.WCB.0000067721.64998.F5
Asgari S, Bergsneider M, Hamilton R et al (2011) Consistent changes in intracranial pressure waveform morphology induced by acute hypercapnic cerebral vasodilatation. Neurocrit Care 15:55–62. https://doi.org/10.1007/s12028-010-9463-x
Yoshihara M, Bandoh K, Marmarou A (1995) Cerebrovascular carbon dioxide reactivity assessed by intracranial pressure dynamics in severely head injured patients. J Neurosurg 82:386–393
Ainslie PN, Shaw AD, Smith KJ et al (2014) Stability of cerebral metabolism and substrate availability in humans during hypoxia and hyperoxia. Clin Sci (Lond) 126:661–670. https://doi.org/10.1042/CS20130343
Sekhon MS, Ainslie PN, Menon DK et al (2020) Brain hypoxia secondary to diffusion limitation in hypoxic ischemic brain injury postcardiac arrest. Crit Care Med 48:378–384. https://doi.org/10.1097/CCM.0000000000004138
Hoiland RL, Robba C, Menon DK, Sekhon MS (2020) Differential pathophysiologic phenotypes of hypoxic ischemic brain injury: considerations for post-cardiac arrest trials. Intensive Care Med 46:1969–1971. https://doi.org/10.1007/s00134-020-06200-5
Du T, Mestre H, Kress BT et al (2021) Cerebrospinal fluid is a significant fluid source for anoxic cerebral oedema. Brain. https://doi.org/10.1093/brain/awab293
Nakayama S, Migliati E, Amiry-Moghaddam M et al (2016) Osmotherapy with hypertonic saline attenuates global cerebral edema following experimental cardiac arrest via perivascular pool of aquaporin-4. Crit Care Med 44:e702–e710. https://doi.org/10.1097/CCM.0000000000001671
Nakayama S, Amiry-Moghaddam M, Ottersen OP, Bhardwaj A (2016) Conivaptan, a selective arginine vasopressin V1a and V2 receptor antagonist attenuates global cerebral edema following experimental cardiac arrest via perivascular pool of aquaporin-4. Neurocrit Care 24:273–282. https://doi.org/10.1007/s12028-015-0236-4
Oddo M, Vespa P, Menon DK (2019) Boosting the injured brain with supplemental energy fuels. Intensive Care Med 45:872–875. https://doi.org/10.1007/s00134-018-05517-6
NICE-SUGAR Study Investigators, Finfer F, Chittock DR et al (2009) Intensive versus conventional glucose control in critically ill patients. N Engl J Med 360:1283–1297. https://doi.org/10.1056/NEJMoa0810625
Vespa P, McArthur DL, Stein N et al (2012) Tight glycemic control increases metabolic distress in traumatic brain injury: a randomized controlled within-subjects trial. Crit Care Med 40:1923–1929. https://doi.org/10.1097/CCM.0b013e31824e0fcc
Cho J, Kee Y, Spincemaille P et al (2018) Cerebral metabolic rate of oxygen (CMRO2) mapping by combining quantitative susceptibility mapping (QSM) and quantitative blood oxygenation level-dependent imaging (qBOLD). Magn Reson Med 80:1595–1604
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21:1133–1145. https://doi.org/10.1097/00004647-200110000-00001
Harris JJ, Attwell D (2012) The energetics of CNS white matter. J Neurosci 32:356–371. https://doi.org/10.1523/jneurosci.3430-11.2012
Binks AP, Cunningham VJ, Adams L, Banzett RB (2008) Gray matter blood flow change is unevenly distributed during moderate isocapnic hypoxia in humans. J Appl Physiol (1985) 104:212–217. https://doi.org/10.1152/japplphysiol.00069.2007
Martin E, Rosenthal RE, Fiskum G (2005) Pyruvate dehydrogenase complex: metabolic link to ischemic brain injury and target of oxidative stress. J Neurosci Res 79:240–247
Beckstead JE, Tweed WA, Lee J, MacKeen WL (1978) Cerebral blood flow and metabolism in man following cardiac arrest. Stroke 9:569–573. https://doi.org/10.1161/01.str.9.6.569
Hoedemaekers CW, Ainslie PN, Hinssen S et al (2017) Low cerebral blood flow after cardiac arrest is not associated with anaerobic cerebral metabolism. Resuscitation 120:45–50. https://doi.org/10.1016/j.resuscitation.2017.08.218
