
Abstract
The pyridazine ring is endowed with unique physicochemical properties, characterized by weak basicity, a high dipole moment that subtends π-π stacking interactions and robust, dual hydrogen-bonding capacity that can be of importance in drug-target interactions. These properties contribute to unique applications in molecular recognition while the inherent polarity, low cytochrome P450 inhibitory effects and potential to reduce interaction of a molecule with the cardiac hERG potassium channel add additional value in drug discovery and development. The recent approvals of the gonadotropin-releasing hormone receptor antagonist relugolix (24) and the allosteric tyrosine kinase 2 inhibitor deucravacitinib (25) represent the first examples of FDA-approved drugs that incorporate a pyridazine ring. In this review, the properties of the pyridazine ring are summarized in comparison to the other azines and its potential in drug discovery is illustrated through vignettes that explore applications that take advantage of the inherent physicochemical properties as an approach to solving challenges associated with candidate optimization.
Graphical Abstract

Similar content being viewed by others
Introduction
The pyridazine ring (1) is endowed with unique physicochemical properties that can render it an attractive heterocycle for drug design, advocated as a less lipophilic and advantageous substitute for the ubiquitous phenyl ring (2) or deployed as a replacement for homologous azines and azoles, either as a scaffolding element or a pharmacophoric moiety [1,2,3]. The physicochemical properties inherent to 1 and its fused homologues 12 and 13 distinguish it from the other azines 3–11 in a fashion that can be advantageous when deployed judiciously and make a compelling case for broader application of 1 as a privileged structural element in drug design [1, 3,4,5]. However, perhaps surprisingly, this heterocycle is not represented in the top 100 drugs of the total of 1175 molecules that were marketed prior to 2014 and there are only three drugs that contain 1 that have been approved by health authorities around the world [6,7,8,9,10]. The three approved pyridazine-containing drugs are the monoamine oxidase (MAO) inhibitor minaprine (14), the non-peptidic human gonadotropin-releasing hormone (GnRH) receptor antagonist relugolix (24) and the allosteric inhibitor of tyrosine kinase 2 (TYK2) deucravacitinib (25), all of which, interestingly, are based on a 3-aminopyridazine heterocycle as the core scaffolding element (Table 1) [6, 11, 12]. Minaprine (14) was approved in France in 1972 as an atypical antidepressant but was withdrawn in 1996 due to an unacceptable incidence of convulsions [10]. Relugolix (24) was approved by the U.S. Food and Drug Administration (FDA) in December, 2020 as a therapeutic agent for the treatment of advanced prostate cancer while deucravacitinib (25) was licensed by the FDA in September, 2022 to treat moderate-to-severe plaque psoriasis [11, 12]. Two marketed drugs contain the phthalazine heterocycle (12), the closely related antihypertensive agents hydralazine (16), which was approved by the FDA in 1953, and dihydralazine (17), which is licensed in Europe but has not been approved in the U.S., while the isomeric cinnoline ring system 13 is not currently represented in any marketed therapeutic agent, although a cinnolin-4(1H)-one moiety is embedded in the antibacterial agent cinoxacin (15) [6, 13, 14]. Other approved drugs that embed a pyridazine heterocycle include the pyridazine-3-one derivatives 18–21 and the fused imidazo[1,2-b]pyridazine ring system that is a structural feature of both the multi-targeted tyrosine kinase inhibitor ponatinib (22) and the survival of motor neuron 2-directed RNA splicing modifier risdiplam (23) (Table 1) [6].

In this review, we will focus on applications of 1, 12 and 13 and select homologs in drug design, with an emphasis on highlighting those examples where the physicochemical properties of the heterocyclic ring are used to advantage in molecular recognition or are exploited to solve a challenge encountered in the progression of a drug candidate into development. As a consequence, fused ring systems with a nitrogen atom at the bridgehead or molecules incorporating pyridazine-3-one moieties will be discussed only in an incidental fashion. It should be noted that the latter were heavily explored in the 1980s and 1990s as a pharmacophore for phosphodiesterase 3 (PDE3) inhibition, with cardiac insufficiency typically the primary therapeutic indication although many of these compounds also inhibited blood platelet aggregation [15]. Levosimendan (26), which incorporates an unusual phenylcarbonohydrazonoyl dicyanide moiety, emerged from those studies as drug that has been approved for marketing outside of the United States (U.S.) [16]. However, the biochemical pharmacology associated with 26 is complex, with the PDE3 inhibition secondary to its effect on sensitizing the cardiac contractile protein troponin C to Ca2+ and the opening of adenosine triphosphate (ATP)-dependent K+ channels located in vascular smooth muscle cells and on mitochondrial membranes [16, 17]. Adding further to the scenario of complicated pharmacology is the production of the active metabolite 27 in vivo [16, 17].

Physicochemical properties of pyridazine
The key physicochemical properties of 1 that impinge upon its productive deployment in drug design encompass the topology of the ring and its substituent vectors, the dipole moment, which manifests effects in both intermolecular and intramolecular interactions, the low intrinsic basicity (pKa), the H-bond acceptor potential (pKBHX), the H-bond donating potential of the ring C-H bonds, the topological polar surface area (TPSA), lipophilicity (cLog P), and the effects of electron withdrawal at the C-3 and C-6 positions, which modulates the properties of substituents [1, 5, 18,19,20,21,22,23,24,25,26,27,28,29,30,31]. The physicochemical data for 1 are compiled in Table 2 along with comparative data for benzene (2), the family of azines 3–11, phthalazine (12), cinnoline (13) and the azole isosteres of 1, 1,3,4-oxadiazole (28), 1,3,4-thiadiazole (29) and 1H-1,3,4-triazole (30), where the relevant data are available [32,33,34,35,36,37].

The hexagonal ring topology associated with 1 and the homologous azines 3–11 reflects the geometry of the six most prevalent ring systems found in 1175 drug molecules that were marketed prior to 2014, with benzene (538), pyridine (54), piperidine (54), piperazine (51), cyclohexane (38) and pyran (32) represented in a combined 767 (65%) compounds [7]. Pyrimidine (n = 32, 11th) and pyrazine (n = 6, 50th) are the only other azines represented in this list. The prevalence of this topological geometry has persisted, with benzene (n = 727), pyridine (n = 86), piperidine (n = 76), piperazine (n = 65) and cyclohexane (n = 47) identified as the top five rings found in molecules listed in the FDA Orange Book prior to January 2020 [9]. The pyran ring (n = 39) appeared as the 7th most prevalent ring with a frequency identical to that of cyclopropane (n = 39), while pyrimidine (n = 32, 11th), morpholine (n = 15, 25th) and pyrazine (n = 6, 50th) rings were also represented in the top 50 compounds [9]. However, an analysis of the 1034 drugs in clinical trials in the U.S. that were listed in the FDA Orange Book prior to January 2020 identified pyridazine (1) as the most prevalent ring system that was deemed as not represented in a marketed drug, with 9 examples, while the oxidized homolog 4,5-dihydropyridazin-3(2H)-one was the third most common ring system (n = 6), just behind the 4-membered oxetane ring (n = 7) [9]. Only the top nine ring systems were present in more than one drug candidate, with the vast majority of the novel ring systems represented only as singletons [9]. These data are suggestive of a rising appreciation of the advantageous properties of the pyridazine heterocycle in drug design that is beginning to be reflected in the landscape of emerging drugs, where the diversity of ring systems under evaluation is greater than that found in marketed drugs [9]. The prevalence of the hexagonal topology in marketed and investigational drugs clearly identifies it as a privileged geometry for the design of both scaffolds and pharmacophoric elements. In this context, 1 offers four vectors for decoration that, when presented in a divalent mode, allows sampling of ortho-, meta- and para-substituted topological dispositions. The physicochemical properties and exit vectors in 1 offer complementarity to its diazine isomers pyrimidine (4) and pyrazine (5), with both presumed to enjoy a higher prevalence in marketed drugs and investigational compounds as a reflection of their occurrence in nature, which is considerably higher than that for 1 [1].
The dipole moment of pyridazine (1) is the largest of the three diazine heterocycles and the magnitude is amplified by the additional nitrogen atoms present in 1,2,3-triazine (8) and 1,2,3,4-tetrazine (9) but not in 1,2,4-triazine (7), while the dipole moment is abrogated completely by the symmetry inherent to 1,2,4,5-tetrazine (11) [25, 38]. Fusion of a phenyl ring enhances the dipole moment, as exemplified by the increased values associated with phthalazine (12) and cinnoline (13), which also demonstrates an independence of the effect of ring fusion topology. The dipole moment values of monocyclic azine heterocycles are of importance in drug design because π-stacking interactions with aromatic rings, 9‑methyladenine and amide bonds, an important aspect of biological recognition, have been strongly correlated with dipole moments [38,39,40,41,42,43,44]. However, that correlation does not extend to fused azines, where other factors dominate, or to the stacking of heterocycles and fused heterocycles on salt bridges, although even in that context, 1 demonstrated the strongest calculated interaction energy amongst the ten azines studied [38, 45]. In a computational analysis of the stacking interactions between a wide range of heterocycles and the side chains of phenylalanine, tyrosine and tryptophan, where toluene, 4-methyl phenol and 3-methyl indole were used as structurally simplified surrogates, respectively, to facilitate the calculations, dipole interactions provided a reasonable correlation [41]. However, a more sophisticated analysis using symmetry-adapted perturbation theory (SAPT) revealed a prominent role for both electrostatic effects and dispersion interactions in the association [41].
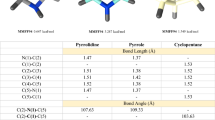
The basicity of 1 is modest (pKa = 2.0), particularly compared to pyridine (3) (pKa = 5.2), although it is more basic that the homologous diazines 4 and 5 and, interestingly, basicity is enhanced moderately by the phenyl ring fusion introduced in 12 and 13. This limits the opportunities for salt formation with 1 and its fused homologs to strong acids when compared with the more basic pyridine (3). However, 3-aminopyridazines exhibit enhanced basicity and minaprine (14) has been formulated as a dihydrochloride salt that is crystalline and exhibits excellent aqueous solubility, in contrast to the free base which is an oil [1, 46].
The H-bonding properties of 1 further emphasize its uniqueness within the azine series, with a pKBHX value that approaches that of pyridine (3) and which is enhanced in the fused-ring homolog phthalazine (12) [26,27,28]. Because of the low basicity associated with 1, H-bonding interactions are preserved at lower pH values than for 3, where protonation will obviate the H-bond acceptor properties of the single nitrogen atom. The robust H-bond accepting properties associated with 1 have been attributed to the adjacent lone pair effect, often referred to as the α effect, and the presence of lone pairs of electrons on adjacent atoms facilitates dual H-bonding interactions with biological targets that is a unique property within the azine series [27, 47,48,49,50,51]. While the H-bonding properties of the nitrogen atoms in 1 are identical, ring substitution will remove the degeneracy, as exemplified by the calculated pKBHX values for 4-dimethylaminopyridazine (31) where the H-bond accepting properties are enhanced in an asymmetrical fashion [27]. The pKBHX value calculated for N-1 of 31 is comparable to that measured for 4-dimethylaminopyridine (32, pKBHX = 2.80) but the basicity of the latter (pKa = 9.70) is considerably higher than that predicted for 31 for which the pKa value is expected to be similar to that measured for 4-aminopyridazine (pKa = 6.8) [27]. The C-3 hydrogen atom of 1 has been calculated to be a better C-H-bond donor than the C-2 H atom of pyridine (3) and both the C-2 and C-4 hydrogen atoms of pyrimidine (4) [28]. This is reflected in the predicted differences of the pKa values of the C-3 and C-2 C-H bonds, respectively, for the two heterocycles which are depicted in Fig. 1, with the difference attributed to the reduced aromaticity associated with the additional nitrogen atom present in 1 (the aromaticity index (IA) for 1 is 79 while for 3 the IA value is calculated to be 86, which compares to a benchmark of 100 for benzene) [52, 53]. However, in the case of 1, the C-4 hydrogen is calculated to be more acidic than that at C-3 while in pyridine (3) both the C-3 and C-4 hydrogen atoms are predicted to possess lower pKa values than the C-2 hydrogen atom [51]. The index of the electron withdrawing effect at the carbon atoms adjacent to the ring nitrogen atoms of 1–4, designated as \({{{\mathrm{C}}}}_{{{\mathrm{X}}}}^{{{{\mathrm{Ph}}}}}\) and derived from NMR studies, exhibits a reasonable correlation with the calculated C-H hydrogen-bond donor energies (R2 = 0.72) and the calculated bond dissociation energies (R2 = 0.86) that are compiled in Table 1 and plotted in Fig. 2 [54].

Calculated pKa values for C-H bonds in 1 and 3

Plot of electron withdrawing effect (\({{{\mathrm{C}}}}_{{{\mathrm{X}}}}^{{{{\mathrm{Ph}}}}}\)) versus the calculated C-H hydrogen-bond donor energy (A) and calculated bond dissociation energy (B) for pyridazine, pyridine, pyrimidine C-2, pyrimidine C-4, and benzene

The calculated lipophilicity data compiled in Table 1 indicate that introducing heteroatoms into a phenyl ring leads to a reduction in the cLog P value but there is no significant correlation with either TPSA or dipole moment [29, 55,56,57]. A matched molecular pair (MMP) analysis of the effect on measured Log D of adding an azine ring to a molecule is summarized in Fig. 3A whilst the effect on measured Log D of replacing a benzene ring with an azine heterocycle is captured in Fig. 3B [30, 55]. The effects of these molecular edits on the measured Log D values exhibits dependence on the point of attachment to the azine ring, with the greatest reduction in lipophilicity occurring when the azine nitrogen atoms are remote from the point of attachment and, thus, more exposed.

The strong H-bond accepting properties of 1 in the absence of overt basicity, the high dipole moment that contributes to reduced lipophilicity, and the electron deficiency at the C-3 and C-6 positions that can influence the properties of substituents, suggest potential advantage in drug design when deployed with the appropriate consideration. In a study of phenyl ring replacements assessed in the context of several drug developability parameters, a C-3-substituted pyridazine ring offered the optimal compromise of properties [55]. The 2.17 unit reduction in Log D7.4 associated this topological arrangement relative to benzene contributed to enhanced aqueous solubility and reduced human serum albumin (HSA) binding although, perhaps not surprisingly, membrane permeability was attenuated. Metabolic clearance in human liver microsomes (HLM) and rat liver microsomes (RLM) was reduced slightly, although the effects were within the margin of error, whilst inhibitory effects toward several cytochrome P450 (CYP 450) enzymes were not significant [55, 58]. This analysis confirmed an earlier study that assessed the pedigree of 19 heteroaromatic and heteroaliphatic rings for their potential to favorably affect aqueous solubility, HSA binding and CYP 450 inhibition. In this study, the presence of a pyridazine ring 1 in a molecule offered superior performance compared to pyridine (3), which fared poorly in the CYP 450 assay, as might be anticipated, and was, perhaps surprisingly based on its basicity, a less effective molecular edit in the solubility analysis [59]. This result was consistent with earlier studies that classified 1 as a poor CYP 450 inhibitor despite its potential to coordinate with metal ions [60,61,62]. Interestingly, the analysis of heterocycle pedigree separated the oxadiazole 28 from its less polar 1,2,4-isomer based on advantageous effects on aqueous solubility and protein binding, although it carried the burden of being more likely to interfere with CYP 450 function, observations that supported the results of a complementary study that focused specifically on analyzing the properties of this MMP of oxadiazole isomers [34, 59].
Pyridazines and intermolecular H-bonding in molecular recognition
The robust H-bonding potential of the ring nitrogen atoms in 1 has found gainful application in drug design and there are circumstances where both heteroatoms simultaneously engage a target protein in dual H-bonding interactions. This augments the potential applications of 1 which can, in principle, be mimicked by the azole homologs 28–30 and subtends the potential for bioisosteric relationships (vide infra). A chemotype where the dual H-bonding effects of 1 and 29 are prominent is in the family of allosteric inhibitors of kidney-type glutaminase (GLS1) represented by 33–38 in Fig. 4 [63, 64]. Glutaminase converts glutamine to glutamate and the altered metabolism of cancer cells leads to an increased reliance on GLS as an important source of glutamic acid [63, 64]. Bis-2-[5-(phenylacetamido)-1,3,4-thiadiazol-2-yl]ethyl sulfide (BPTES, 33) is the prototype GLS1 inhibitor, with two of these molecules found to bind across the dimer interface of a tetrameric form of the enzyme. Glutaminase exists in two isoforms, designated kidney glutaminase (KGA) and glutaminase C (GAC), that arise from alternative splicing but share in common the first 550 residues of the amino terminus [63, 64]. This class of allosteric GLS1 inhibitor binds to the enzyme activation loop, which is defined by residues Gly315-Glu325, stabilizing an open and inactive conformation of the enzyme [63,64,65,66,67]. In the cocrystal structures with KGA and GAC, 33 adopts a U-shaped conformation but the binding modes differ with respect to the orientation of the thiadiazole ring which in GAC engages the backbone N-Hs of Phe322 and Leu323 in both of the enzyme monomers whilst in KGA, the heterocycle is oriented to project the sulfur atom toward Phe322 and Leu323 [65,66,67,68]. In both cases, the N-Hs exocyclic to the thiadiazole rings establish H-bonds with the carbonyl moiety of Leu323 while the side chain amine of Lys320 associates with one of the terminal phenyl rings [67, 68]. The conformational flexibility associated with 33 appears to contribute to its relatively modest inhibitory potency since under the conditions associated with serial room temperature crystallography, it has been found to bind to GAC in an extended form that sacrifices some of the key drug-target interactions [67, 68].

Structure of BPTES (33) and related GLS inhibitors with associated inhibitory potency data
The structure-activity relationships (SARs) summarized in Fig. 4 indicate that the sulfide moiety of 33 can be replaced with a methylene (34) and that a pyridazine ring is an effective substitute for the thiadiazole ring, in both a symmetrical (35) and unsymmetrical configuration (36) [63, 69]. However, despite the structural resemblance and conservation of the key H-bonding elements, the oxadiazole ring system in 37 is incompatible with potent GLS inhibition, perhaps a reflection of geometrical differences between this heterocycle and 1 and 29 [5, 63, 64]. Two GLS inhibitors, telaglenastat (38, CB-839) and IPN60090 (39), have been advanced into clinical trials and both are based on a pyridazine heterocycle that is, interestingly, deployed differentially since the latter incorporates an amidotriazole ring as one of the dual H-bond acceptor elements and exploits a pyridazine ring as more of a scaffolding element [63, 64, 69, 70]. Although 38 continues to undergo clinical evaluation in several cancer indications, 39 appears to have been abandoned [71,72,73,74].
An interesting and productive example of taking advantage of the unique H-bonding properties of 1 and the homologs 28 and 29 is provided by a series of mechanism-based inhibitors of the cysteine protease cathepsin L (Cat L) that were pursued based on the potential involvement of this enzyme in osteoarthritis, osteoporosis, autoimmune disorders and cancer [75]. As part of the optimization campaign, avoiding inhibition of cathepsins B (Cat B) and L2 (Cat L2) was given priority since they were considered to be important and potentially problematic anti-targets [75]. An analysis of the X-ray cocrystal structure of the lead inhibitor 40 bound to Cat L indicated that Met70 and Asp71, which are located close to the methyl substituent bound to the meta-position of the phenylalanine moiety that occupies the S2 pocket, presented their backbone N-Hs in a topologically aligned fashion. This was viewed as an opportunity to further optimize for potency over Cat B where Met70 is replaced by a proline. The concept pursued in the context of a less potent (~30-fold) phenyl substituent at P3 was to introduce heterocycles at the 3-position of the phenylalanine ring that would be able to simultaneously engage the N-Hs of both Met70 and Asp71 of the Cat L enzyme, with oxadiazole, thiadiazole and pyridazine amongst the heterocycles explored [75]. The inhibitory profiling data compiled in Table 3 for 40–46 indicate that the effects of this kind of molecular edit were more pronounced for inhibition of Cat S than for Cat L or Cat L2, with the selectivity over Cat B preserved [75]. For the isomeric pyridazines 44 and 45, there was a clear difference in inhibitory potency toward 3 of the 4 enzymes that favored the topology presented by 44 and which was anticipated in the design process, despite the preservation of one H-bond acceptor with the potential to engage the enzyme in 45. However, differences in the conformation at the pseudo-biphenyl junction of the azine 45 influenced by the presence of the hydrogen atom at C-3 may play a contributory role. The reduced enzyme inhibitory potency observed with 45 was essentially mirrored by the pyridine 46. Further optimization of the series focused on the 1,3,4-oxadiazole chemotype, with the introduction of a basic amine designed to engage the side chain acids of Asp71 and Asp114 that, in the context, of 47 offered an eightfold enhancement of inhibitory potency toward Cat L while Cat S inhibition was reduced by 30-fold [75]. The X-ray cocrystal structure of with 47 with Cat L indicated engagement of Asp71 and Asp114 by the primary amine while the 1,3,4-oxadiazole nitrogen atoms interacted with the N-Hs of both Met70 and Asp71 (Fig. 5A). As an extension of that cocrystal structure data, the proposed dual H-bond bonding interactions between the pyridazine of 44 and Cat L are depicted in Fig. 5B.

A X-ray cocrystal structure of Cat L with 47 (3HWN). B Proposed binding mode for 44 to Cat L depicting the dual H-bonds from the enzyme protein to the pyridazine ring based on the cocrystal structure data obtained with 47


A pyridazine heterocycle is the structural hallmark of a series of human rhinovirus (HRV) capsid inhibitors that originate with R-61837 (48) and encompass BTA-188 (50) and the clinically-evaluated homologs pirodavir (49) and vapendavir (51) [76,77,78,79,80,81,82,83,84,85,86]. An X-ray cocrystal structure of 48 with HRV14 revealed that the compound bound in a lipophilic pocket below the depression that encircles the fivefold axes of the icosahedral protein shell of the virus [77]. Modeling poses suggested that the pyridazine nitrogen atom of 48 that is distal from the piperazine scaffold engaged the HRV capsid protein via a H-bonding interaction with an intervening H2O molecule that, in turn, interacted with the backbone N-Hs of Leu106 and Asn219, with details of the drug-target interactions captured in Fig. 6. In addition, the pyridazine ring of 48 appeared to π-stack with the phenol ring of Tyr197 [77]. Pirodavir (49) was advanced into clinical trials where frequent intranasal administration demonstrated beneficial effects on reducing virus shedding in experimental infections [80]. However, the drug was ineffective at reducing clinical symptoms in naturally-occurring rhinovirus infections when administered intranasally six times a day, with dosing initiated within 2 days of symptom onset [81]. The limited clinical efficacy associated with 49 has been attributed to rapid hydrolysis of the ester moiety in vivo to the carboxylic acid, which is essentially inactive as an antiviral agent [87]. This observation inspired the design of ester bioisosteres that would be more metabolically stable in vivo, with the oxime BTA-188 (50) an early iteration that was further refined into the 3-ethoxybenzisoxazole moiety found in vapendavir (BTA-798, 51) [82,83,84,85,86]. Vapendavir (51) demonstrated better metabolic stability than 50, which translated into an improved in vivo PK profile in preclinical species and targeted exposure in humans in a Phase 1 clinical study [85]. Although 51 was claimed to demonstrate clinical efficacy in an experimental rhinovirus infection, specific details have not been disclosed; however, development of the drug is continuing following its licensing in 2021 to Altesa Biosciences [88].

Key contacts between 48 and the HRV capsid protein in the X-ray cocrystal structure (1R09)

An application of the pyridazine ring where the unique H-bonding properties are hypothesized to play a role in molecular recognition has been provided by an investigation of peptide nucleic acids (PNAs) that assemble into a triplex structure with a double-stranded ribonucleic acid (dsRNA) hairpin construct HRPC [89]. This study focused on targeting a cytosine-guanosine (C-G) inversion in HRPC with PNAs that incorporate H-bond acceptors designed to engage the exocyclic NH2 of cytosine. A systematic study of azine heterocycles installed at the PN moiety of the 9-mer PNA NH2-Lys-MTMTMPNTMMCONH2, where M is the 2-aminopyridine-based PNA 52 that is believed to engage a G-C pair by the complementary H-bonding interactions depicted in Fig. 7. In the PNA construct 53, the lysine moiety at the amino terminus contributes to binding affinity whilst also promoting cell permeability [89].

Proposed molecular recognition of a H-bonded G-C pair by the 2-aminopyridine-based PNA element 52
The results of the survey of the azine-based elements 55–63 are summarized in Fig. 8, which includes the prototype 54 and two control elements, the acetamide 64 and phenylacetamide 65. The association constants (Ka) for HRPC were determined using isothermal calorimetry (ITC) while thermal melting temperatures (Tm) were measured by an ultraviolet method (UV) at 300 nM [89]. Both assays indicated that the majority of the constructs formed a triple helical structure with HRPC but there was a poor correlation between the Ka and Tm values. The pyridazine derivative 63 formed the most stable triple complex based on the high Ka and Tm values and was markedly superior to the isomer 62, reflecting a preference for the specific topology presented by 63. While the reduced Ka and Tm values for the phenyl homolog 65 provided a reference point for the absence of H-bond acceptors, the SARs presented in Fig. 7 did not provide a clear insight into the effects of the H-bonding interactions of individual heterocycles. This was attributed to a molecular association that also depended on π-stacking interactions, a hypothesis further explored by incorporating 54, 58, 59, 61 and 63 into four PNA constructs which were assessed for their association with complementary HRP hairpin sequences. In this study, the pyridazine derivative 63 was consistently superior to the other four azine-based molecules, a result reinforced by further studies of additional PNA sequences and HRP constructs. However, aspects of the H-bonding properties of the azine heterocycles in PNAs remained imprecise and the high Ka and Tm values recorded for the simple acetamide 64 suggested that the presence of the larger 6-membered rings in 54–63 and 65 may incur a steric penalty [89]. Nevertheless, the pyridazine ring in 63 appeared to provide the optimal compromise of properties as a PNA motif with complementarity to a C-G inversion, with the dual H-bonding effects summarized in Fig. 9 hypothesized to play a role in molecular recognition.

Azine heterocycles and control motifs explored as PN in the context of the PNA NH2-Lys-MTMTMPNTMMCONH2 with association constants (Ka) measured by ITC and Tm measured by UV at 300 nM

Hypothesized mode of interaction of the pyridazine-containing PNA residue 63 with the exocyclic amine moiety of cytosine (C) in a C-G inversion
An interesting and practically useful application of the H-bonding effects of a pyridazine-based heterocycle on kinase inhibitor selectivity has been observed in the context of a series of phthalazine-derived p38 mitogen activated protein kinase (MAPK) inhibitors, of which 67 and 68 are seminal representatives [90,91,92]. These compounds and the isoquinoline 66 are competitive inhibitors of several kinases that function by binding to the ATP recognition pocket and engaging with the backbone of the hinge residues through H-bonding interactions. The drug-target interactions that have been proposed to stabilize the complex between 66 and cKit are depicted in Fig. 10A, with the isoquinoline nitrogen atom hypothesized to accept a H-bond from the backbone N-H of Cys673 while the adjacent C-H can engage the amide C = O of the same residue in a C-H H-bonding interaction [91]. The isoquinoline 66 is a potent MAPK inhibitor, IC50 = 0.6 nM, that also exhibits high affinity for cKit, IC50 = 42 nM. In contrast, the molecularly matched phthalazine 67 retains the potent MAPK inhibition but is an order of magnitude less effective toward inhibiting cKit. Optimization of 66 afforded 67 which is a potent MAPK inhibitor with high selectivity over inhibition of cKit, Kdr, Lck and JNK1-3. An X-ray co-crystal structure revealed that the phthalazine nitrogen atoms of 67 are engaged in dual H-bonding interactions with the N-Hs of Met109 and Gly110 of MAPK, as summarized in Fig. 10B [90, 91]. The absence of the Cα substituent in Gly110, a residue present in only 40 representatives of the kinome, facilitated an inversion of the topology of the amide moiety, presumably induced by the presence of the phthalazine nitrogen atom, that projected the N-H into the ATP binding pocket [93]. This kind a conformational change is energetically less favorable in kinase inhibitors with hinge residues that possess larger Cα substituents and thus, in the absence of the topological inversion, one of the phthalazine nitrogen atoms would encounter a repulsive interaction with the C = O of Cys673 in cKit.

Structure, potency and proposed binding mode of 66 with cKit (A) and structure, potency and binding mode of 68 with MAPK from an X-ray cocrystal structure (B) (3DS6)


Pyridazines and Intramolecular H-bonding
The potential of a pyridazine ring to engage in intramolecular H-bonding can be advantageous in drug design, with one demonstration illustrated by SAR studies associated with splicing modulators of survival motor neuron-2 (SMN2) which have been explored as potential therapeutics for the treatment for spinal muscular atrophy (SMA) [94,95,96,97]. The pyridazine 69 was identified as a lead pre-mRNA splicing modulator that stimulated synthesis of the full length SMN2 RNA with inclusion of exon 7 in a cell-based screen [95, 96]. In this assay, which was conducted in high throughput screening (HTS) mode, 69 activated the SMN2 reporter to 1700% of the dimethyl sulfoxide (DMSO) control with an EC50 value of 3.5 µM. This effect was confirmed in an enzyme-linked immunosorbent assay (ELISA) assay that assessed full length SMN2 production in SMNΔ7 mouse myoblasts where the EC50 value was 0.6 µM and SMN protein levels were increased by 2.5-fold [95, 96]. SAR studies conducted to examine the role of the pyridazine scaffold of 69 revealed it to be a critical structural element, with the isomeric pyridines 70 and 71, pyrimidines 72 and 73 and pyrazine 74 all failing to demonstrate a significant effect on mRNA splicing in the cell-based assay, as summarized in Fig. 11 [96]. The two topologically complementary thiazoles 75 and 76 were also inactive but the thiadiazole 77 exhibited potent splicing modulation, with a several-fold advantage over 69, providing a compelling example of bioisosterism between pyridazine (1) and thiadiazole (29) rings that was subsequently explored in greater detail [96, 97]. Interestingly, the oxadiazole analog does not appear to have been evaluated, although the N-hydroxy piperidine derivatives are a known chemotype that display antibacterial activity [98, 99].

SARs associated with variation of the pyridazine core of the SMN2 splicing modulator 69

As the SAR survey for this chemotype evolved, particularly instructive insights were obtained from molecular edits made to the benzothiophene heterocycle [96]. Replacement of the benzothiophene ring with a 2-substituted naphthyl homolog (78) fully preserved the cellular spicing modulatory activity. However, the introduction of an ortho hydroxy substituent (79) resulted in a 20-fold increase in potency, a unique SAR point since other ortho substituents generally led to reduced potency, while preserving a 2.4-fold elevation of SMN protein levels in the ELISA-based cellular assay. This observation was confirmed with the truncated analog 80 where methylation of the phenol to afford the anisole 81 resulted in a 50-fold reduction in splicing modulation potency. These results were interpreted as a preference for a planar arrangement between the pyridazine heterocycle and the pendent aryl ring, stabilized by an intramolecular H-bonding interaction between the phenol moieties of 79 and 80 and the proximal pyridazine nitrogen atom. This hypothesis was reinforced by the single crystal X-ray structures of 80 and 81 which indicated a planar topography (3o twist out of the plane) for the phenol 80 based on an intramolecular H-bond at a distance of 2.54 Å between the O and N atoms (Fig. 12A) while in 81, the anisole ring was disposed at an out of plane angle of 29o (Fig. 12B) [96, 97]. These observations reflect the conformational properties of the lead hit 69 in which the narrower bond angles associated with the thiophene ring compared to naphthalene and an energetically productive interaction between the low-lying C-S σ*orbital of the benzothiophene and the lone pair of electrons of the proximal pyridazine nitrogen atom would favor a coplanar topography (vide infra) [100]. The disposition of the aminopiperidine ring in the solid state structures of 80 and 81 differed, with that displayed by the anisole 81 considered to be the active conformation since the 4-O-linked analogs, which offered an efficacy advantage, are heterocyclic ethers that are known to display a strong propensity to adopt this conformation in order to avoid unfavorable non-bonded interactions [101].

Single crystal X-ray structures of 80 (A) and 81 (B)
The 2-(pyridazin-3-yl)phenol moiety in 80 was preserved throughout the subsequent optimization program which was focused on avoiding a human ether-à-go-go-related gene (hERG) cardiac ion channel liability whilst maintaining biological potency and efficacy and optimizing central nervous system (CNS) penetrance. Branaplam (82) was the culmination of that effort and, although advanced into clinical trials as a potential therapeutic for SMA, development has recently been redirected to focus on Huntington’s disease following the demonstration that the drug lowers the level of huntingtin protein in patient-derived neuronal and non-neuronal cells [102, 103]. In branaplam (82), the unique construction of the molecule on a pyridazine scaffold controls the conformation of both the phenol and tetra-methylated piperidine rings and, thus, the topographical presentation of the molecule to its target (vide infra).

3-Amino-6-phenylpyridazine has been identified as an important structural element in ligands that bind to the bromodomains (BRDs) of SWI/SNF-related matrix-associated actin-dependent regulator of chromatin A2 (SMARCA2, BRM) and SMARCA4 (BRG1) and the fifth bromodomain of (polybromo-1) bromodomain 5 (PBRM1(5)) [104,105,106,107,108]. Screening of a library of 43,000 compounds in a competition assay evaluating the binding of a biotinylated peptide containing four acetylated lysine residues to the SMARCA4 bromodomain identified the fused tricyclic aminopyridazine derivative 83 as a molecule with modest affinity, IC50 = 5.3 µM, that also bound to the SMARCA2 and PBRM1 bromodomains, IC50 values = 4.3 and 3.1 µM, respectively, but not to BRD4 [104]. An X-ray cocrystal structure of 83 with SMARCA4 illuminated the binding mode, revealing that the aminopyridazine moiety engaged Asn1540 in a dual H-bonding interaction that mimics the acetamide moiety of acetylated lysine, as summarized in the two-dimensional representation presented in Fig. 13A [104, 106,107,108,109]. The second pyridazine nitrogen atom engaged Tyr1497 through the intermediacy of a H2O molecule, although the two molecules of H2O typically found in the binding pocket were absent [104, 107, 108]. A molecular edit that involved excision of the NH of 83, a functionality not intimately involved in drug-target interactions, resulted in the structurally simpler and conformationally more mobile 84, which exhibited demonstrable, although tenfold weaker, affinity for SMARCA4, IC50 = 57.1 µM. Drawing inspiration from the X-ray cocrystal structural data obtained for PFI-3 (87) bound to SMARCA4, the phenol 85 was evaluated and found to be a substantially more potent bromodomain ligand, with IC50 values of 30, 37 and 8.9 nM toward SMARCA4, SMARCA2 and PBRM1(5), respectively, reflecting a remarkable 1900-fold potency increase in affinity for SMARCA4 [104, 105]. The X-ray cocrystal structure of 85 with SMARCA4 revealed a preserved binding mode with respect to recognition of the aminopyridazine moiety, with the phenol ring almost coplanar with the heterocyclic core (the measured dihedral angle is ~17o). This conformation projected the hydroxy substituent toward the nitrogen atom of the pyridazine ring, with an O to N distance of 2.60 Å that is compatible with an intramolecular H-bonding interaction. The phenol of 85 engaged the OH of Tyr1497 directly, displacing the H2O molecule observed in the cocrystal structure of 83 as well as displacing an additional H2O molecule from the binding pocket. In this arrangement, the intramolecular H-bond in 85 pre-organizes the molecule into the bound, planar topography discovered with 83 (Fig. 14A) whilst correctly orienting the topology of the phenolic hydroxy substituent with respect to the aminopyridazine core to allow the interaction with Tyr1497 (Fig. 14B). The poor solubility and low metabolic stability associated with 85 rendered it an unsuitable probe for in vivo studies, liabilities addressed by GNE-064 (86) which maintained the bromodomain binding profile of the progenitor whilst also improving the aqueous solubility by tenfold, enhancing metabolic stability and moderating the affinity for plasma proteins [104].

Key drug-target interactions between 83 and the SMARCA4 protein (A) (7TD9) and between 85 and the SMARCA protein (B) (7TAB)

Conformation of 83 (A) and 85 (B) abstracted from the cocrystal structures with SMARCA4 protein

The 3-amino-6-phenylpyridazine moiety has been further exploited as the basis for the design of pan inhibitors of SMARCA and polybromo-1 bromodomains in molecules that include 88–90 and the proteolysis-targeting chimeras (PROTACS) 91 and 92 designed to facilitate degradation of these proteins [105, 109, 110]. The binding conformation observed with 85 in SMARCA4 was preserved with these molecules and extended to both PBRM1(5) and to the complicated, multi-component assembly that formed the bromodomain of human SMARCA2 bound to 91 and the complex formed by the von Hippel-Lindau gene product pVHL and elonginC and elonginB that is recognized by a ubiquitin E3 ligase (Fig. 15) [105, 109]. In the majority of these structures, the dihedral angle between the pyridazine and phenol rings was typically small at <5o.

Structure of 91 abstracted from the ternary complex with SMARCA2 and pVHL:ElonginC:ElonginB. For the 6-aminopyridazin-3-yl)phenol moiety, the measured O to N distance is 2.55 Å and the dihedral angle between the pyridazine and phenolic rings is 1.06o (6HAX)

Intramolecular H-bonds in pyridazine-3-carboxamide derivatives
The preferred conformation of pyridazine-3-carboxamide derivatives is depicted in Fig. 16, where the favored topology is stabilized by an intramolecular H-bond between the amide N-H and the proximal heterocyclic nitrogen atom of the core heterocycle which is reinforced by the anti-parallel alignment of the pyridazine and amide dipoles [111]. The conformational preference is illustrated by the single crystal X-ray structure of 93 presented in Fig. 17 where the torsion angle between the pyridazine ring and the amide C-N bond is approaching planarity at 4.2o [112]. This topological arrangement was also observed in a series of pyridazine-3-carboxamide-based allosteric inhibitors of the hepatitis C virus (HCV) NS5B RNA-dependent, RNA polymerase (RdRp) represented by 94-99 [111, 113]. When optimally configured, the 3-carboxamide moiety in this series contributes to a substantial increase in antiviral potency compared to the chloride 96 and the methyl ester 97, an SAR point explained by the introduction of productive drug-target interactions. In the cocrystal structures of 98 and 99 with HCV NS5B, the pyridazine-3-carboxamide oxygen atom acts as a H-bond acceptor for the phenolic hydroxy of Tyr555 and the side chain hydroxy of Ser288, with oxygen-to-oxygen distances of 2.71 and 2.81 Å, respectively, for 98 and 2.66 and 2.98 Å, respectively, for 99. The amide N-H appears to donate a H-bond to the oxygen atom of Tyr191 where the nitrogen to oxygen distances are 3.30 Å for 98 and 3.14 Å for 99. The pyridazine nitrogen atoms, particularly the one distal from the amide substituent, engage the backbone carbonyl oxygen atom of Phe193 through the intermediacy of a H2O molecule, whilst the dihedral angle between the pyridazine ring and the amide C-N bond (N-C-C-N) is 3.25o for 98 and 12.02o for 99 [113]. The key drug -target interactions between 99 and the HCV GT1a and GT1b NS5B enzymes in the cocrystal structures are depicted in Fig. 18A and B, respectively. For the pyrazine homolog 95, the preferred conformation depicted in Fig. 19 presents only a single nitrogen atom in a topology that is suitable to engage the H2O molecule which, along with the reduced pKBHX value associated with this heterocycle, may account for the tenfold reduced antiviral potency [111, 113]. In the pyridazine series, expansion of the 3-carboxamide N-substituent from the simple methyl found in 94 to those in 98 and 99 led to enhanced potency.