Lemiale V, Huet O, Vigué B et al (2008) Changes in cerebral blood flow and oxygen extraction during post-resuscitation syndrome. Resuscitation 76:17–24. https://doi.org/10.1016/j.resuscitation.2007.06.028
Bogaert YE, Rosenthal RE, Fiskum G (1994) Postischemic inhibition of cerebral cortex pyruvate dehydrogenase. Free Radic Biol Med 16:811–820
Ikeda K, Liu X, Kida K et al (2016) Thiamine as a neuroprotective agent after cardiac arrest. Resuscitation 105:138–144. https://doi.org/10.1016/j.resuscitation.2016.04.024
Chouchani ET, Pell VR, Gaude E et al (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515:431–435. https://doi.org/10.1038/nature13909
Paulin Beske R, Henriksen HH, Obling L et al (2022) Targeted plasma metabolomics in resuscitated comatose out-of-hospital cardiac arrest patients. Resuscitation. https://doi.org/10.1016/j.resuscitation.2022.06.010
Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM (2002) Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J Neurochem 80:207–218
Sugawara T, Fujimura M, Morita-Fujimura Y et al (1999) Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 19:RC39
Stanimirovic DB, Markovic M, Micic DV et al (1994) Liposome-entrapped superoxide dismutase reduces ischemia/reperfusion “oxidative stress” in Gerbil Brain. Neurochem Res 19:1473–1478
Hayashida K, Bagchi A, Miyazaki Y et al (2019) Improvement in outcomes after cardiac arrest and resuscitation by inhibition of S-nitrosoglutathione reductase. Circulation 139:815–827. https://doi.org/10.1161/CIRCULATIONAHA.117.032488
Minhas JS, Rook W, Panerai RB et al (2020) Pathophysiological and clinical considerations in the perioperative care of patients with a previous ischaemic stroke: a multidisciplinary narrative review. Br J Anaesth. https://doi.org/10.1016/j.bja.2019.10.021
Oshima T, Karasawa F, Satoh T (2002) Effects of propofol on cerebral blood flow and the metabolic rate of oxygen in humans. Acta Anaesthesiol Scand 46:831–835
Hoffman WE, Albrecht RF, Miletich DJ et al (1986) Cerebrovascular and cerebral metabolic effects of physostigmine, midazolam, and a benzodiazepine antagonist. Anesth Analg 65:639–644
Bernard SA, Gray TW, Buist MD et al (2002) Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 346:557–563
Holzer M, Sterz F, Darby JM et al (2002) Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 346:549–556. https://doi.org/10.1056/NEJMoa012689
Dankiewicz J, Cronberg T, Lilja G et al (2021) Hypothermia versus normothermia after out-of-hospital cardiac arrest. New Engl J Med 384:2283–2294. https://doi.org/10.1056/nejmoa2100591
Lascarrou JB, Merdji H, le Gouge A et al (2019) Targeted temperature management for cardiac arrest with nonshockable rhythm. N Engl J Med 381:2327–2337. https://doi.org/10.1056/NEJMoa1906661
Sandroni C, Nolan JP, Andersen LW et al (2022) ERC-ESICM guidelines on temperature control after cardiac arrest in adults. Intensive Care Med 48:261–269. https://doi.org/10.1007/s00134-022-06620-5
Damian MS, Ellenberg D, Gildemeister R et al (2004) Coenzyme Q10 combined with mild hypothermia after cardiac arrest: a preliminary study. Circulation 110:3011–3016. https://doi.org/10.1161/01.CIR.0000146894.45533.C2
Holmberg MJ, Andersen LW, Moskowitz A et al (2021) Ubiquinol (reduced coenzyme Q10) as a metabolic resuscitator in post-cardiac arrest: a randomized, double-blind, placebo-controlled trial. Resuscitation 162:388–395. https://doi.org/10.1016/j.resuscitation.2021.01.041
Tsai MS, Huang CH, Tsai CY, et al (2014) Combination of intravenous ascorbic acid administration and hypothermia after resuscitation improves myocardial function and survival in a ventricular fibrillation cardiac arrest model in the rat. In: Academic emergency medicine. Blackwell Publishing Ltd, pp 257–265
Knapp J, Roewer J, Bruckner T et al (2015) Evaluation of cyclosporine a as a cardio- and neuroprotective agent after cardiopulmonary resuscitation in a rat model. Shock 43:576–581. https://doi.org/10.1097/SHK.0000000000000357