The preferred topology of pyridazine-3-carboxamide derivatives

Structure and single crystal X-ray structure of 93

Cocrystal structure of 99 bound to HCV GT1a NS5B (3QGH) (A) and to the Bartenschlager HCV GT 1b NS5B construct (3QGG) (B) highlighting key drug-target contacts

Preferred conformation of N-methylpyrazine-2-carboxamide
A pyridazine-3-carboxamide moiety is prominent in inhibitors of stearoyl-coenzyme A desaturase-1 (SCD), an iron-containing, fatty acid desaturase that catalyzes the introduction of a double bond between carbons 9 and 10 of the stearic acid chain of stearoyl-CoA to form oleoyl-CoA [114]. The up-regulation of SCD1 has been associated with several metabolic disorders that includes diabetes, fatty liver disease and obesity, and SCD1 also appears to play an essential role in the growth of tumors [114, 115]. A HTS campaign identified the piperazinyl pyridine 100 as a lead inhibitor with modest potency that was enhanced by>50-fold with the relatively minor structural modifications inherent to 101, a compound that exerted an inhibitory effect on SCD1 in a HepG2 cell-based assay [114, 116]. In this chemotype, replacing the pyridine ring with a pyridazine heterocycle (102) provided an additional threefold boost in inhibitory potency in both assays, ultimately leading to the identification of XEN103 (103), an orally bioavailable compound that demonstrated a dose-dependent inhibition of SCD1 activity in rat liver measured 4 h post dosing, with an ED50 value of 0.8 mg/kg [114, 116]. The SARs delineated by 104–107 demonstrate the uniqueness of the pyridazine heterocycle in the setting of this pharmacophore, with only the pyridine 104, the topological isomer of 100, preserving the SCD1 inhibitory activity, although with a threefold reduction in potency in the biochemical assay and a >20-fold decline in the cell-based system [116, 117]. Although not definitive, the SARs are suggestive of a role for intramolecular H-bonding between the amide N-H and the heterocycle in the expression of SCD1 inhibitory activity. While this pyridazine-based chemotype spawned many homologous inhibitors of SCD1, a broad range of additional inhibitory chemotypes have been identified that do not depend on a pyridazine ring or a well-defined bioisostere; however, these have not been reconciled with the structure of the enzyme which would provide critical insight into drug-target interactions while illuminating the SAR observations [114, 118]. The clinical development of systemically bioavailable SCD1 inhibitors has met with significant challenges due to on-target toxicity arising from inhibition of the enzyme in the skin and eye [114]. The liver-selective SCD1 inhibitor MK-8245 (108), which was designed to take advantage of organic anion transporter proteins for selective delivery to the liver, was an attempt to address the systemic toxicity problem and, although advanced into clinical trials, was subsequently abandoned [114, 119, 120].

The versatility of the pyridazine-3-carboxamide moiety in drug design is further illustrated by its presence in several exploratory molecules that address a range of therapeutic areas and which express varied modes of action [121]. Notable examples include the dCTP pyrophosphatase 1 inhibitor 109, the histone deacetylase (HDAC) inhibitor 110, the antibacterial agent 111, the potent hematopoietic prostaglandin D synthase (HPGDS) inhibitor 112, the androgen receptor degrader ARV-110 (113), the anaplastic lymphoma kinase (ALK) inhibitor ensartinib (114), which is approved for marketing in China, the rapidly accelerated fibrosarcoma (RAF) kinase inhibitor 115 and the potent human immunodeficiency virus-1 (HIV-1) integrase inhibitor 116 [122,123,124,125,126,127,128,129,130]. In mouse and human LMs, the t1/2 value for the dCTP pyrophosphatase 1 inhibitor 109 was short at approximately 3 min, which contrasted with observations with structurally similar SCD1 inhibitors [116, 121]. Metabolite identification studies indicated that oxidation occurred on both benzene rings and the core, with oxidation of the pyridazine nitrogen atoms also observed [121]. Interestingly, in this piperazine-based chemotype, replacing the pyridazine ring with a pyrazine heterocycle led to reduced metabolic stability. However, in this example, targeted metabolic stability was achieved with 117 which reflected 3 molecular edits, including replacing the piperazine ring with a 2,6-diazaspiro[3.3]heptane moiety [121]. For ensartinib (114), four phase I metabolites were detected, which included hydroxylation of the pyridazine and halogenated phenyl rings, both of which produced chemically reactive intermediates, and demethylation and oxidation of the piperazine moiety, although the latter pathway did not produce a chemically reactive metabolite [128].

Whilst the available data do not provide insight into the potential importance of intramolecular interactions between the amide N-H-and the pyridazine core in 109–117, one chemotype where this phenomenon does appear to be of value is in inhibitors of the non-receptor tyrosine kinase TYK2 that act at an allosteric Janus homology 2 (JH2) pseudokinase domain which is catalytically inactive [131,132,133,134]. A key observation is provided by comparison of the nicotinamides 118 and 120 with their pyridazine congeners 119 and 121, respectively, which resulted from optimization of a lead nicotinamide-based inhibitor identified by HTS [131]. In these molecules, the methylated amide is important for kinase inhibitory selectivity with respect to both the Janus (JAK) family members and the broader kinome. Consequently, deuteration of the methyl amide was introduced to slow oxidative demethylation in vivo which delivers the primary amide, a similarly potent, but markedly less selective TYK2 inhibitor. The measured high performance liquid chromatography (HPLC) Log P data indicate that the pyridazines are more lipophilic than the pyridine analogs, despite the larger dipole moment associated with the former. This is presumably a function of an intramolecular interaction between the methyl amide N-H and the pyridazine nitrogen atom. The close proximity between these functionalities can be observed in the cocrystal structure of deucravacitinib (25) bound to TYK2, where the distance between the heterocyclic and amide nitrogen atoms is 2.68 Å and the torsion angle between the amide carbonyl bond and the pyridazine ring is 8.4o [132, 135, 136]. The nicotinamide analog of 25, compound 122, shows much reduced membrane permeability in Caco-2 cells, with a Pc value of <15 nm/s compared to 70 nm/s for 25 [132]. The key drug-target interactions between 25 and TYK2 are summarized in Fig. 20, which highlights the donor-acceptor-donor H-bonding pattern between the inhibitor and the backbone of the hinge which is comprised of the residues Glu688, Tyr689 and Val690. Additional interactions include H-bonds between the triazole heterocycle and Arg738 and the anisole oxygen atom and Lys642 while the Cα proton of the cyclopropyl carboxamide is proximal to the C = O moieties of Val690 and Glu691 [131, 132]. Deucravacitinib (25) was approved for marketing by the FDA on September 10th, 2022 as a therapeutic agent for the treatment of moderate to severe plaque psoriasis and is the first de novo deuterated drug to be licensed for the U.S. market [137].

Key drug-target interactions between deucravacitinib (25) and the TYK2 enzyme from the cocrystal structure (A) (6NZP) rendered in a two-dimensional representation (B)


The 3-carboxypyridazine-based chemotype continues to be of interest in the design of allosteric TYK2 inhibitors, with 123 recently characterized as a potent, selective and efficacious degrader of the protein with a DC50 value of 14 nM and efficacy that amounts to 78% degradation of the enzyme in Jurkat cells [138].

An alternate TYK2 JH2 inhibitor chemotype that was identified by HTS is based on a bicyclic imidazo[1,2-b]pyridazine heterocycle that also embeds a pyridazine moiety, with 124 representative [139, 140]. In the cocrystal structure of this compound bound to the TYK2 enzyme, the secondary amide moiety is oriented by engaging in an intramolecular H-bonding interaction with one of the core pyridazine nitrogen atoms, as depicted in the abstracted structure presented in Fig. 21. Interestingly, the silhouette outlined by this chemotype has some analogy with that found in NDI-034858 (125) which is based on a pyrazolo[1,5-a]pyrimidine core and has recently completed Phase 2 clinical trials [141, 142]. The design of this bicyclic heterocyclic core preserves the potential for an intramolecular H-bonding interaction between a nitrogen atom of the core heterocycle and the pendent amide N-H whilst also providing an interesting example of a bioisosteric relationship between pyridazine and pyrimidine rings.

The conformation of the 124 abstracted from the cocrystal structure with the JH2 domain of TYK2. The distance between the amide nitrogen and the proximal nitrogen atom of the core imidazo[1,2-b]pyridazine heterocycle is 2.93 Å while the dihedral angle between the amide carbonyl moiety and the core heterocycle is –2.32o (6NSL)

An X-ray cocrystal structure of the potent of checkpoint 1 kinase (CHK1) inhibitor 126 revealed that the amide moiety, the thieno[3,2-c]pyridine and the chlorophenyl ring adopted an overall planar arrangement that optimally presented the primary amide to the carbonyl and N-H moieties of the hinge backbone residues Glu85 and Cys87, respectively [143, 144]. This conformation is stabilized by an intramolecular interaction between the amide carbonyl oxygen atom and the low lying σ* orbital associated with the thienyl sulfur atom, with an oxygen-to-sulfur distance of 2.86 Å that is well below the sum of the van der Waals radii of the two atoms [100]. The topological isomer 127 is over 1500-fold less potent than 126, attributed to a distortion of the secondary amide moiety, which is intimately involved in target recognition, from a coplanar arrangement. This is due to a combination of the loss of the stabilizing O-to-S interaction and the introduction of allylic 1,3-strain between the carbonyl oxygen and the thienyl 3-H atom [145]. The thieno[2,3-d]pyridazine-based inhibitor 128 offers an intramolecular H-bond between the amide N-H and the core as an additional element stabilizing the planar conformation (Fig. 22A) while the added nitrogen atom in the core allows engagement of a second interfacing H2O molecule in the binding pocket (Fig. 22B), which may explain the slightly enhanced CHK1 inhibitory potency [143].

A Conformation of the imidazo[1,2-b]pyridazine 128 abstracted from the cocrystal structure with CHK1 (3PA4). The distance between the amide oxygen and thienyl sulfur atoms is 2.77 Å while that between the amide nitrogen atom and the proximal nitrogen atom on the fused heterocyclic core is 2.72 Å. The dihedral angle between the amide and the core heterocycle is 3.4o. B Key drug target interactions between 128 and CHK1

Conformational aspects of 3-alkoxy and 3-alkoxymethyl pyridazines
The conformation of 3-alkoxy-pyridazine, 3,6-dialkoxy-pyridazine and other azine and azole ether derivatives is influenced by non-bonded interactions between the lone pairs of electrons on the ring nitrogen atoms and those on the proximal oxygen substituents, with calculated energy differences that can be substantial [101]. The preference for the conformation designated as anti in Fig. 23 is a function of a destabilization of the syn conformer by in-plane repulsive interactions between the lone pairs of electrons on the ether oxygen and the adjacent ring nitrogen atoms [101]. In single crystal X-ray structures of both simple and complex pyridazinyl ethers, the anti conformation is prevalent, as illustrated by 129 and 130 in Fig. 24 [146, 147]. These molecules are planar around the heterocyclic ether junction, with dihedral angles of 4.5o for 129 and 1.7o for 130.

Energetics associated with the preferred conformation of 3-alkoxy and 3,6-dialkoxy -pyridazines

Structures and single crystal X-ray structures of 129 (A) (RAPPAA) and 130 (B) (NATDOC)
The stereochemical influence of alkoxy pyridazines and alkoxy phthalazines has found application in the design of chiral catalysts of the type represented by 131 and 132 that provide scaffolds on which to convene OsO4 and an olefin to promote asymmetric dihydroxylation [148,149,150,151,152,153]. In these molecules, the interaction between the pyridazine and phthalazine cores and the ether oxygen atoms confers an element of conformational control that contributes to the cup shape of the molecule that defines their three-dimensional structures and provides an asymmetric, enzyme-like environment as the site of catalysis, as exemplified by the shape of 132 presented in Fig. 25 [101, 150,151,152,153].

Single crystal X-ray structure of 132 [150] (TORNIX)

3-Alkoxypyridazines have found application in the agricultural arena where a series of 6-(benzyloxy)pyridazin-3-amine derivatives has been studied for their potential to function as weed killers [154]. Compounds of the type depicted in Fig. 26 were assessed through laboratory-based tests on the Chinese cabbage Brassica campestris L (rape), a dicotyledonous plant, and barnyard grass where they displayed moderate herbicidal properties.

Generic structure of a series of 6-(benzyloxy)pyridazin-3-amine derivatives explored for their potential to function as herbicides
A 3-methoxypyridazine moiety featured in a series of pyrazole-based cyclooxygenase inhibitors, explored for their effects on arachidonic acid-induced aggregation of human blood platelets, that emerged from a broader survey of the chemotype [155,156,157]. The single crystal X-ray structure of 133 contained two conformers of the chlorophenyl ring in the unit cell, with the pyridazine ring oriented as depicted in the presented structure of 133, presumably reflecting a preferred dipole alignment with the pyrazole ring, while the MeO substituent adopted the more stable anti conformation [101, 157]. Optimization in this series identified 134 as a potent inhibitor of arachidonic acid-induced human blood platelet aggregation, although the more lipophilic anisole homolog 135 was threefold more active [157].
The homologous 3-alkoxymethylpyridazine derivatives also appear to express a conformational preference based on dipole arrangements and the avoidance of non-bonded interactions, analogous to the effect that has been noted for 3-alkoxymethylpyridines [158, 159]. An illustration of this phenomenon is provided by the single crystal X-ray structure of 3,6-bis(methoxymethyl)pyridazine (136) depicted in Fig. 27 where the methoxymethyl substituents are oriented anti to avoid unfavorable non-bonded interactions between the lone pairs of electrons on the ring nitrogen atoms and the exocyclic oxygen atoms. The methoxymethyl substituents also adopt a relatively planar topographical arrangement with respect to the plane of the pyridazine ring, with N-C-C-O dihedral angles that reflect deflections of 18.8o and 8.0o. This contrasts with (methoxymethyl)benzene derivatives which favor an orthogonal topography to avoid unfavorable allylic 1,3-strain [145, 159].

Structure (A) and single crystal X-ray structure (B) of 136 (GEYSEI)
However, alkoxymethyl azines can sample the orthogonal topographical arrangement providing an element of chameleonic behavior with respect to conformational flexibility, as illustrated by the pyridine-based casein kinase 1 (CK1) inhibitor 137 which binds to the ATP recognition site of the enzyme and exhibits 75- and 12-fold selectivity for CK1γ over CK1α and CK1δ, respectively [160]. The X-ray cocrystal structures of 137 with CK1γ and CK1δ reveal similar binding interactions with the hinge region, with the pyridine nitrogen atom engaging the backbone N-H of Leu119 in CK1γ and of Leu85 in CK1δ. The amide moiety makes key interactions in both cocrystal structures, with the C = O engaging the protonated side chain amine of Lys72 while the N-H engages the side chain carboxylate of Asp185 and a H2O molecule that bridges to the hydroxy of Tyr90 the backbone N-H of Asp185 in CK1γ. In CK1δ, the amide oxygen atom engages the protonated amine moiety of Lys38 directly and the side chain carboxylate of Glu52 and the hydroxy of Tyr56 via the intermediacy of a H2O molecule while the amide N-H directly engages the side chain carboxylate of Asp149. However, there are significant differences in the conformations around the alkoxymethyl pyridine moiety of the two structures which distinguishes the projection of the difluorophenyl ring, further complicated by the observation of two different conformations of the difluorophenyl ring in the CK1δ structure. In the available structure of 137 with CK1δ, the alkoxymethyl pyridine moiety adopts a relatively planar conformation, with an N-C-C-O torsion angle that is distorted by just 7.7o from coplanarity, that favorably aligns the dipoles of the pyridine ring and C-O bond in an anti topology, with the ether oxygen atom interfacing with the backbone carbonyl oxygen atom of Gly88 via a H2O molecule (Fig. 28). In contrast, in the cocrystal structure of 137 with CK1γ, the torsion angle deviates by 69.4o from a planar topography, with the difluorophenyl ring stacked parallel to Pro333 in a face-to-face-type of interaction [160]. The effect of the introduction of the oxygen atom can be seen by comparing the profiles of the MMP of 138 and 139 where the presence of the ether oxygen atom in the latter compound enhances CK1δ and CK1γ inhibitory activity by almost 10-fold and 5-fold, respectively, presumably a reflection of a combination of increased access to the more planar topography and the introduction of a H-bond acceptor in 139 [160]. Both the potency and the enzyme inhibitory selectivity of 139 were enhanced compared to the pyrimidine analog 140. While the pyridazine analogs were not prepared as part of this SAR survey, the conformational preferences of alkoxymethyl pyridazines would be anticipated to be similar to those of the alkoxymethyl pyridines.

Conformation of 137 when bound to CK1γ (A) (4HGS) and CK1δ (B) (4HGT)


The kind of conformational flexibility inherent to alkoxymethyl azines may have value in circumstances where a ligand engages a protein through H-bonding interactions mediated by the intermediacy of a H2O molecule and drug design is focused on introducing structural elements that can act as bioisosteres of the ligand/H2O complex. An illustrative example is provided by the isoquinoline 141, which is a potent inhibitor (IC50 = 2.34 nM) of human hematopoietic prostaglandin (PG) D2 synthase (hH-PGDS) identified by a screening campaign [161]. An X-ray cocrystal structure of 141 bound to the enzyme revealed that the isoquinoline nitrogen atom engaged the protein via the intermediacy of a H2O molecule that interfaced with the backbone amide oxygen atom of Leu199 and the side chain hydroxy of Thr159, as depicted in Fig. 29A and B. The dihedral angle between the isoquinoline heterocycle and the phenyl ring in the cocrystal structure was ~45o. This observation stimulated the design of ligands capable of displacing the bound H2O molecule in an effort to take advantage of the potency enhancement that can result, with the naphthalenyl alcohol 142 and the homologous amine 143 synthesized and evaluated as a test of the concept [161,162,163]. However, both 142 and 143 were characterized as poor inhibitors of hH-PGDS, with 632- and 361-fold increases in the IC50 values, respectively, compared to the progenitor 141. Despite the reduced potency, both compounds were cocrystallized with the enzyme, with the structures revealing that the H2O molecule had indeed been displaced, as depicted for 142 in Fig. 29C, D and 143 in Fig. 29E, with the OH and NH2 moieties engaging the protein directly. However, the measured dihedral angles between the OH and NH2 moieties and the naphthalene core were 21o and 27o, respectively, well below the lowest energy conformation of 90o, indicating that the binding of these molecules to hH-PGDS introduces conformational strain and an associated energy penalty (Fig. 29F, G) [161]. In addition, the dihedral angles between the naphthalene and phenyl rings increased to 62o in 142 and 64o in 143. Conceivably, the cinnolines 144 and 145 or their isoquinoline homologs (which would be isomers of 142 and 143) would have been capable of relieving some of the energetic burden given the reduction in non-bonded interactions that would be experienced in the more planar conformation targeted.

Key binding interactions between hH-PGDS and 141 (A, B) (4EE0), the alcohol 142 (C, D) (4EDY), the amine 143 (E) (4EC0) and the conformations of 142 and 143 (F and G) as abstracted from the cocrystal structures

Pyridazines and sulfur interactions
An intramolecular interaction between a lone pair of electrons and the low lying σ* orbital of C-S bonds and some N-S bonds has been shown to provide conformational bias, and the nitrogen atoms of pyridazine have been shown to participate in this phenomenon [100, 164]. In the single crystal X-ray structure of 3-methyl-6-(thiophen-2-yl)pyridazine (146), the molecule is stabilized in a planar topography by a energetically productive N-to-Sσ* interaction, as evidenced by a torsion angle between the thiophene and pyridazine rings of 0.2o and an N-to-S distance of 2.89 Å that is less than the sum of the respective van der Waals radii of 3.35 Å (Fig. 30A) [100, 164]. Similarly, in the single crystal X-ray structure of 3,6-di(thiophen-2-yl)pyridazine (147), the planes of the thiophene rings and the pyridazine core are close to planarity, with S-C-C-N angles of 10.3o and 12.7o, stabilized by close N-to-S contacts of 2.93 and 2.94 Å, respectively (Fig. 30B) [165].

Conformation of 146 (A) (ZUHTED) and 147 (B) (IHEMOX) in the single crystal X-ray structures with the nitrogen to sulfur distances marked all of which are less than the sum of the van der Waals radii of nitrogen and sulfur atoms

For the 7,10-di-2-thienyl-acenaphtho[1,2-d]pyridazine (148), the molecule presents a symmetrical topography in which the torsion angles for both thiophene-pyridazine relationships are 30.9o, a distortion from planarity that is presumably a function of the allylic 1,3-strain encountered by the C-3 hydrogen atoms on the thiophene and the acenaphthylene rings which are forced into proximity in this rigid system [166]. The distance between the proximal thienyl and core hydrogen atoms in 148 is 2.33 Å, which is slightly shorter than the 2.41 Å and 2.42 Å in 147, while the nitrogen-to-sulfur distances are equivalent at 2.99 Å each (Fig. 31A) [166]. The presence of the CH3 substituents in 1,6-dimethyl-7,10-di-2-thienyl-acenaphtho[1,2-d]pyridazine (149) amplifies the allylic 1,3-type strain such that the thiophene rings are distorted from the plane of the pyridazine by 45.2o and 55.1o, respectively (Fig. 31B) [166]. However, while the nitrogen to sulfur distances in 149 are 20% longer than those in 148 at 3.08 Å, this remains below the sum of the van der Waals radii for nitrogen and sulfur, indicative of a stabilizing interaction [100].

Conformation of 148 (A) (FUBNEW) and 149 (B) (FUBLOE) in the single crystal X-ray structures
An example where nitrogen-to-sulfur interactions appear to play a role in the topography of a pyridazine-based molecule is illustrated by 68, the initial screening lead in the Novartis series of SMN splicing modulators [96, 97]. The detailed and extensive SAR studies that have been conducted with this chemotype define a planar pharmacophore at the benzothiophene-pyridazine junction where the ortho-substituted phenols in 72, 73 and 75 provide an intramolecular H-bonding interaction to favor a planar topography. Although there are no X-ray structural data available for 68, the observations discussed above suggest that, in this context, the sulfur atom is functioning as the equivalent of a phenolic OH when engaging the pyridazine nitrogen atom [100].
A stabilizing nitrogen lone pair-to-sulfur C-Sσ* interaction also appears to influence the conformation of the thiazole moiety in the potent PDE4 inhibitor 150 [167,168,169]. NMR studies of 150 dissolved in either CDCl3 or DMSO-d6 that assessed nOe enhancements between the peri proton of the phthalazine ring and the thiazole hydrogen atoms were consistent with a coplanar conformation. An nOe enhancement in the NMR experiment was observed only for the thiazole C-3 proton, consistent with a topology in which the thiazole sulfur atom is oriented toward the phthalazine nitrogen atom [169]. The weaker PDE4 inhibitory potency associated with the phenyl analog 151 maybe a function of the combination of reduced polarity and unfavorable non-bonded interactions that would distort the phenyl ring from a planar arrangement with the phthalazine core. The triazole 152 retains reasonable potency and in the solid state this molecule exhibits a planar topography between the triazole and phthalazine rings, as depicted in Fig. 32. The torsion angle between the two heterocycles in 152 is 4o and the C-5 hydrogen atom of the triazole ring projects toward the phthalazine nitrogen atom, with the distance between the phthalazine nitrogen and triazole C-5 carbon atoms measured as 2.71 Å, a potentially stabilizing C-H H-bonding interaction [28, 169]. However, in solution (CDCl3, DMSO-d6, CD3OD were used to mitigate the effects of the dielectric constant of the solvent) the triazole ring of 152 and that of the poorly active imidazole 153 appear to more readily occupy a conformation in which the azole rings approach orthogonality, providing a potential explanation for their lower inhibitory potency [169].

Conformation of 152 in the solid state (JASGIU)

The pyridazine ring and effects on potency
The deployment of pyridazine rings and the fused homologs phthalazine, cinnoline and related compounds to replace other azines and fused azines can exert significant effects on biochemical potency. Not surprisingly, molecular edits of this type can either enhance or erode potency, dependent upon the circumstance, and for reasons that are not always apparent. In an analysis of the SwissBioisostere database in which seven azine replacements for a 4-pyridazinyl heterocycle were queried, of the 159 examples surveyed, 25 (16%) resulted in reduced potency, 75 (47%) maintained potency and 59 (37%) examples resulted in enhanced potency (Table 4) [170, 171]. This study was conducted against the backdrop of a survey of azine replacements for the pyridazine moiety of the insecticide dimpropyridaz (154), which acts by an unknown mechanism, explored in the context of the simplified analog 155 [171]. Whilst 155 was lethal toward 50% of green peach aphids (M. persicae) at a concentration of 0.78 mg/L, 156–159 were inactive at concentrations as high as 200 mg/L., reflecting a precise specificity not only for the pyridazine ring in this context, but also for the topology inherent to 154 and 155. In an effort to understand this observation, differences in the calculated HOMO and LUMO energies were analyzed since this had proved to be illuminative in earlier studies of insecticides [171]. The pyridazine 156 and pyrimidines 158 and 159 possessed larger ΔE values than the pyridazine 155 and whilst pyrazine 157 had a similar ΔE value, the ELUMO was the lowest of the cohort, data that collectively provided some insight into the observed SARs [171].

In the examples culled from the SwissBioisostere database where a pyridazine heterocycle provided a potency advantage over other azines, the magnitude of the effect was generally modest, a phenomenon that can extend to cinnolines and phthalazines [33, 171]. As an illustrative example, the cinnoline 161 is a twofold more potent inhibitor of leucine rich repeat kinase 2 (LRRK2) than the quinoline homolog 160, an SAR point reproduced in the MMP of inhibitors of Bruton’s tyrosine kinase (BTK) 162 and 163 that recapitulate a similar molecular edit [172,173,174]. Notably, in the X-ray cocrystal structure of 163 with BTK, only one of the cinnoline nitrogen atoms is engaged in an intermolecular H-bonding interaction, interfacing with the enzyme a via a H2O molecule, as depicted in Fig. 33A [173]. However, the cinnoline nitrogen atom proximal to the carboxamide does appear to be aligned appropriately to facilitate an intramolecular H-bond with the primary amide N-H, with a dihedral angle between the amide carbonyl bond and the core heterocycle of 14o and a nitrogen-to-nitrogen distance of 2.72 Å (Fig. 33B) [173]. The preferred conformations of 160–163 also reflect favorable intramolecular H-bonding interactions between the amide carbonyl oxygen atoms and the ortho amine substituents.

A Bound conformation of 163 with BTK depicting the key drug-target interactions (4Z3Z). B The conformation of 163 abstracted from the cocrystal structure—the dihedral angle between the amide carbonyl and the core heterocycle is 14o with a nitrogen-to-nitrogen distance of 2.72 Å

Although not a perfect MMP, the thieno[2,3-c]pyridazine heterocycle in the muscarinic M4 positive allosteric modulator (PAM) 165 offers a several-fold potency advantage over the thieno[2,3-b]pyridine scaffold in 164 without a compromise of agonistic efficacy [175]. As a consequence, this heterocycle was used as the vehicle for additional studies that led to the identification of 166 as a clinical candidate for the treatment of schizophrenia [176].

The query of the SwissBioistere database summarized in Table 4 suggested that replacing a 4-substituted pyridazine with another azine was more likely to lead to an increase in potency rather than a decrease [171]. However, there are several examples where replacing an azine heterocycle with a pyridazine ring has resulted in a decline in potency than can be significant in magnitude. As an illustration of this phenomenon, replacing the phenyl ring in the potent phosphoinositide 3-kinase δ (PI3Kδ) inhibitor 167 with the pyridazine in 168 resulted in an 800-fold reduction in potency that was unique to this heterocycle since the analogous pyridine 169, pyrazine 170 and substituted pyridine 171 fully preserved the inhibitory potency of the progenitor [177]. In this series, the pyrazole heterocycle of the potent inhibitor 172 was shown by an X-ray cocrystal structure to engage the enzyme in dual a H-bond donor/acceptor interaction with Asp787 and Lys779, respectively. Whilst 168, 169 and 170 are all capable of adopting a geometry suitable for engaging Lys779, it was not abundantly clear why the pyridazine 161 was a uniquely poor inhibitor of PI3Kδ, with the introduction of unfavorable, repulsive interactions within the binding pocket offered as the potential underlying reason [177].

In a series of potent, CNS-penetrant PDE10A inhibitors compiled in Table 5 that were of interest as a therapeutic approach to the treatment of schizophrenia, a cocrystal structure revealed that the pyridine nitrogen atom of the lead molecule 173 engaged the backbone of the protein via a H-bonding interaction with an intermediary H2O molecule, with the key interactions depicted in Fig. 34A [178]. Interestingly, in this cocrystal structure the conformation of the quinoline ether moiety does not benefit from an optimal alignment of the dipoles of the heterocycle and the ether element, further demonstrating the chameleonic conformational behavior of this type of substructure (Fig. 34B) [159]. Against the backdrop of that observation, the 500-fold reduction in potency observed with the phenyl analog 174 is unsurprising, while the pyridazine 175 and pyrimidine 176 exhibited progressively weaker PDE10A inhibitory potency (Table 5). The variation in inhibitory potency between the pyridine 173, the pyridazine 175 and pyrimidine 176 was found to be dependent on the H-bonding potential of the azine, a phenomenon explored by calculating the H-bonding potential for these heterocycles. The rank order of potency was predicted by the calculated energies, as presented in Fig. 35A, although the absolute potency differences were not correlated. The contention that H-bonding is a critical determinant of potency is supported by the simple plot of IC50 values against the pKBHX values of the three azine heterocycles that is presented in Fig. 35B, where the correlation is compelling. Assuming an absence of steric, dipole or lipophilicity effects associated with the molecular edits that differentiate 173, 175 and 176, the potency results in this pharmacophore provide some insight into expectations of the effects of azine modification in a circumstance where a single H-bond interaction is important. In this example, replacing the pyridine of 173 with the pyridazine in 176 results in a 25-fold reduction in potency; however, as discussed above, a pyridazine would presumably offer advantage where dual H-bonding is an important element of molecular recognition. In this series, CYP 450 inhibition did not present as a significant issue, attributed to the effect of conjugation with the pyrazole ring, and PF-2545920 (178) was the analog ultimately advanced into clinical trials [178].

A Cocrystal structure of inhibitor 173 with the PDE10A enzyme (3HQZ). B Conformation of 173 abstracted from the cocrystal structure with PDE10A

Plots of PDE10A IC50 value against computed H-bonding potential (A) and pKBHX (B) for pyridine, pyrimidine and pyridazine heterocycles

Pyridazines as pharmacophoric elements
Cocrystal structural data for the dual Rho-associated protein kinase 1 (ROCK1) and ROCK2 inhibitor 179 revealed that the pyridine nitrogen atom of the molecule engaged both kinases via a single H-bonding interaction to hinge residues, accepting a H-bond from the backbone N-H of Met156 in ROCK1 and of Met172 in ROCK2, with N-N distances of 2.82 Å and 2.86 Å, respectively [179]. In each case, the enzyme-inhibitor complexes were further stabilized by an additional H-bonding interaction between the amide C = O of 179 and the side chain ammonium moiety of Lys105 in ROCK1 and Lys121 in ROCK2. While 179 offered promise as a lead inhibitor of the ROCK enzymes, inhibition of CYP 2C9 (IC50 = 2.04 µM), 2D6 (IC50 = 0.20 µM) and 3A4 (IC50 = 0.08 µM) detracted from its drug-like qualities and provided an important focus for the optimization campaign, which explored the introduction of steric encumbrance around the pyridine nitrogen atom and the effects of azine replacements. In the context of the more potent analog 180, substitution of the pyridine with a pyridazine heterocycle (181) resulted in an improved CYP 450 inhibition profile (CYP 2C9 and 2D6 IC50 > 20 µM, 3A4 IC50 = 16.7 µM) but at the expense of a substantial erosion of kinase inhibitory potency, which amounted to 553-fold for ROCK1 and 58-fold for ROCK2 [179]. The pyrimidine analog 182 offered improved performance in both kinase inhibition assays, although this compound was an order of magnitude weaker than 180, while CYP 450 inhibition was also mitigated (CYP 2C9 IC50 = 6 µM, 2D6 IC50 = 11.6 µM and 3A4 IC50 = 19.6 µM). As a consequence, a pyrimidine heterocycle was preserved in the more refined tool molecule 183 that offered enhanced selectivity over protein kinase A (PKA) inhibition. In this specific context, the 44- to 260-fold superiority of the pyrimidine 182 over the pyridazine 181 is not reflected in the pKBHX values for these heterocycles.

Another example from the kinase inhibitor arena where time-dependent CYP 3A4 inhibition (TDI) emerged as a problem was in a series of inhibitors of protein kinase C θ (PKC θ), an enzyme that is selectively expressed in T-cells, platelets, and skeletal muscle [180]. The lead molecule 184 was under study for its potential to treat transplant rejection but incubation of this compound with CYP 3A4 for 30 min resulted in a loss of 64% of the capacity of the enzyme to metabolize midazolam. Whilst CYP 3A4 inhibition could be partially abrogated by structural adjustment to the ethanolamine moiety, this approach ultimately proved to be an inadequate solution that was further exacerbated by the inherently poor aqueous solubility of analogs that retained the (trifluoromethoxy)benzene terminus. As part of an effort to enhance the polarity of the chemotype, the (2-methoxy)pyridine analog 185 was prepared and found to be a potent PKC θ inhibitor with a much improved CYP 3A4 TDI profile (94% residual activity remained after 30 min of incubation). However, in this chemotype the introduction of a pyridazine heterocycle (186) was associated with a substantial, >50-fold loss of inhibitory potency compared to the pyridine 185 [180].

A pyridazine heterocycle was found to be a poor substitute for the 4-ethoxybenzyl moiety of the potent sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (187) and the thienyl ring of canagliflozin (188), drugs that lower plasma glucose levels in vivo by inhibiting glucose resorption and thereby promoting excretion [181]. Installed in the structural backdrop of 187, the pyridazine homolog 189 was almost 100-fold less potent while 190 was associated with a further sixfold weakening of inhibitory potency.

In the agricultural chemistry arena, attempts to replace the pharmacophoric pyridine heterocycle of the thiazole-based insecticide 192, an analog of tyclopyrazoflor 191, were successful in the context of the pyrimidine 193, which exhibited improved potency toward killing the green peach aphid M. persicae [182]. However, the pyridazine congener 194 was found to be inactive, even at the highest tested dose of 200 mg/L. Interestingly, in this chemotype attempts to replace the scaffolding thiazole moiety with a pyridine heterocycle, an established bioisostere, were also not successful [182].
Pyridazine heterocycles as scaffolding elements
The pyridazine heterocycle has also been explored as a replacement for phenyl- and azine-based scaffolding elements, with the effects on potency dependent on the circumstance under study. In the muscarinic M1 PAMs compiled in Table 6, the prototype biphenyl derivative 195 exhibited modestly potent potentiation of the effects of acetylcholine (ACh) acting at the muscarinic M1 receptor but was associated with a high associative binding to both rat (98%) and human (98.4%) plasma proteins [183]. Replacing the central phenyl ring with azines was explored as an approach to enhancing polarity with a view to reducing plasma protein binding, an enterprise that met with only modest success and which necessitated further optimization [183, 184]. The SARs presented in Table 6 reveal specificity for both azine identity and topology, with pyridine 197 a more potent M1 potentiator than its isomer 196, while pyrimidines 198 and 199 and the pyrazine 200 were poorly active. The pyridazine 201 preserved the biochemical potency of the prototype 195; however, the increased polarity did not significantly affect the binding of the molecule to human plasma proteins [183]. In a follow up study where replacement of the terminal phenyl ring of 195 with a pyrazole (202) enhanced potency (Table 7), a pyridine heterocycle scaffolding element was tenfold less potent in both topologies (203 and 204) and the less basic pyridazine 205 fared even more poorly, reducing potency by 250-fold, a remarkably different result to that observed in the biphenyl series based on the data compiled in Table 6 [184].
In the series of cannabinoid receptor antagonists 206–210 compiled in (Table 8), pyridine 206 provided the targeted CB1-selective profile and potency while azine variants of the pyridine core revealed that potent CB1 affinity was only preserved with the pyrazine 209 [185]. In this setting, the results were interpreted in favor of the preferred conformation for 209 that is depicted in Fig. 36, where the amide topology is favored by an intramolecular H-bond between the amide N-H and the pyrazine nitrogen atom. The pyridines 207 and 208 will favor this amide topology for the same reason but 207 will present the chlorophenyl ring and ether substituents in an altered topology that does not complement the receptor binding pocket. The isomeric pyridine 206 exhibits no specific conformational preference since the ring nitrogen atom cannot engage in intramolecular H-bonding but is believed to engage in an intermolecular H-bonding interaction with the CB1 receptor, explaining the potency enhancement when compared to 208. The conformational argument also explains the poor activity associated with the pyridazine 210 in the context of this pharmacophore [185].

Preferred conformation of 209 that is favored by the depicted intramolecular H-bond
The potent prolylhydroxylase domain-1 (PHD-1) inhibitor 211 (IC50 = 34 nM) presented in Table 9 is unusual in that it engages the catalytic Fe2+ atom via a monodentate interaction involving the N-1 nitrogen atom of the [1,2,4]triazolo[1,5-a]pyridine core while the N-3 nitrogen atom engages with the hydroxy of Tyr313 via the intermediacy of a H2O molecule, as depicted in Fig. 37 [186]. In addition, the nitrogen atom of the benzonitrile moiety engages the protein via a H-bonding interaction with the side chain amide of Asn315 in a pocket that is created by movement of the side chain of Arg367, a residue that is often directly engaged by carboxylic acid moieties present in other PHD-1 inhibitors, while the phenyl ring π-stacks with Tyr287. Decoration of the phenyl ring in the positions ortho and meta to the nitrile was restricted to small substituents while attempts to replace this scaffolding element with azines led to reduce potency, with the pyrimidine 214 superior to the pyridines 212 and 213. In this context, both the pyrazine 215 and the pyridazine 216 experienced substantial, 40-fold declines in potency, which does not appear to correlate with the effect of the electron withdrawing properties of the azine ring on the H-bond basicity of the nitrile.