Cour M, Loufouat J, Paillard M et al (2011) Inhibition of mitochondrial permeability transition to prevent the post-cardiac arrest syndrome: a pre-clinical study. Eur Heart J 32:226–235. https://doi.org/10.1093/eurheartj/ehq112
Cour M, Abrial M, Jahandiez V et al (2014) Ubiquitous protective effects of cyclosporine A in preventing cardiac arrest-induced multiple organ failure. J Appl Physiol 117:930–936. https://doi.org/10.1152/japplphysiol.00495.2014.-Opening
Argaud L, Cour M, Dubien PY et al (2016) Effect of cyclosporine in nonshockable out-of-hospital cardiac arrest the CYRUS randomized clinical trial. JAMA Cardiol 1:557–565. https://doi.org/10.1001/jamacardio.2016.1701
Daniele SG, Trummer G, Hossmann KA et al (2021) Brain vulnerability and viability after ischaemia. Nat Rev Neurosci 22:553–572. https://doi.org/10.1038/s41583-021-00488-y
Xu J, Pan H, Xie X et al (2018) Inhibiting succinate dehydrogenase by dimethyl malonate alleviates brain damage in a rat model of cardiac arrest. Neuroscience 393:24–32. https://doi.org/10.1016/j.neuroscience.2018.09.041
Murphy E, Kohr M, Sun J et al (2012) S-nitrosylation: a radical way to protect the heart. J Mol Cell Cardiol 52:568–577
Cerchiari EL, Hoel TM, Safar P, Sclabassi RJ (1987) Protective effects of combined superoxide dismutase and deferoxamine on recovery of cerebral blood flow and function after cardiac arrest in dogs. Stroke 18:869–878
Balu R, Rajagopalan S, Baghshomali S et al (2021) Cerebrovascular pressure reactivity and intracranial pressure are associated with neurologic outcome after hypoxic–ischemic brain injury. Resuscitation 164:114–121. https://doi.org/10.1016/j.resuscitation.2021.04.023
Fergusson NA, Hoiland RL, Thiara S et al (2021) Goal-directed care using invasive neuromonitoring versus standard of care after cardiac arrest: a matched cohort study. Crit Care Med 49:1333–1346. https://doi.org/10.1097/CCM.0000000000004945
Sekhon MS, Griesdale DE, Ainslie PN et al (2019) Intracranial pressure and compliance in hypoxic ischemic brain injury patients after cardiac arrest. Resuscitation 141:96–103
Annoni F, Su F, Peluso L et al (2023) Hypertonic sodium lactate infusion reduces vasopressor requirements and biomarkers of brain and cardiac injury after experimental cardiac arrest. Crit Care 27:161. https://doi.org/10.1186/s13054-023-04454-1
Tas J, Beqiri E, Van Kaam RC et al (2021) Targeting autoregulation-guided cerebral perfusion pressure after traumatic brain injury (COGiTATE): a feasibility randomized controlled clinical trial. J Neurotrauma 38:2790–2800. https://doi.org/10.1089/neu.2021.0197
Coppler PJ, Emler J (2021) Optimizing cerebral oxygen delivery after cardiac arrest. Resuscitation 169:220–222. https://doi.org/10.1016/j.resuscitation.2021.10.011
Richter J, Sklienka P, Setra AE et al (2021) Is jugular bulb oximetry monitoring associated with outcome in out of hospital cardiac arrest patients? J Clin Monit Comput 35:741–748. https://doi.org/10.1007/s10877-020-00530-x
Richter J, Sklienka P, Chatterjee N et al (2021) Elevated jugular venous oxygen saturation after cardiac arrest. Resuscitation 169:214–219. https://doi.org/10.1016/j.resuscitation.2021.10.011
Hoiland RL, Rikhraj KJK, Thiara S et al (2022) Neurologic prognostication after cardiac arrest using brain biomarkers. JAMA Neurol 79:390–398. https://doi.org/10.1001/jamaneurol.2021.5598
Rosenthal G, Hemphill JC, Sorani M et al (2008) Brain tissue oxygen tension is more indicative of oxygen diffusion than oxygen delivery and metabolism in patients with traumatic brain injury. Crit Care Med 36:1917–1924. https://doi.org/10.1097/CCM.0b013e3181743d77