Key drug-target interactions between PHD-1 and inhibitor 211 in the cocrystal structure (A) and as a two-dimensional representation (B) (5V1B)
In the trifluoromethyl dihydrothiazine-based β-secretase (BACE1) inhibitors, the pyrimidine 217 and pyrazine 218 offered similar potency in both biochemical and cell-based assays; however, the pyridazine 219 was close to an order of magnitude less potent in the biochemical assay and 20-fold less effective in the cellular assay [187]. Interestingly, in this context the installation of both a pyrazine and pyridazine heterocycle resulted in a significant increase in the measured Log D values compared to the pyrimidine progenitor 217.

Applications of the electron withdrawing properties of pyridazines
The electron deficient 3-position of pyridazine can be exploited to modulate the properties of substituents, acidifying an attached N-H or activating a carbonyl or nitrile moiety. One chemotype where this physicochemical property of the pyridazine ring has been exploited is in the context of inhibitors of fatty acid amide hydrolase (FAAH), a serine hydrolase that cleaves fatty acid amides to release the parent carboxylic acid [188,189,190]. FAAH preferentially hydrolyzes amides derived from arachidonic and oleic acids and inhibitors offer potential in the treatment of pain and sleep disorders by enhancing levels of fatty acid amides which act on a variety of endogenous receptors [191]. α-Keto heterocycles are designed to present an electrophilic carbonyl moiety to the enzyme as a pseudo substrate that reacts to form a stable, covalent hemi-acetal intermediate with the catalytic serine hydroxy, as depicted for 220 in Fig. 38 [188, 189, 192]. The SARs summarized in Fig. 39 reflect a clear dependence of potency on both the electron withdrawing properties of the heterocycle deployed and its point of attachment to the ketone [190, 193, 194]. The latter phenomenon is illustrated by the poor activity associated with the oxazole 222 compared to the topological isomers 220 and 221, in which the ketone is installed at the more electron deficient 2-postion of the heterocycle. However, the thiazole 223 is fourfold weaker than its oxygen isostere 222 despite being the stronger electron withdrawing moiety, an SAR point that may reflect geometrical differences associated with the longer carbon-sulfur bonds and the geometrically altered topology [3]. The 1,2,4-oxadiazole 224 and 1,3,4-thiadiazole 225 exhibit 16-fold and 30-fold enhanced potency, respectively, compared to the matched prototypes 220 and 223, whilst the poor activity associated with the triazole 226 was attributed to the presence of the acidic N-H [193, 194]. In this chemotype, the performance of the pyridazine 227 is comparable to that of the thiazole 223, which has similar electron withdrawing properties, but inferior to the oxazoles 220 and 221, despite being the more electron deficient at C-3 compared to C-2 of 220 and 221. In this example, the geometrical attributes of pyridazinyl ring may account for the reduced potency compared to the more active azole analogs.

A Key drug-target interactions between a humanized rat FAAH and 220. B Two-dimensional representation of the key interactions between 220 and FAAH (2WJ2)

SARs associated with a series of α-keto heterocycle-based FAAH inhibitors
Whilst aminomethyl nitriles, aryl nitriles and several heteroaryl nitriles have found application in the design of mechanism-based cysteine protease inhibitors, derivatives of pyridazine-3-carbonitrile appear to be notably absent [195,196,197,198,199]. Pyridazine-3-carbonitrile offers a nitrile with electrophilicity that is comparable to that of pyridine-2-carbonitrile, although both are calculated to be much less reactive toward CH3SH than pyrimidine-2-carbonitrile (Fig. 40) [200]. The chemical susceptibility of these three heterocyclic motifs toward thiols has been studied in some detail and all were shown to form thiazoline adducts when incubated with glutathione (GSH) or N-acetyl cysteine (NAC) in a process catalyzed by HLM [200, 201]. However, the independence of the effect of HLM from the CYP 450 cofactor NADP(H) was suggestive of the involvement of microsomal enzymes like γ-glutamyl-transpeptidase [200, 201]. As depicted in Scheme 1, incubation of a series of heteroaryl nitriles 228 with GSH and HLM afforded the two thiazoline adducts 229 and 230, with the former produced when 228 was incubated with NAC. The formation of the thiazoline heterocycle was proposed to proceed by an initial attack of the thiol moiety of GSH or NAC on the nitrile in a thio-Pinner-type of reaction process to afford a thioimidate intermediate which cyclized with the loss of NH3 to afford the thiazoline, with subsequent cleavage of the glycine moiety by a peptidase in the case of the GSH adduct [200, 201]. The reaction of nitriles with cysteine has been shown to correlate well with the calculated reactivity toward CH3SH, which can be modulated by structural modifications that affect nitrile electronics, and this was observed with the series of substrates represented by 228 [198, 200, 201]. The susceptibility of pyridazine-3-carbonitrile to thiol addition suggests that the deployment of this structural motif in circumstances where chemical reactivity is viewed as a potential liability may require some design finesse in order to optimally tune the inherent electrophilicity [198,199,200,201].

Calculated energetics (kcal/mol) associated with the reaction of aryl and heteroaryl nitriles with CH3SH

Reaction of heteroaryl nitriles with GSH and NAC to form thiazoline derivatives
Although there may be concerns around the potential for pyridazine-3-carbonitriles to be inherently reactive toward biological nucleophiles, this motif has found gainful application in drug design in circumstances that do not rely upon nitrile activation. A recent and interesting example is provided by a series of allosteric activators of pantothenate kinase (PanK) that function by blocking feedback inhibition of the enzyme by CoA thioesters [202,203,204]. PanK catalyzes the first and rate-determining step in the biosynthesis of Coenzyme A (CoA), thereby regulating the cellular levels of this cofactor which is important for mitochondrial function and both fatty acid and folate metabolism. There are four known isoforms of PanK in human cells that are encoded by three genes and they are differentially expressed in tissues. Inactivating mutations in PanK2, the major isoform of the enzyme found in neurons, results in depressed levels of CoA that precipitates pantothenate kinase-associated neurodegeneration (PKAN). PKAN is a rare disease in which there is progressive degeneration of specific regions of the CNS that manifests as a neurological movement disorder involving changes in muscle tone, involuntary abnormal movements, and postural disturbances [202,203,204]. Modulators of PanK enzymes, designated as pantazines, have been discovered by HTS campaigns and these molecules act as allosteric activators by associating with the pantothenic acid binding site of one protomer of the dimeric enzyme in a fashion that restrains the other protomer in an active conformation that is resistant to feedback inhibition by CoA thioesters. With CNS penetration a key consideration, optimization of the lead modulator PZ-2789 (231) focused on monitoring cLog P values and maximizing lipophilic efficiency (LipE, which is defined as pIC50 – cLog P) while maintaining physicochemical properties compatible with blood-brain barrier permeability [202,203,204]. Replacing the urea moiety with an amide (232) removed a H-bond donor and improved potency by tenfold whilst editing of the pyridine ring to a pyridazine further enhanced inhibitory potency by 60-fold, affording PZ-2891 (233) as a potent PanK3 modulator with a high LipE value of 6.65 based on a cLog P value of 2.35 [202, 203]. An X-ray cocrystal structure of 233 bound to PanK3 revealed that molecular recognition was dominated by two key H-bonding interactions, both of which involved the side chain guanidine moieties of arginine residues within the enzyme. The amide carbonyl moiety engaged in a dual H-bonding interaction with the side chain guanidine terminus of Arg207, which is known to engage the amide carbonyl and CO2H terminus element of pantothenic acid monophosphate (Fig. 41) [202, 203, 205]. Both nitrogen atoms of the pyridazine heterocycle formed H-bonds with the guanidine moiety of Arg306 of the other protomer (designated as Arg306’), as depicted in Fig. 41 [202, 203, 205]. This observation adds to the repertoire of intermolecular H-bonding interactions of the pyridazine ring with protein targets in molecular recognition (vide supra). The 60-fold difference in potency between the pyridine 232 and the pyridazine 233 provides an index of the value of the additional H-bonding interaction in the pyridazine derivative. In this context, the pyridazine ring is functioning in a fashion similar to that of a carboxylate moiety presenting a similar planar geometry but in the absence of the burden of a formal negative charge, an observation that may portend additional opportunity in drug design [206]. This interaction also has some analogy to sulfone mimicry of a carboxylate where the two sulfone oxygen atoms can emulate the H-bonding interactions, also in the absence of a formal charge, but with a very different geometry to that of the planar carboxylate moiety and pyridazine heterocycle [207, 208]. Additional stabilization of the complex between 233 and PanK3 was conferred by a π-stacking interaction between Trp341’ and the pyridazine ring, while the nitrile nitrogen atom was positioned 3.12 Å from the carbonyl carbon atom of Gly302’, possibly reflective of a multipolar interaction and/or dipole interaction [209, 210]. The value of the pyridazine ring in this context is further underscored by the relatively modest potency associated with the benzonitrile 234 while the importance of amide topology is defined by the ketone 235 which is inactive [202, 203]. Modification of the isopropyl substituent, which mimics the gem-dimethyl element of pantothenic acid, to a cyclopropyl ring (236) was required in order to enhance metabolic stability, a molecular edit also associated with a reduction in the cLog P value from 2.3 to 1.9 [205].

Key interactions between 233 (PZ-2891) and the PanK3 enzyme (6H4K)


A cinnoline-3-carbonitrile has found application in the design of inhibitors of scytalone dehydratase, an enzyme in the plant fungal pathogen Magnaporthe grisea that catalyzes two steps in the melanin biosynthesis pathway [211]. The quinazoline 237 and the benzotriazine 238 were identified as potent scytalone dehydratase inhibitors with Ki values of 0.15 nM and 0.22 nM, respectively. Modeling of these compounds in the active site of the enzyme recognized the presence of an extended H-bonding network that included two H2O molecules, with that bridging between Tyr30 and Tyr50 and engaging the nitrogen atom of the heterocyclic inhibitor viewed for its potential to be displaced by incorporation of a nitrile substituent to the core scaffold (Fig. 42A). If the design principle was successful, the nitrile nitrogen atom would be able to engage the tyrosine hydroxy substituents directly by H-bonding and the energetics of the drug-target association would benefit from the entropic advantage of displacing the H2O molecule, as depicted in Fig. 42B [162, 163, 212, 213]. The isoquinoline and cinnoline nitriles 239 and 240 were found to be considerably more potent inhibitors of scytalone dehydratase, with advantages of 22-fold and 28-fold, respectively, over 238 [211]. Comparison of the matched molecular pairs 241 and 242, which lack both the nitrile substituent and the heterocyclic nitrogen atom capable of H-bonding with the H2O molecule, further emphasized the success of the design principle since the inhibitory potency declined by more than 20,000-fold for each. A cocrystal structure of the cinnoline 240 with scytalone dehydratase confirmed that the nitrile moiety had indeed displaced the H2O molecule, as depicted in Fig. 43, with the nitrile nitrogen atom directly engaging both tyrosine hydroxy substituents, as had been predicted [211].

Proposed key interactions between scytalone dehydratase and 237 (A) and 239 (B)

Key interactions between scytalone dehydratase and 240 in the cocrystal structure (3STD)

Pyridazine heterocycles and the modulation of solubility properties
The dipole moment and H-bonding properties of the pyridazine ring contributes to the lower Log P/D value compared to a phenyl ring, which can confer beneficial physicochemical properties to a molecule [1, 27]. An interesting illustrative example is provided by a study of analogs of navitoclax (243), a large (MW = 974.5) and highly lipophilic molecule (cLog P = 12.4) that binds to members of the antiapoptotic B-cell lymphoma 2 (Bcl-2) family of proteins and prevents them from sequestering apoptotic proteins [214]. In the study compiled in Table 10 that exploited 244 as the prototype, the effect of replacing the central phenyl ring with heterocycles was explored with a view to enhancing aqueous solubility. The pyridazine analog 245 (cLog P = 8.7) helped in that purpose, with an almost threefold enhancement in aqueous solubility, although at the expense of a 5- to 10-fold erosion of binding affinity. Interestingly, the isomeric pyrimidine 246 (cLog P = 8.8), whilst retaining binding potency toward Bcl-2 and Bcl-xL, exhibited tenfold lower solubility than 245 and was, remarkably, less soluble than the phenyl prototype 244 (Table 10) [214].

A pyridazine ring emerged as the optimal heterocycle in a survey of replacements for the pyridine in the lead cannabinoid CB2 receptor agonist 247, with 252 preserving the binding affinity and efficacy of the prototype whilst providing some improvement in solubility compared to the pyrazine 251 and triazine 253 (Table 11) [215]. Although still poor, the solubility of 252 was enhanced by 20-fold to 120 µg/mL by formation of a salt with HCl and this core was selected as the vehicle for further study in a search for compounds that could effectively treat inflammatory pain. The isoquinoline derivative 254 met the targeted criteria, with potent and selective CB2 agonism, good aqueous solubility as the di-hydrochloride salt and targeted metabolic stability in RLM, although clearance in vivo was characterized as moderate-high. Nevertheless, these properties collectively translated into efficacy in rat models of inflammatory pain at doses ranging from 0.1–1 mpk per os (PO) without overt effects on the CNS via the modest inherent CB1 agonist activity at doses of up to 300 mpk [215].

The pyridazine ring in the protein kinase CK2 inhibitor 258 conferred much higher solubility in aqueous buffer than either of the two pyridine homologs 256 and 257 while, interestingly, none of these modifications were deleterious to the potent kinase inhibition observed with the phenyl prototype 255, as summarized in Table 12 [216]. However, the more polar compounds 256–258 showed much reduced anti-proliferative activity in A549 lung cancer cells which was attributed to reduced membrane permeability.
Solving hERG and TDI CYP inhibition issues by the introduction of a pyridazine heterocycle
The ataxia telangiectasia and Rad3-related protein (ATR) kinase regulates the S and G2 checkpoints and DNA repair following exposure of cells to drugs that damage DNA and inhibitors of this enzyme sensitize cancer cells to cytotoxic agents [217]. A combination of virtual and high-throughput screening identified the imidazo[1,2-c]pyrimidine derivative 259 as a potent lead ATR inhibitor that exhibited remarkable kinase selectivity with respect to the 76 kinases tested. Docking of 259 in a homology model of ATR that was constructed based on PI3Kδ suggested that the morpholine oxygen atom engaged the backbone N-H of Val2380, which is part of the hinge of the kinase, while the indole adopted a conformation that was distorted about 30o from the plane of the imidazopyrimidine core, which facilitated its projection into an affinity pocket. However, the imidazo[1,2-c]pyrimidine core failed to provide a path forward, attributed to unfavorable intramolecular interactions between the morpholine and imidazopyrimidine rings which restricted the conformational mobility of the former. A topological rearrangement of the nitrogen atoms in the core of 259 relieved this constraint and, as compiled in Table 13, the imidazo[4,5-c]pyridine-based 260 demonstrated 400-fold increased ATR inhibitory potency compared to the progenitor [217]. The enhanced potency was attributed, in part, to the improved vectors that allowed the morpholine ring to adopt a conformation where the CH3 substituent could more effectively fill a lipophilic pocket while the imidazole nitrogen atom provided a site for derivatization to access a second binding pocket. However, 260 and close analogs suffered from both hERG and CYP 3A4 inhibition, with the latter time-dependent, which precluded further advancement. Installation of an azaindole that was designed take advantage of a H-bond interaction with Lys2327, provided 261 as a potent ATR inhibitor that expressed activity in the cell-based pChk1 assay. However, both hERG and CYP 3A4 TDI remained a problem, with susceptibility to aldehyde oxidase (AO) and P-gp-mediated extrusion from cells appearing as additional challenges. The ortho-fluorine substituent in 262 reduced the basicity of the azaindole core and abrogated the hERG inhibition while also lowering the susceptibility to metabolism by AO and recognition by P-gp; however, this compound was positive in the CYP 3A4 TDI assay. The solution was to replace the C-F of 262 with a nitrogen atom to provide the 1H-pyrrolo[2,3-d]pyridazine derivative 263, which embeds a pyridazine heterocycle. This molecular edit resolved all four of the liabilities and provided a compound that, although moderately orally bioavailable in rats, exhibited low Cl and provided a useful tool molecule to enable further study (Table 13) [217].

Inhibitors of hypoxia-inducible factor (HIF) prolyl hydroxylase 1–3 inhibitor (PHD) stabilize the enzyme and stimulate red blood cell (RBC) production in vivo via activation of the erythropoietin receptor (EpoR), offering a potential approach to the treatment of anemia [218]. A HTS campaign identified 264 as a potent PHD2 inhibitor in which the hydroxypyrimidine, which is likely deprotonated at physiological pH, and the pendent amide C = O combine into a Fe2+ binding moiety that together function as a carboxylic acid bioisostere [218, 219]. Interestingly, this molecule is absent the overt carboxylic acid moiety that is found in many PHD2 inhibitors and which engages Arg383, with modeling studies suggesting that the pyridine ring of 264 projects in the channel toward this residue. The quality of 264 as a lead was further underscored by its PK profile in rats where it was characterized by low clearance, a t½ of 3.3 h and oral bioavailability of 67%. More importantly, 264 was devoid of the hERG inhibition (IC50 > 30 µM) that had plagued an earlier series of pan-PHD1–3 inhibitors constructed on a 1,3,8-triazaspiro[4.5]decane-2,4-dione core scaffold [220]. Gem-demethylation of the benzylic position increased potency fourfold, providing a vehicle with which to probe pyridine replacement SARs, with the pyrazole 265 and pyridazine 266 suitable substitutes that were 7- and 2-fold more potent, respectively, while the profile of the pyrimidine 267 was much less attractive. Pyrazole 265 exhibited a good PK profile in the rat, with F = 99% and a t½ of 4.2 h. However, the t1/2 values for 265 in the cynomolgus monkey and the dog were exceptionally long, at >90 h, reflective of the low turnover of the compound in LMs from rat, dog, cynomolgus monkey and humans. With concern around the long t½ values, additional bulk was installed at the benzylic position along with the introduction of potential metabolic soft spots in the guise of the MeO substituents found in 268. Modeling studies suggested that one of the anisole rings could interact with Arg322 in a π-cation interaction while both MeO substituents had the potential to engage the enzyme via H-bonding with the N-H of Trp258 and the side chain amide NH of Asn318. However, hERG inhibition was found to be a liability for pyrazole 268, with an IC50 value of 900 nM, but this was completely resolved with the molecular edit that afforded the pyridazine MK-8617 (269), hERG IC50 > 30 µM. Equally important, 269 was free of the burden of CYP inhibition, with the IC50 values for all enzymes tested >60 µM with the exception of 2C8, IC50 = 1.6 µM, while additional preclinical profiling supported advancing this compound into clinical trials [218].

Antagonists of the histamine H3 receptor have been studied for their potential as therapeutics in the treatment of CNS disorders that are associated with deficiencies in neurotransmitter release, conditions that include Alzheimer’s disease, attention-deficit/hyperactivity disorder (ADHD) and sleep disorders [221, 222]. The urea 270 was identified as a novel and potent histamine H3 antagonist from a screening campaign but was also characterized as a potent hERG inhibitor in a high-throughput Rb efflux assay, with inhibition subsequently confirmed in an IonWorks assay [222]. A broad-based SAR survey indicated that potency in both assays followed similar trends but a careful focus on controlling lipophilicity provided an advance in the guise of 271, where the separation in potency in the H3 receptor and IonWorks assays was amplified from 2-fold to 950-fold. Appending a pyridazine heterocycle (272) further modulated hERG inhibition in a productive direction, with H3 antagonist potency fully restored in the bromo homolog 273 where the pyridazine continued to confer resistance against hERG inhibition. The improvement in the hERG inhibitory profile of this series was attributed to the reduction in cLog P values from 6.56 for 270 to 2.98 for both 272 and 273, although across the series the correlation was not absolute [221]. Additional profiling of 273 indicated that the molecule combined good kinetic aqueous solubility (>250 µM) with high Caco-2 membrane permeability and while orally bioavailable in the rat, the t1/2 was short at 1 h, attributed to rapid metabolism. Notably, in this study attempts to deploy pyridine heterocycles in place of the pyridazine were unsuccessful, presenting unresolvable challenges in balancing histamine H3 affinity with reduced hERG inhibitory potency.
The PI3K pathway has been shown to be activated in a broad range of human tumors and signaling can be initiated indirectly by the activation of receptor tyrosine kinases (RTKs) or by inactivation of the tumor suppressor phosphatase and tensin homolog PTEN. GSK-1059615 (274) is a potent inhibitor of PI3Kα (IC50 = 2 nM) that inhibits the phosphorylation of Akt kinase in cells (EC50 = 40 nM), profiling data that subtended its advancement into clinical trials (Table 14) [223]. Follow-on studies sought a refined inhibitor with enhanced potency, higher selectivity and improved PK properties, with optimization guided by a process exploiting structure-based drug design (SBDD) principles. The X-ray cocrystal of 274 with PI3Kγ, used as a surrogate for the α isoform of the enzyme, indicated that polar elements of the thiazolidinone ring engaged with the side chain ammonium terminus of the catalytic Lys833. This analysis also identified the potential for installing larger groups in this region of the pharmacophore, which was viewed as a useful approach to enhancing potency. The succession of structural modifications began by replacing the thiazolidinone heterocycle with a simple phenyl ring (275), which resulted in a large reduction in potency that was restored in a stepwise fashion through the progression of pyridine 276 and indazole 277, with elements of these two hybridized to provide 278 as a potent enzyme inhibitor that translated into good cellular potency (Table 14) [223]. However, it was the molecular edit to the sulfonamide 279 that set the stage for additional structural manipulation that arrived at 280 as a compound demonstrating targeted potency in the in vitro assays. However, 280 exhibited CYP 450 inhibition at unacceptable levels, a liability solved by introduction of a CH3O substituent ortho- to the sulfonamide moiety and manipulation of the 4-pyridine heterocycle to the pyridazine found in GSK2126458 (281). These structural changes resulted in an improved CYP inhibition profile, with IC50 values toward CYP 1A2, 2C9, 2C19, 2D6 and 3A4 all >25 µM. In the X-ray cocrystal structure with PI3Kγ, the sulfonamide of 281, which is acidic and 87% deprotonated at physiological pH based on a pKa value of 6.56, engaged the side chain of Lys833, while the structural attributes allowed read through-type access to a lipophilic pocket that is filled by the fluorinated aryl ring. GSK2126458 (281) is a pan PI3K inhibitor with a preclinical PK and toxicological profile deemed suitable for advancement into clinical study [223].

An example of the advantage of a pyridazine over other heterocycles for abrogating CYP inhibition is provided by the series of urea-containing nicotinamide phosphoribosyltransferase (NAMPT) inhibitors 282–286 compiled in Table 15 [224]. NAMPT catalyzes the rate-determining step in the biosynthesis of nicotinamide adenine dinucleotide (NAD) from nicotinamide, with inhibitors of this enzyme anticipated to abrogate the growth of cancer cell lines where the cellular metabolic state is elevated. The pyridine 282 was identified as a potent NAMPT inhibitor that exhibited anti-proliferative activity toward the A2780 ovarian cancer cell line but was also a potent inhibitor of CYP 2C9. The SARs around pyridine variation summarized in Table 15 indicated that in this specific context, the pyridazine homolog 285 retained NAMPT inhibitory potency in the biochemical assay while moderating CYP 2C9 inhibition by tenfold, although cellular antiproliferative activity was also reduced. In contrast, the pyrimidine 283 and pyrazine 284 were poor NAMPT inhibitors, although CYP 450 the liability was also attenuated with these molecular edits. Interestingly, the optimal solution arrived at for this series was the aminopyridine 286 and an X-ray cocrystal structure revealed that this molecule, like many pyridine derivatives, is a mechanism-based enzyme inhibitor. Thus, the pyridyl nitrogen atom forms an alkylated adduct by reacting with phosphoribosyl pyrophosphate (PRPP) in the active site of the enzyme to produce what is, presumably, a more potent inhibitory species that drives the observed cellular activity [224].
A scaffold hop from a pyridine to a pyridazine was probed in an effort to reduce the CYP 450 inhibition liability associated with a series of spingosine-1-phosphate 2 (S1P2) antagonists pursued for their potential to treat idiopathic pulmonary fibrosis [225]. The pyridinyl urea 287 exhibited potent antagonism of S1P2-mediated Ca2+ flux in CHO cells expressing the human receptor, with a reasonable LipE value of 4.4 that translated into effective inhibition of IL8 production in human ling fibroblasts. However, liability profiling revealed that 287 was a potent inhibitor of CYP 450 2C9, 2C19 and 3A4 (83, 100 and 83%, respectively, at a concentration of 10 µM). The matched pyridazine analog 288 maintained intrinsic S1P2 antagonism that was associated with an improved LipE value due to the reduction in cLog P from 3.6 to 3.0. This was attributed to the effects of the more polar pyridazine ring which translated into more potent inhibition of IL8 production in the cell-based assay while being associated with reduced CYP 450 inhibition liability (at a concentration of 10 µM, CYP 2C9, 2C19 and 3A4 were inhibited by 58, 61 and 23%, respectively). As a consequence, the pyridazine core was adopted for further optimization which identified GLPG2938 (289) as a development candidate that did not present a significant CYP inhibition liability, with IC50 values of >33 µM for CYP 450 2C19 and >100 µM for CYP 450 1A2, 2C9, 2D6 and 3A4 [225].


The piperidinyl urea PF-3485 (290), which traces its origins to a lead discovered by a HTS campaign, is a time-dependent, mechanism-based inhibitor of FAAH that is accepted by the enzyme as a substrate, presenting the urea moiety to the catalytic Ser241 hydroxy which reacts to expel 3-aminopyridine and leave behind an inactive, carbamoylated enzyme [226,227,228,229]. The Ser241-Ser217-Lys142 catalytic triad of FAAH distinguishes it from other hydrolases which use a Ser-His-Asp motif and this difference confers an ability for FAAH to process both esters and amides with similar efficiency [226]. The early SARs in this series indicated that a 3-aminopyridine heterocycle was twofold more reactive than either a 2-aminopyridine or a 3-aminopyridazine and this motif was used for additional studies of 290 [229]. Introducing unsaturation at the piperidine 4-postion afforded 291, which was a twofold more potent inhibitor of human FAAH and twofold more potent toward rat FAAH, as measured by the second order rate constant kinact/Ki which is not sensitive to substrate concentration or incubation time (Table 16). However, the pyridine moiety in 2901 conferred potent CYP inhibition, with IC50 values toward 2D6 and 3A4 of less than 10 µM. Replacing the pyridine heterocycle of 291 with either a pyrazine (292) or a pyridazine (293) abrogated the CYP liability whilst also enhancing FAAH inhibitory potency by twofold and the latter compound, PF-04457845 (293), was selected as a clinical candidate for evaluation as a potential therapy to treat inflammatory pain [230].
The histamine H1 antagonist 295, patterned after dimethindene (294), was advanced into clinical trials for the treatment of insomnia [231, 232]. However, in further studies with the chemotype, a detailed analysis of the metabolism of 295 and the indene-based pyrazine homolog 297 revealed that both were metabolized predominantly (>90%) by CYP 2D6, a polymorphic enzyme with heterogeneity across the population that can be a source of PK variability. Against this backdrop, a series of analogs was screened of using an assay designed to assess the contribution of CYP 2D6 to metabolism, with a cut-off of a 60% bias targeted as an objective. An analysis of the data indicated that substitution at C-6 of the indene core contributed to reduced susceptibility to metabolism by CYP 2D6 and the data obtained from a cadre of 10 compounds narrowed interest to 297 and the pyridazine 298, both of which were right on the cusp of the targeted criterion. However, interest in 297 and 298 strengthened with the observation that amongst the cohort of compounds evaluated, these compounds were uniquely susceptible to formation of a stable N-glucuronide, a known metabolic pathway for antihistamines [233]. The presence of an alternative pathway for metabolism relieved concern about the dependence on CYP 2D6 and 297 and 298 were studied in further detail where they were compared to 295 [232]. The profiles of the three compounds, which included the PK results from microdosing studies conducted in normal healthy volunteers (NHVs), are compiled in Table 17. All three compounds exhibited high affinity for the histamine H1 receptor and exhibited no significant binding to a panel of receptors and enzymes, with >1000-fold selectivity over the monoamine histamine H3, serotonin 5-HT2A, and muscarinic M1 and M3 receptors, although analogs of 297 bearing a CH3O substituent at the 3-position of the pyrazine ring exhibited significant binding to the histamine H2 receptor. Liability profiling of 297 and 298 revealed that the inhibitory effects on CYP 450 2D6 and 3A4 was evident only at concentrations >10,000-fold higher than histamine H1 affinity while the margin over hERG inhibition was >500-fold [232]. An additional targeted property for an alternative clinical candidate to 295 was a longer t1/2 in humans and in vitro studies projected that the systemic clearance of 297 and 298 would be lower than for 295, a prediction that was confirmed in the human microdosing studies. However, the half-life and AUC values for 295 and 297 exhibited higher variability than 298 for reasons that were not clear but contributed to the selection of this pyridazine derivative as the preferred clinical candidate [232].


Pyridazines and metal binding
Although pyridazines, phthalazine and cinnoline derivatives have not been associated with potent CYP 450 inhibition, unlike pyridine and some of its homologs, these heterocycles will bind to metals when appropriately configured, typically when additional metal binding elements pendent to one of the azine nitrogen atoms are present that enhance the metal coordination capability [60,61,62, 234, 235]. Bipyridazine (299) and homologs can form complexes with metals under experimental conditions in vitro but applications in which the coordination of pyridazine or its fused homologs to metals in the design of enzyme inhibitors has been limited [234,235,236,237,238]. One example is provided by the cinnoline-based HIF-PHD inhibitor 300 that engages the catalytic Fe2+ ion of the hydroxylating enzyme via the nitrogen atom proximal to the amide (Fig. 44) [239,240,241,242]. Cinnoline 300 is a potent HIF-PHD inhibitor, IC50 = 155 nM, that was originally studied an inhibitor of the chicken enzyme.

Structure of 300 bound to the HIF-PHD enzyme from the cocrystal structure (A), with the key drug-target interactions delineated two dimensionally (B) (6NMQ)
Pyridazines and bioisosterism
In the foregoing discussion, the bioisosteric relationship between pyridazines, phenyl rings, azines, diazoles has been highlighted, where effective mimicry can be subtended by shape, H-bonding effects or dipole moments, which often act in combination. The relationship between the dipole moment values of pyridazine, pyridine, pyrimidine, 1,3,4-oxadiazole and 1,3,4-thiadiazole and fluorinated phenyl rings is summarized in Fig. 45 and provides insight into the potential for functional mimicry based on this physicochemical parameter [243]. The bioisosteric similarity between pyridazine (1) and the fused bicyclic heterocycles imidazo[1,2-a]pyrimidine (302), imidazo[1,2-b][1,2,4]triazine (303) and 8-fluoroimidazo[1,2-a]pyridine (304) has been recognized in the context of studies of a series of ligands that bind to the benzodiazepine site of γ-amino butyric acid A (GABAA) ligand-gated chloride channel receptors and act as PAMs to potentiate the effects of γ-aminobutyric acid (GABA) [244]. The landscape of allosteric GABAA receptor ligands includes molecules incorporating the pyrazolo[1,5-d][1,2,4]triazine (305) and 5H-imidazo[4,5-c]pyridazine (306) heterocycles, more overt derivatives of pyridazine that support the proposed molecular mimicry. Clinical candidates that expressed a non-sedating anxiolytic profile were sought based on the concept of identifying selective GABAA α2/α3 PAMs that avoided functional activity at the α1 receptor, although potent binding affinity was typically evident with these compounds [244]. The 1,2,4-triazolo[4,3-b]pyridazine derivative L-838417 (307) was the prototype GABAA α2/α3 PAM that helped to define an understanding around receptor selectivity and although a potent ligand for GABAA α1, α2, α3 and α5 receptors, partial functional activity was expressed only at α2, α3 and α5 subtypes [244, 245]. The poor preclinical PK profile of 307 prevented progression beyond that of a tool molecule but the homologs MRK-409 (308) and TPA023 (MK-0777, 309) were suitable candidates for development and both were advanced into clinical trials. Clinical experience with 308 revealed sedation in humans, unanticipated based on preclinical studies but attributed to a higher sensitivity of human GABAA α1 receptors to the low efficacy (18%) recorded in the in vitro assays with this compound [244]. The relatively small structural changes implemented in 309 were sufficient to eliminate efficacy at the α1 receptor and clinical evaluation of this compound suggested an anxiolytic profile in the absence of sedation; however, the observation of cataracts in dogs in long term toxicology studies led to the compound being abandoned [244, 246]. The imidazo[1,2-b][1,2,4]triazine derivative TPA023B (310) offered higher efficacy at GABAA α2, α3 receptors than both 308 and 309 and was well-tolerated in clinical study where it did not cause sedation at a GABA receptor occupancy of 52 and 46% measured at 5 and 24 h post-dose, respectively; however, this compound was ultimately abandoned for business reasons [244, 247, 248]. The imidazo[4,5-c]pyridazine derivative PPF-06372865 (CVL-865, darigabat, 311) originated with a screening campaign that identified multiple chemotypes for which the structure-property relationships were expanded and triaged based on the potential to deliver the targeted profile of high receptor affinity, functional receptor selectivity, metabolic stability in LMs and CNS penetration based on permeability across MDCK cells [244, 249, 250]. In preclinical studies, darigabat (311) demonstrated anxiolytic and anticonvulsant activity in mouse models of disease and was advanced into clinical trials where it demonstrated efficacy in reducing the response to intermittent photic stimulation in photosensitive epilepsy patients, leading to development being focused on this indication and panic disorder [244, 251].

Dipole moment values associated with fluorinated benzenes and select azine and azole heterocycles


In this extensive series of GABAA agents, the imidazo[1,2-a]pyrimidine derivative 312 exhibited a receptor and efficacy profile similar to the core aza homolog 310, providing an additional chemotype for study [252]. It was in this structural context that bioisosterism between a ring nitrogen atom and a C-F was explored as part of the effort to avoid α1 agonism, with the results summarized by the matched series of analogs 313–315 [253]. The receptor affinity differences between 313 and 314 were relatively modest at 6- to 10-fold and although this might be viewed as exhibiting some consistency with a H-bonding interaction, a more sophisticated explanation focused the structure-function analysis on the physicochemical properties of the three core heterocycles that encompassed the complexion of the electrostatic surface, pKa and dipole moments [26]. The dipole moments of 313 and 315 were both larger than for 314, indicating that the C-H to C-F molecular edit represented a useful bioisosteric similarity that was reflected in maps of the electrostatic surface [253]. The effects of fluorination on the measured pKa value aligned with the aza analog 313 but lipophilicity values were quite different, with 315 > 314 > 313. This may underlie the effects on functional efficacy of the three compounds which varied, with 315 exhibiting 104% of the response to chlorodiazepoxide (CDZ) at the α3 receptor where it profiled as a stronger agonist than both 313 and 314 [253].

It was against this backdrop where small structural changes could exert a significant effect on the complexion of binding affinity and, in particular, receptor function that alternate GABAA ligands were sought, with a focus that was expanded to include the identification of compounds with selective inverse agonism (negative allosteric modulators or NAMs) at the α5 receptor [244, 254,255,256,257,258,259]. GABAA α5 receptor NAMs were sought based on preclinical experimental evidence that suggested that molecules with this phenotype would function as cognition enhancers without producing the anxiety or convulsant/proconvulsant effects associated with ligands for the other GABAA receptor subtypes. The initial iteration of this initiative evaluated structural variation of 307 and 308 and led to the characterization of α51A (318), derived from 317, as a functionally selective α5 receptor NAM meeting the targeted profile [244, 254,255,256]. In preclinical studies, 318 demonstrated good GABAA receptor occupancy in mice and rats following oral administration and improved the performance of rats in a variant on the Morris water maze model of assessing cognition enhancement [255]. A single 2 mg dose of 318 to NHVs resulted in 50% receptor occupancy, as measured in a positron emission tomography (PET) experiment using [11C]-flumazenil. However, clinical development of 318 had to be abandoned following the observation of kidney toxicity in preclinical studies and attention was redirected toward the back-up compound MRK-016 (319) in which the core heterocycle is rearranged to that of a pyrazolo[1,5-d][1,2,4]triazine [244, 257, 258]. In preclinical studies, 319 enhanced cognitive performance in behavioral tests and exhibited anti-depressant activity but clinical development of the compound was ultimately abandoned due to poor tolerability in elderly patients and inter-patient variability in the PK profile [257, 258].

The pyridazine 321, which was determined to be the active principle in a sample of 320 evaluated as part of the screening campaign designed to identify additional GABAA modulator chemotypes, profiled as a GABAA receptor ligand with modest affinity for the α1, α3 and α5 receptors that was largely preserved following removal of the ester moiety [259]. The cyano derivative 322 offered higher potency at all three receptors, with increased inverse agonism at α5 and only weak functional inverse agonism at the α1 and α3 receptors. Although small in size and with a reasonable cLog P/D value of 2.7, in vitro and in vivo profiling of 322 revealed that the PK properties were sub-optimal and inadequately addressed by the judicious introduction of fluorine substituents [259]. However, more extensive structural modifications designed based on the premise that, in this series, the pyridazine ring was bioisosteric with the imidazo[1,2-a]pyrimidine 302 and imidazo[1,2-b][1,2,4]triazine 303 heterocycles by the analogy depicted in Fig. 46, although the distances between the two nitrogen atoms are lengthened compared to 1 [260]. This exercise led to the identification of a family of potent GABAA ligands in which the functional selectivity profiles were sensitive to small changes in the structure, as illustrated by the profiling data associated with 324–327 [260]. Thus, 324 profiled as an antagonist of the α1 and α5 receptors but was a moderate partial agonist at the α2 and α3 subtypes, predicting the observed efficacy in rat and squirrel monkey models of anxiety at doses below 1 mg/kg and the absence of ataxia and sedation at doses as high as 10 mg/kg. The rearranged pyridine topology in 325 and 326 reduced the propensity for metabolic N-oxidation while the absence of a fluorine substituent from the other pyridine ring in 324 enhanced the α2/3 agonist activity; however, this compound was an α5 agonist. Quite remarkably, restoration of the fluorine substituent in 326 provided a compound that expressed functional activity only via the α2 receptor [260]. This kind of SAR profile is quite common in allosteric modulators where small structural changes can exert a significant impact on the biochemical pharmacological profile of a molecule [261,262,263].