Hlatky R, Valadka AB, Gopinath SP, Robertson CS (2008) Brain tissue oxygen tension response to induced hyperoxia reduced in hypoperfused brain. J Neurosurg 108:53–58. https://doi.org/10.3171/JNS/2008/108/01/0053
Ainslie PN, Hoiland RL (2014) Transcranial Doppler ultrasound: valid, invalid, or both? J Appl Physiol 117:1081–1083. https://doi.org/10.1152/japplphysiol.00854.2014
Jha RM, Elmer J (2019) Transcranial Dopplers after cardiac arrest: should we ride this wave? Resuscitation 141:204–206
van den Brule JMD, Vinke E, van Loon LM et al (2017) Middle cerebral artery flow, the critical closing pressure, and the optimal mean arterial pressure in comatose cardiac arrest survivors—an observational study. Resuscitation 110:85–89. https://doi.org/10.1016/j.resuscitation.2016.10.022
Doepp F, Reitemeier J, Storm C et al (2014) Duplex sonography of cerebral blood flow after cardiac arrest—a prospective observational study. Resuscitation 85:516–521. https://doi.org/10.1016/j.resuscitation.2013.12.021
Rafi S, Tadie J, Gacouin A et al (2019) Doppler sonography of cerebral blood flow for early prognostication after out-of-hospital cardiac arrest: DOTAC study. Resuscitation 141:188–194. https://doi.org/10.1016/j.resuscitation.2019.05.024
Ainslie PN, Cotter JD, George KP et al (2008) Elevation in cerebral blood flow velocity with aerobic fitness throughout healthy human ageing. J Physiol 586:4005–4010. https://doi.org/10.1113/jphysiol.2008.158279
Cardim D, Griesdale DE, Ainslie PN et al (2019) A comparison of non-invasive versus invasive measures of intracranial pressure in hypoxic ischaemic brain injury after cardiac arrest. Resuscitation 137:221–228. https://doi.org/10.1016/j.resuscitation.2019.01.002
Rasulo FA, Calza S, Robba C et al (2022) Transcranial Doppler as a screening test to exclude intracranial hypertension in brain-injured patients: the IMPRESSIT-2 prospective multicenter international study. Crit Care. https://doi.org/10.1186/s13054-022-03978-2
Ito H, Ibaraki M, Kanno I et al (2005) Changes in the arterial fraction of human cerebral blood volume during hypercapnia and hypocapnia measured by positron emission tomography. J Cereb Blood Flow Metab 25:852–857. https://doi.org/10.1038/sj.jcbfm.9600076
Cardim D, Griesdale DE (2018) Near-infrared spectroscopy: unfulfilled promises. Br J Anaesth 121:523–526. https://doi.org/10.1016/j.bja.2018.05.058
Hoiland RL, Griesdale DE, Sekhon MS (2020) Assessing autoregulation using near infrared spectroscopy: more questions than answers. Resuscitation 156:280–281. https://doi.org/10.1016/j.resuscitation.2020.07.035
Hoiland RL, Sekhon MS, Cardim D et al (2020) Lack of agreement between optimal mean arterial pressure determination using pressure reactivity index versus cerebral oximetry index in hypoxic ischemic brain injury after cardiac arrest. Resuscitation 152:184–191. https://doi.org/10.1016/j.resuscitation.2020.03.016
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
CR reports speaker fees from Edwards Life Sciences and Masimo. DKM reports consulting fees from Neurotrauma Sciences, consulting fees and research support from Lantmannen AB and PressuraNeuro, and research support from GlaxoSmith Kline. GC reports research grants from Neuroptics and Integra, consulting fees from Neuroptics, Integra, and Invex, and honoraria from Neuroptics and Integra. CR and CS are members of the Editorial Board of Intensive Care Medicine. GC is the Editor-in-Chief of Intensive Care Medicine. RLH and MSS report no conflicts of interest, financial or otherwise.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hoiland, R.L., Robba, C., Menon, D.K. et al. Clinical targeting of the cerebral oxygen cascade to improve brain oxygenation in patients with hypoxic–ischaemic brain injury after cardiac arrest. Intensive Care Med 49, 1062–1078 (2023). https://doi.org/10.1007/s00134-023-07165-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-023-07165-x