Proposed bioisosteric relationship between pyridazine and fused-ring heterocycles that project two proximal lone pairs of in the context of allosteric GABAA receptor ligands with approximate distances between the nitrogen atoms abstracted from representative single crystal X-ray structures

The successful substitution of the imidazo[1,2-a]pyrimidine heterocycle in 313 by 8-fluoroimidazo[1,2-a]pyridine 315 illustrates the potential for a C-halogen to mimic a heterocyclic nitrogen atom, a design concept that has been extended to the alkaloid epibatidine 327 [264,265,266,267,268]. Epibatidine (327), which was isolated in trace amounts in extracts of the skin of an Ecuadorian frog, is a potent nicotinic acetyl choline (ACh) receptor modulator that expresses non-opioid-mediated analgesic activity in mice where its potency is superior to that of morphine. However, the therapeutic potential of 327 is limited by its toxicological profile, which has stimulated the synthesis and evaluation of analogs with potentially improved pharmacological properties [264,265,266]. The pyridazine analog 328, which exists as two conformational isomers in the single crystal X-ray structure, retained agonist efficacy at nicotinic ACh α3β4, α4β2 and α7 receptors but was 320-, 4- and 80-fold less potent than 327, respectively (Table 18) [267]. However, 328 was a more potent agonist than nicotine (329) at all three of the ACh receptors assessed. That much of the profile of 327 was retained in 328 was rationalized based on the similarity in distance between the nitrogen atom of the 7-azabicyclo[2.2.1]heptane core and the pyridine and pyridazine ring nitrogen atoms, which is 5.51 Å for the former and 5.38 Å for the latter.


The biochemical pharmacological profiles of a series of azine analogs of UB-165 (330), the 9-azabicyclo[4.2.1]non-2-ene homolog of 327, are compiled in Table 19 [268]. These compounds, 331–334, are based on the ring system found in the neurotoxic natural product anatoxin a (335) and were prepared in optically pure form. UB-165 (330) profiled similarly to 327 and 335, with receptor affinity values that fell between those of the two natural products. Of the azine variants, the pyrimidine 332 exhibited the highest affinity for (α4)2(β2)3 receptors, with enhanced selectivity over the α3β4 subtype. In this series, the pyrazine 333 and pyridazine 334 demonstrated low affinity for all three receptors evaluated but with selectivity for the (α4)2(β2)3 subtype [268].
An example where implementation of a bioisosteric relationship between a C-F moiety and a heterocyclic nitrogen atom fared less effectively is provided by the KCNQ2 blockers 338 and 339, which are based on the anthroquinone prototypes XE991 (336) and DMP-543 (337) [269]. These compounds were studied for their potential to block Kv7.2/7.3 channels and stimulate ACh release as an approach to therapeutics designed to enhance cognition. While 336 and 337 were effective Kv7.2/7.3 channel blockers in patch clamp experiments (81 and 31% inhibition at 1.5 µM, respectively), the former demonstrated poor metabolic stability while the latter suffered from low aqueous solubility, potent inhibition of CYP 3A4 and modestly potent hERG inhibition [269]. However, neither the pyridazine 338 or the pyrimidine 339 were active in the patch clamp assay, although 338 was inactive in the hERG assay and exhibited weaker CYP 3A4 inhibition, a profile that inspired its combination with the 3,5-difluoropyridine moiety found in JDP107 (340). JDP107 (340) combined Kv7.2/7.3 channel blockade (73% inhibition at 1.5 µM) that was comparable to 336 with a low inhibitory effect on the hERG cardiac channel, although CYP 3A4 inhibition remained a significant liability, which can probably be attributed to the presence of the pyridine ring [269].

Toxicity and metabolism of pyridazines
The pyridazine heterocycle is not characterized as a structural alert and there is little evidence to suggest that the N-N bond is labile to toward reductive metabolism in vivo, although some azine bonds can be reduced by microbiota in the gut [270,271,272,273,274,275,276,277,278,279]. In vitro and in vivo metabolism studies indicated that the nitrogen atoms of pyridazines can be oxidized to an N-oxide or subject to direct glucuronidation or ribosylation, while the carbon atoms of the ring are susceptible to oxidation [128]. Pyridazines, phthalazines and cinnolines not bearing substituents adjacent to the ring nitrogen atoms may be susceptible to metabolism by aldehyde oxidase (AO) [280,281,282,283,284,285,286]. However, there are circumstances where modification of a susceptible pyridine to a pyridazine can be a useful tactic to confer resistance to oxidation by AO, as exemplified by 341 and 342 where the latter is stable in rat cytosol [287].

A pyridazine can be a useful replacement for a para-substituted phenyl ring where enhanced metabolic stability is LMs is sought, although in one analysis, thiadiazole, (2,4)-1H-imidazole, oxadiazole, pyrazine and piperazine offered superior performance while pyrimidine and, particularly, (2,5)-1H-imidazole, were less effective as determined by the mean change (Δ) values summarized in Fig. 47 [55, 58].

Effect on metabolic stability in LMs of replacement of a para-substituted benzene ring with a heterocycle
Anilines can be oxidized by CYP 450 enzymes into reactive iminoquinones, diiminoquinones and iminoquinone methides that are activated Michael acceptors capable of reacting with proteins to precipitate inactivation of an enzyme or trigger an immune response after haptenization of a protein, events that have the potential to subtend a variety of toxicological events in vivo [270, 271]. In a systematic study of the bioactivation potential of several series of heteroaryl amines in HLM, where an aniline was used as the prototypical benchmark and GSH trapping of a reactive intermediate was used as the analytical readout, a pyridazine ring provided the best resistance to the generation of adducts [288]. As summarized in Fig. 48, an aminopyrazine offered minimal advantage over an aniline while aminopyrimidines and aminopyrimidines provided improved stability but all 3 were inferior to an aminopyridazine ring. Detailed analyses indicated that there was no correlation between bioactivation potential and cLog D7.4, TPSA or stability in HLM. Interestingly, there was no association between the intensity of GSH adducts formed and the HOMO, LUMO or HOMO-LUMO energies which would reflect the inherent chemical reactivity of the quinone-based oxidized products. However, there was a correlation between the calculated difference in BDEs for the first hydrogen atom abstraction from a substrate and the intensity of the GSH adducts formed when compared to the benchmark reference substrate. For those substrates with a ΔBDE of >3 kcal/mol, 75% failed to form a GSH adduct, which contrasts with substrates with a ΔBDE of <3kcal/mol where 87% afforded adducts [289]. For 1,4-disubsituted pyridazines, the calculated ΔBDE values were >4 kcal/mol and none of the substrates studied were observe to form GSH adducts. In a matched pairs-type of analysis, comparison of the three kinase inhibitors 343–345 highlighted the benefits of incorporating an heteroarene since the progressive introduction of nitrogen atoms into the phenyl ring scaffold reduced the amount of GSH adduct formed although metabolic stability was reduced for both 343 and 345 [288]. These observations were reproduced in four additional comparisons where, in each case, a pyridazine ring provided complete protection against GSH adduct formation compared to a phenyl-substituted prototype.

Experimental propensity for the formation of GSH conjugates for a series of heteroarylamines compared to aniline when incubated in HLM
Aminopyridazines can also associated with a reduced propensity to score as mutagenic in an Ames test conducted in the presence of activation by liver S9 microsomal preparation, as illustrated by studies of an N-phenylated pyrazole-based P2Y12 antagonist chemotype, platelet aggregation inhibitors pursued for their potential as antithrombotic agents [289]. An extensive optimization campaign refined the screening hit 346 into the advanced candidate 347 but concerns around the potential for the hydrolytic release in vivo of the aniline 348, which was mutagenic in the Ames S9 assay, led to a further round of molecular refinement designed to address the liability. In this example, concerns around the aniline would be associated not only with the production of quinoid derivatives but also with the potential for CYP 450-mediated oxidation of the aniline nitrogen atom to a chemically reactive aryl nitrenium species, a bioactivation process that heterocyclic amines can also participate in [290]. The potential replacements for the aniline moiety in 348 compiled in Fig. 49, which were probed with a view to reducing lipophilicity, were assessed in the Ames S9 mutagenicity assay, with those testing negative marked in green and those profiling as mutagenic colored in red. 3-Aminopyridazine, 2-aminopyridine and 2 aminopyrazine were clean in the assay whilst 3-aminopyridine and 2-aminothiazole were positive. While other scaffolding elements, including N-aminopiperidine and cyclohexane, were assessed in the final selection campaign, the 3-aminopyridazine SAR216471 (349) provided the best combination of biochemical pharmacological and PK properties and was selected for development [289, 291]. The binding affinity of 349 for the P2Y12 receptor was slightly better than that of the aniline 347 whilst inhibition of arachidonic acid (AA)-induced blood platelet aggregation in rabbit platelet-rich plasma (PRP) was improved sixfold, a profile that extended to human PRP where the EC50 value was 100 nM.

Mutagenicity of a series of heteroaryl amines in an Ames II Salmonella assay in which they were incubated in the presence of S9 liver fraction compared to the aniline prototype. Compounds colored green were classified as non-mutagenic while those colored red were classified as mutagenic

Epilogue
The physicochemical properties of the pyridazine ring (1) subtend practical and gainful applications in drug design and development where it can offer beneficial effects in establishing drug-target interactions whilst resolving off-target liabilities. Proffered as a privileged structure in 2011 by the late Professor Camille G. Wermuth, an exponent of the applications of the pyridazine ring in drug design, there appears to be a developing appreciation of its potential based on the rising prevalence in clinical candidates and preclinical compounds of interest [1, 9, 292]. In turn, these studies have illuminated deeper insight into the utility of 1 in molecular recognition and liability resolution that has furthered the understanding of its properties while stimulating additional creative applications in drug design. The most prominent physicochemical properties of 1 that subtend its value in molecular recognition appear to be the robust H-bond accepting properties and the large dipole moment, while the absence of an overt association with CYP 450 inhibition and the demonstrated potential to abrogate interference with the hERG cardiac potassium channel indicate its potential in liability resolution. The proximity of the lone pairs of electrons on the two adjacent nitrogen atoms of 1 confers a unique structural attribute that underlies both the strength of the H-bonding interactions and the ability to simultaneously engage dual H-bond donors [1, 26, 27, 293]. The early exploitation of the dual H-bonding effects of 1 and its fused ring homologs focused on applications in binding pockets where adjacent backbone amide N-Hs were projecting in a parallel orientation. However, the more recent observation of a dual H-bonding interaction between the two nitrogen atoms of a substituted pyridazine with the side chain guanidine terminus of an arginine residue in pantothenate kinase is suggestive of the potential for broader application, possibly as a carboxylic acid isostere without the burden of an overt negative charge or the susceptibility to metabolic modification to potentially problematic acyl glucuronides or CoA esters [75, 90,91,92,93, 203, 294,295,296,297,298,299,300]. Moreover, the juxtaposition of this observation with the bioisosteric relationship that has been proposed between 1 and fused bicyclic heterocycles of the type represented by 302, 303 and others that convene two nitrogen atoms in close proximity with their lone pairs of electrons aligned, provides additional insight into the design of innovative drug-target interactions [293].
The dipole moment of 1 contributes to π-stacking interactions, with the examples described herein further embellished by observations from cocrystal structures of the allosteric glutamate antagonist EVT-101 (350) and the STING-activating non-nucleotide cGAMP mimetic SR-711 (351) where the pyridazine rings are closely associated with tyrosine residues in the binding pocket [301, 302].

The recent licensing of the allosteric TYK2 inhibitor deucravacitinib (25) brings to patients an effective therapeutic agent for the treatment of moderate-to-severe plaque psoriasis that appears to be the second therapeutic agent incorporating a pyridazine heterocycle to receive FDA approval after relugolix (24). In 25, the effects of several of the unique attributes of the pyridazine heterocycle are apparent, including an intramolecular H-bond to enhance membrane permeability and conformational effects of the dipole moments of the pyridazine and amide moieties which favor the enzyme-bound conformation. Other notable pyridazine derivatives that are currently in clinical development include the mRNA splicing modulator branaplam (82), the androgen receptor degrader ARV-110 (113) and the ALK inhibitor ensartinib (114) [102, 103, 126, 128]. The most recently-described clinical candidate that is constructed on a pyridazine core is provided by the potent and selective monocarboxylate transporter 4 (MCT4) inhibitor AZD0095 (352) that blocks lactate export from tumor cells and is being evaluated for its potential anti-tumor and immune modulating properties [303]. These compounds hold individual promise in their respective disease categories while illustrating the unique properties and applications of the pyridazine ring in drug design that collectively advocate for its broader deployment in candidate optimization. The successful applications of heterocyclic rings in compound optimization is very much dependent on the structure, shape and properties of a targeted binding site and its complementarity to the physicochemical attributes of a specific ring system. Thus, there is no simple prescription for the selection of a specific heterocycle although a detailed understanding of the physicochemical properties will subtend informed design and decision making.

Abbreviations
- AA:
-
arachidonic acid
- ACh:
-
acetylcholine
- ADHD:
-
attention-deficit/hyperactivity disorder
- ALK:
-
the anaplastic lymphoma kinase
- ALL:
-
acute lymphoblastic leukemia
- AO:
-
aldehyde oxidase
- ATP:
-
adenosine triphosphate
- ATR:
-
ataxia telangiectasia and Rad3-related
- BACE1:
-
β-secretase-1
- Bcl-2:
-
B-cell lymphoma-2
- BPTES:
-
Bis-2-[5-(phenylacetamido)-1,3,4-thiadiazol-2-yl]ethyl sulfide
- BRD:
-
bromodomain
- BTK:
-
Bruton’s tyrosine kinase
- CAT B:
-
cathepsin B
- CAT L:
-
cathepsin L
- CAT S:
-
cathepsin S
- CDZ:
-
chlorodiazepoxide
- CHK1:
-
checkpoint 1 kinase
- CK1:
-
casein kinase 1
- CML:
-
chronic myeloid leukemia
- CNS:
-
central nervous system
- CoA:
-
Coenzyme A
- CYP 450:
-
cytochrome P450
- DMSO:
-
dimethyl sulfoxide
- dsRNA:
-
double-stranded ribonucleic acid
- ELISA:
-
enzyme-linked immunosorbent assay
- FAAH:
-
fatty acid amide hydrolase
- FDA:
-
U.S. Food and Drug Administration
- GABA:
-
γ-aminobutyric acid
- GAC:
-
glutaminase C
- GLS1:
-
kidney-type glutaminase
- GnRH:
-
gonadotropin-releasing hormone
- GSH:
-
glutathione
- H-bond:
-
hydrogen bond
- HCV:
-
hepatitis C virus
- hERG:
-
human ether-à-go-go-related gene
- hH-PGDS:
-
human hematopoietic prostaglandin (PG) D2 synthase
- HIF:
-
hypoxia-inducible factor
- HIV-1:
-
human immunodeficiency virus-1
- HLM:
-
human liver microsomes
- HPLC:
-
high performance liquid chromatography
- HRV:
-
human rhinovirus
- HSA:
-
human serum albumin
- HTS:
-
high throughput screening
- IA :
-
aromaticity index
- ITC:
-
isothermal calorimetry
- JH2:
-
Janus homology 2
- KGA:
-
kidney glutaminase
- LipE:
-
lipophilic efficiency
- MAO:
-
monoamine oxidase
- MAPK:
-
p38 mitogen activated protein kinase
- MCT4:
-
monocarboxylate transporter 4
- MMP:
-
matched molecular pair
- MW:
-
molecular weight
- NAC:
-
N-acetyl cysteine
- NAD:
-
nicotinamide adenine dinucleotide
- NAM:
-
negative allosteric modulator
- NAMPT:
-
nicotinamide phosphoribosyltransferase
- NHV:
-
normal healthy volunteer
- NSCLC:
-
non-small cell lung cancer
- PAM:
-
positive allosteric modulator
- PanK:
-
pantothenate kinase
- PBRM5:
-
fifth bromodomain of (polybromo-1) bromodomain 5
- PDE:
-
phosphodiesterase
- PET:
-
positron emission tomography
- PHD-1:
-
prolylhydroxylase domain-1
- PI3K:
-
phosphoinositide 3-kinase
- PKC θ:
-
protein kinase C θ
- PNA:
-
peptide nucleic acids
- PO:
-
per os
- PROTACS:
-
proteolysis-targeting chimeras
- PRP:
-
platelet-rich plasma
- PRPP:
-
phosphoribosyl pyrophosphate
- RAF:
-
rapidly accelerated fibrosarcoma
- RdRp:
-
RNA-dependent, RNA polymerase
- RLM:
-
rat liver microsomes
- ROCK:
-
Rho-associated protein kinase
- RTK:
-
receptor tyrosine kinase
- S1P2:
-
spingosine-1-phosphate 2
- SAPT:
-
symmetry-adapted perturbation theory
- SAR:
-
structure-activity relationship
- SBDD:
-
structure-based drug design
- SCD:
-
stearoyl-coenzyme A desaturase
- SGLT2:
-
sodium-dependent glucose cotransporter 2
- SMA:
-
spinal muscular atrophy
- SMARCA:
-
SWI/SNF-related matrix-associated actin-dependent regulator of chromatin A
- SMN2:
-
survival motor neuron-2
- T m :
-
thermal melting temperature
- TPSA:
-
topological polar surface area
- TYK2:
-
tyrosine kinase 2
- U.S.:
-
United States
- UV:
-
ultraviolet
References
Wermuth CG. Are pyridazines privileged structures? Med Chem Comm. 2011;2:935–41. https://doi.org/10.1039/C1MD00074H
Subbaiah MAM, Meanwell NA. Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design. J Med Chem. 2021;64:14046–128. https://doi.org/10.1021/acs.jmedchem.1c01215
Meanwell NA. A synopsis of the properties and applications of heteroaromatic rings in medicinal chemistry. Adv Het Chem. 2017;123:245–361. https://doi.org/10.1016/bs.aihch.2016.11.002
Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, et al. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J Med Chem. 1988;31:2235–46. https://doi.org/10.1021/jm00120a002
Schnur D, Hermsmeier MA, Tebben AJ. Are target-family-privileged substructures truly privileged? J Med Chem. 2006;49:2000–9. https://doi.org/10.1021/jm0502900
go.drugbank.com (accessed October 19, 2022). https://go.drugbank.com/
Taylor RD, MacCoss M, Lawson AD. Rings in drugs. J Med Chem. 2014;57:5845–59. https://doi.org/10.1021/jm4017625
Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem. 2014;57:10257–74. https://doi.org/10.1021/jm501100b
Shearer J, Castro JL, Lawson ADG, MacCoss M, Taylor RD. Rings in clinical trials and drugs: present and future. J Med Chem. 2022;65:8699–712. https://doi.org/10.1021/acs.jmedchem.2c00473
Fung M, Thornton A, Mybeck K, Wu JH, Hornbuckle K, Muniz E. Evaluation of the characteristics of safety withdrawal of prescription drugs from worldwide pharmaceutical markets -1960 to 1999. Drug Inf J. 2001;35:293–317. https://doi.org/10.1177/0092861501035001
Markham A. Relugolix: first global approval. Drugs. 2019;79:675–9. https://doi.org/10.1007/s40265-019-01105-0
Hoy SM. Deucravacitinib: first approval. Drugs. 2022;82:1671–9. https://doi.org/10.1007/s40265-022-01796-y
Cohn JN, McInnes GT, Shepherd AM. Direct-acting vasodilators. J Clin Hypertens. 2011;13:690–2. https://doi.org/10.1111/j.1751-7176.2011.00507.x
Scavone JM, Gleckman RA, Fraser DG. Cinoxacin: mechanism of action, spectrum of activity, pharmacokinetics, adverse reactions, and therapeutic indications. Pharmacotherapy. 1982;2:266–71. https://doi.org/10.1002/j.1875-9114.1982.tb03195.x
Thompson PE, Manganiello V, Degerman E. Re-discovering PDE3 inhibitors- new opportunities for a long neglected target. Curr Top Med Chem. 2007;7:421–36. https://doi.org/10.2174/156802607779941224
Heringlake M, Alvarez J, Bettex D, Bouchez S, Fruhwald S, Girardis M, et al. An update on levosimendan in acute cardiac care: applications and recommendations for optimal efficacy and safety. Expert Rev Cardiovasc Ther. 2021;19:325–35. https://doi.org/10.1080/14779072.2021.1905520
Pathak A, Lebrin M, Vaccaro A, Senard JM, Despas F. Pharmacology of levosimendan: inotropic, vasodilatory and cardioprotective effects. J Clin Pharm Ther. 2013;38:341–9. https://doi.org/10.1111/jcpt.12067
Heinisch G, Frank H. Pharmacologically active pyridazine derivatives. Part 1 Prog Med Chem. 1990;27:1–49. https://doi.org/10.1016/S0079-6468(08)70288-1
Heinisch G, Kopelent-Frank H. Pharmacologically active pyridazine derivatives. Part 2 Prog Med Chem. 1992;29:141–83. https://doi.org/10.1016/S0079-6468(08)70007-9
Lamberth C. Pyridazine chemistry in crop protection. J Het Chem. 2017;54:2974–84. https://doi.org/10.1002/jhet.2945
Asif M. Diverse biologically active pyridazine analogs: a scaffold for the highly functionalized heterocyclic compounds. Rev J Chem. 2018;8:280–300. https://doi.org/10.1134/S2079978018030019
Asif M, Abida, Tauquir AM. Diverse chemical and biological potentials of various pyridazine and pyridazinone derivatives. Chem Int. 2019;5:206–23. https://doi.org/10.5281/zenodo.2525555
Imran M, Asif M. Study of various pyridazine and phthalazine drugs with diverse therapeutical and agrochemical activities. Russ J Bioorg Chem. 2020;46:745–67. https://doi.org/10.1134/S1068162020050167
He Z-X, Gong Y-P, Zhang X, Ma L-Y, Zhao W. Pyridazine as a privileged structure: an updated review on anticancer activity of pyridazine-containing bioactive molecules. Eur J Med Chem. 2021;209:112946. https://doi.org/10.1016/j.ejmech.2020.112946
Gibson S, McGuire R, Rees DC. Principal components describing biological activities and molecular diversity of heterocyclic aromatic ring fragments. J Med Chem. 1996;39:4065–72. https://doi.org/10.1021/jm960058h
Laurence C, Brameld KA, Graton J, Le Questel J-Y, Renault E. The pKBHX database: toward a better understanding of hydrogen-bond basicity for medicinal chemists. J Med Chem. 2009;52:4073–86. https://doi.org/10.1021/jm801331y
Kenny PW, Montanari CA, Prokopczyk IM, Ribeiro JFR, Rodrigues Sartori G. Hydrogen bond basicity prediction for medicinal chemistry design. J Med Chem. 2016;59:4278–88. https://doi.org/10.1021/acs.jmedchem.5b01946
Pierce AC, Sandretto KL, Bemis GW. Kinase inhibitors and the case for CH···O hydrogen bonds in protein-ligand binding. Proteins. 2002;49:567–76. https://doi.org/10.1002/prot.10259
Ertl P, Rohde B, Selzer P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J Med Chem. 2000;43:3714–7. https://doi.org/10.1021/jm000942e
Landry ML, Crawford JJ. LogD contributions of substituents commonly used in medicinal chemistry. ACS Med Chem Lett. 2020;11:72–6. https://doi.org/10.1021/acsmedchemlett.9b00489
Bradamante S, Pagani GA. Electron and charge demands of stabilizing groups in carbanions and nitroanions: the NMR approach. Pure Appl Chem. 1989;61:709–16. https://doi.org/10.1351/pac198961040709
Han YT, Jung J-W, Kim N-J. Recent advances in the synthesis of biologically active cinnoline, phthalazine and quinoxaline derivatives. Curr Org Chem. 2017;21:1265–91. https://doi.org/10.2174/1385272821666170221150901
Szumilak M, Stanczak A. Cinnoline scaffold—a molecular heart of medicinal chemistry? Molecules. 2019;24:2271. https://doi.org/10.3390/molecules24122271
Boström J, Hogner A, Llinàs A, Wellner E, Plowright AT. Oxadiazoles in medicinal chemistry. J Med Chem. 2012;55:1817–30. https://doi.org/10.1021/jm2013248
Li Y, Geng J, Liu Y, Yu S, Zhao G. Thiadiazole—a promising structure in medicinal chemistry. ChemMedChem. 2013;8:27–41. https://doi.org/10.1002/cmdc.201200355
Hu Y, Li C-Y, Wang X-M, Yang Y-H, Zhu H-L. 1,3,4-Thiadiazole: synthesis, reactions, and applications in medicinal, agricultural, and materials chemistry. Chem Rev. 2014;114:5572–610. https://doi.org/10.1021/cr400131u
Aggarwal R, Sumran G. An insight on medicinal attributes of 1,2,4-triazoles. Eur J Med Chem. 2020;205:112652. https://doi.org/10.1016/j.ejmech.2020.112652
An Y, Doney AC, Andrade RB, Wheeler SE. Stacking interactions between 9‑methyladenine and heterocycles commonly found in pharmaceuticals. J Chem Inf Model. 2016;56:906–14. https://doi.org/10.1021/acs.jcim.5b00651
Huber RG, Margreiter MA, Fuchs JE, von Grafenstein S, Tautermann CS, Liedl KR, et al. Heteroaromatic π‑stacking energy landscapes. J Chem Inf Model. 2014;54:1371–9. https://doi.org/10.1021/ci500183u
Harder M, Kuhn B, Diederich F. Efficient stacking on protein amide fragments. ChemMedChem. 2013;8:397–404. https://doi.org/10.1002/cmdc.201200512
Bootsma AN, Doney AC, Wheeler SE. Predicting the strength of stacking interactions between heterocycles and aromatic amino acid side chains. J Am Chem Soc. 2019;141:11027–35. https://doi.org/10.1021/jacs.9b00936
Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed. 2003;42:1210–50. https://doi.org/10.1002/anie.200390319
Salonen LM, Ellermann M, Diederich F. Aromatic rings in chemical and biological recognition: energetics and structures. Angew Chem Int Ed. 2011;50:4808–42. https://doi.org/10.1002/anie.201007560
Persch E, Dumele O, Diederich F. Molecular recognition in chemical and biological systems. Angew Chem Int Ed. 2015;54:3290–327. https://doi.org/10.1002/anie.201408487
Bootsma AN, Wheeler SE. Tuning stacking interactions between Asp−Arg salt bridges and heterocyclic drug fragments. J Chem Inf Model. 2019;59:149–58. https://doi.org/10.1021/acs.jcim.8b00563
Kan JP, Mouget-Goniot C, Worms P, Biziere K. Effect of the antidepressant minaprine on both forms of monoamine oxidase in the rat. Biochem Pharmacol. 1986;35:973–8. https://doi.org/10.1016/0006-2952(86)90085-7
Abraham MH, Duce PP, Prior DV, Barratt DG, Morris JJ, Taylor PJ. Hydrogen bonding. Part 9. Solute proton donor and proton acceptor scales for use in drug design. J Chem Soc. P2 1989:1355-75. https://doi.org/10.1039/P29890001355
Edwards JO, Pearson RG. The factors determining nucleophilic reactivities. J Am Chem Soc. 1962;84:16–24. https://doi.org/10.1021/ja00860a005
Zoltewicz JA, Deady LW. Search for the α effect among heteroaromatic nitrogen nucleophiles. J Am Chem Soc. 1972;94:2765–9. https://doi.org/10.1021/ja00763a039
Taft RW, Anvia F, Taagepera M, Catalán J, Elguero J. Electrostatic proximity effects in the relative basicities and acidities of pyrazole, imidazole, pyridazine, and pyrimidine. J Am Chem Soc. 1986;108:3237–9. https://doi.org/10.1021/ja00272a013
Zhang H, Wu W, Ahmed BM, Mezei G, Mo Y. Adjacent lone pair (ALP) effect: a computational approach for its origin. Chem Eur J 2016;22:7415–21. https://doi.org/10.1002/chem.201600509
Shen K, Fu Y, Li J-N, Liu L, Guo Q-X. What are the pKa values of C–H bonds in aromatic heterocyclic compounds in DMSO? Tetrahedron. 2007;63:1568–76. https://doi.org/10.1016/j.tet.2006.12.032
Bird CW. Heteroaromaticity, a unified aromaticity index. Tetrahedron. 1992;48:335–40. https://doi.org/10.1016/S0040-4020(01)88145-X
Barckholtz C, Barckholtz TA, Hadad CM. C-H and N-H bond dissociation energies of small aromatic hydrocarbons. J Am Chem Soc. 1999;121:491–500. https://doi.org/10.1021/ja982454q
Ritchie TJ, Macdonald SJF. Heterocyclic replacements for benzene: maximising ADME benefits by considering individual ring isomers. Eur J Med Chem. 2016;124:1057–68. https://doi.org/10.1016/j.ejmech.2016.10.029
Chang G, Huard K, Kauffman GW, Stepan AF, Keefer CE. A multi-endpoint matched molecular pair (MMP) analysis of 6-membered heterocycles. Bioorg Med Chem. 2017;25:381–8. https://doi.org/10.1016/j.bmc.2016.11.004
Prasanna S, Doerksen RJ. Topological polar surface area: a useful descriptor in 2D-QSAR. Curr Med Chem. 2009;16:21–41. https://doi.org/10.2174/092986709787002817
Dossetter AG, Douglas A, O’Donnell C. A matched molecular pair analysis of in vitro human microsomal metabolic stability measurements for heterocyclic replacements of di-substituted benzene containing compounds—identification of those isosteres more likely to have beneficial effects. Med Chem Commun. 2012;3:1164–9. https://doi.org/10.1039/C2MD20155K
Ritchie TR, MacDonald SJ, Peace S, Pickett SD, Luscombe CN. The developability of heteroaromatic and heteroaliphatic rings—do some have a better pedigree as potential drug molecules than others? Med Chem Commun. 2012;3:1062–9. https://doi.org/10.1039/C2MD20111A
Leach AG, Kidley NJ. Quantitatively interpreted enhanced inhibition of cytochrome P450s by heteroaromatic rings containing nitrogen. J Chem Inf Model. 2011;51:1048–63. https://doi.org/10.1021/ci2000506
Leach AG. Tactics to avoid inhibition of cytochrome P450s. In: Meanwell N (Ed.) Tactics in Contemporary Drug Design. Topics in Medicinal Chemistry. Berlin, Heidelberg: Springer; 2013. 10.1007/7355_2013_25. 107–58
Gavrilova AL, Bosnich B. Principles of mononucleating and binucleating ligand design. Chem Rev. 2004;104:349–83. https://doi.org/10.1021/cr020604g
Zimmermann SC, Duvall B, Tsukamoto T. Recent progress in the discovery of allosteric inhibitors of kidney-type glutaminase. J Med Chem. 2019;62:46–59. https://doi.org/10.1021/acs.jmedchem.8b00327
Xu X, Meng Y, Li L, Xu P, Wang J, Li Z, et al. Overview of the development of glutaminase inhibitors: achievements and future directions. J Med Chem. 2019;62:1096–115. https://doi.org/10.1021/acs.jmedchem.8b00961
DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, et al. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry. 2011;50:10764–70. https://doi.org/10.1021/bi201613d
Thangavelu K, Pan CQ, Karlberg T, Balaji G, Uttamchandani M, Suresh V, et al. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci USA. 2012;109:7705–10. https://doi.org/10.1073/pnas.1116573109
Huang Q, Stalnecker C, Zhang C, McDermott LA, Iyer P, O’Neill J, et al. Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J Biol Chem. 2018;293:3535–45. https://doi.org/10.1074/jbc.M117.810101
Milano SK, Huang Q, Nguyen T-TT, Ramachandran S, Finke A, Kriksunov I, et al. New insights into the molecular mechanisms of glutaminase C inhibitors in cancer cells using serial room temperature crystallography. J Biol Chem. 2022;298:101535 https://doi.org/10.1016/j.jbc.2021.101535
Li J, Chen L, Goyal B, Laidig G, Stanton TF, Sjogren EB. Heterocyclic inhibitors of glutaminase. World Pat Appl. WO2013/078123, May 30th, 2013. https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013078123&_cid=P20-LBZ5X0-16670-1
Soth MJ, Le K, Di Francesco ME, Hamilton MM, Liu G, Burke JP, et al. Discovery of IPN60090, a clinical stage selective glutaminase-1 (GLS-1) inhibitor with excellent pharmacokinetic and physicochemical properties. J Med Chem. 2020;63:12957–77. https://doi.org/10.1021/acs.jmedchem.0c01398
IPN 60090. Adis Insight. https://adisinsight.springer.com/drugs/800052154, accessed November 23rd, 2022
Riess JW, Frankel P, Shackelford D, Dunphy M, Badawi RD, Nardo L, et al. Phase 1 Trial of MLN0128 (sapanisertib) and CB-839 HCl (telaglenastat) in patients with advanced NSCLC (NCI 10327): rationale and study design. Clin Lung Cancer. 2021;22:67–70. https://doi.org/10.1016/j.cllc.2020.10.006
Meric-Bernstam F, Tannir NM, Iliopoulos O, Lee RJ, Telli ML, Fan AC, et al. Telaglenastat plus cabozantinib or everolimus for advanced or metastatic renal cell carcinoma: an open-label phase I trial. Clin Cancer Res. 2022;28:1540–8. https://doi.org/10.1158/1078-0432.CCR-21-2972
Lee C-H, Motzer R, Emamekhoo H, Matrana M, Percent I, Hsieh JJ, et al. Tannir NM. Telaglenastat plus everolimus in advanced renal cell carcinoma: a randomized, double-blinded, placebo-controlled, phase II ENTRATA trial. Clin Cancer Res. 2022;28:3248–55. https://doi.org/10.1158/1078-0432.CCR-22-0061
Bethel PA, Gerhardt S, Jones EV, Kenny PW, Karoutchi GI, Morley AD, et al. Design of selective cathepsin inhibitors. Bioorg Med Chem Lett. 2009;19:4622–5. https://doi.org/10.1016/j.bmcl.2009.06.090
Andries K, Dewindt B, De Brabander M, Stokbroekx R, Janssen PAJ. In vitro activity of R 61837, a new antirhinovirus compound. Arch Virol. 1988;101:155–67. https://doi.org/10.1007/BF01310997
Chapman MS, Minor I, Rossmann MG, Diana GD, Andries K. Human rhinovirus 14 complexed with antiviral compound R 61837. J Mol Biol. 1991;217:455–63. https://doi.org/10.1016/0022-2836(91)90749-V
Andries K, Dewindt B, Snoeks J, Willebrords R, Van Eemeren K, Stokbroekx R, et al. In vitro activity of pirodavir (R 77975), a substituted phenoxy-pyridazinamine with broad-spectrum antipicornaviral activity. Antimicrob Agents Chemother. 1992;36:100–7. https://doi.org/10.1128/AAC.36.1.100
Jottier WI, De Winter HL, Blaton NM, Peeters OM. Structureof the antiviral drug ethyl4-{2-[1-(6-methyl-3-pyridazinyl)-4-piperidinyl]ethoxy}benzoate. ActaCrystallogr Sect C: Cryst Struct Commun. 1991;C47:1517–20. https://doi.org/10.1107/S0108270190013439
Hayden FG, Andries K, Janssen PA. Safety and efficacy of intranasal pirodavir (R77975) in experimental rhinovirus infection. Antimicrob Agents Chemother. 1992;36:727–32. https://doi.org/10.1128/AAC.36.4.727
Hayden FG, Hipskind GJ, Woerner DH, Eisen GF, Janssens M, Janssen PAJ, et al. Intranasal pirodavir (R77,975) treatment of rhinovirus colds. Antimicrob Agents Chemother. 1995;39:290–4. https://doi.org/10.1128/AAC.39.2.290
Watson KG, Brown RN, Cameron R, Chalmers DK, Hamilton S, Jin B, et al. An orally bioavailable oxime ether capsid binder with potent activity against human rhinovirus. J Med Chem. 2003;46:3181–4. https://doi.org/10.1021/jm0202876
Barnard DL, Hubbard VD, Smee DF, Sidwell RW, Watson KGW, Tucker SPT, et al. In vitro activity of expanded-spectrum pyridazinyl oxime ethers related to pirodavir: novel capsid-binding inhibitors with potent antipicornavirus activity. Antimicrob Agents Chemother. 2004;48:1766–72. https://doi.org/10.1128/AAC.48.5.1766-1772.2004
Brown RN, Cameron R, Chalmers DK, Hamilton S, Luttick A, Krippner GY, et al. 2-Ethoxybenzoxazole as a bioisosteric replacement of an ethyl benzoate group in a human rhinovirus (HRV) capsid binder. Bioorg Med Chem Lett. 2005;15:2051–55. https://doi.org/10.1016/j.bmcl.2005.02.054
Feil SC, Hamilton S, Krippner GY, Lin B, Luttick A, McConnell DB, et al. An orally available 3-ethoxybenzisoxazole capsid binder with clinical activity against human rhinovirus. ACS Med Chem Lett. 2012;3:303–7. https://doi.org/10.1021/ml2002955
Tijsma A, Franco D, Tucker S, Hilgenfeld R, Froeyen M, Leyssen P, et al. The capsid binder vapendavir and the novel protease inhibitor SG85 inhibit enterovirus 71 replication. Antimicrob Agents Chemother. 2014;58:6990–2. https://doi.org/10.1128/AAC.03328-14
Diana GD, Pevear DC. Antipicornavirus drugs: current status. Antivir Chem Chemother. 1997;8:401–8. https://doi.org/10.1177/095632029700800502
Altesa Biosciences, http://altesabio.com/pipeline/. Accessed November 25th, 2022
Brodyagin N, Kumpina I, Applegate J, Katkevics M, Rozners E. Pyridazine nucleobase in triplex-forming PNA improves recognition of cytosine interruptions of polypurine tracts in RNA. ACS Chem Biol. 2021;16:872–81. https://doi.org/10.1021/acschembio.1c00044
Herberich B, Cao G-Q, Chakrabarti PP, Falsey JR, Pettus L, Rzasa RM, et al. Discovery of highly selective and potent p38 inhibitors based on a phthalazine scaffold. J Med Chem. 2008;51:6271–9. https://doi.org/10.1021/jm8005417
Pettus LH, Xu S, Cao G-Q, Chakrabarti PP, Rzasa RM, Sham K, et al. 3-Amino-7-phthalazinylbenzoisoxazoles as a novel class of potent, selective, and orally available inhibitors of p38α mitogen-activated protein kinase. J Med Chem. 2008;51:6280–92. https://doi.org/10.1021/jm8005405
Wu B, Wang H-L, Pettus L, Wurz RP, Doherty EM, Henkle B, et al. Discovery of pyridazinopyridinones as potent and selective p38 mitogen-activated protein kinase inhibitors. J Med Chem. 2010;53:6398–411. https://doi.org/10.1021/jm100567y
Fitzgerald CE, Patel SB, Becker JW, Cameron PM, Zaller D, Pikounis VB, et al. Structural basis for p38α MAP kinase quinazolinone and pyridol-pyrimidine inhibitor specificity. Nat Struct Mol Biol. 2003;10:764–9. https://doi.org/10.1038/nsb949
Calder AN, Androphy EJ, Hodgetts KJ. Small molecules in development for the treatment of spinal muscular atrophy. J Med Chem. 2016;59:10067–83. https://doi.org/10.1021/acs.jmedchem.6b00670
Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol. 2015;11:511–7. https://doi.org/10.1038/nchembio.1837
Cheung AK, Hurley B, Kerrigan R, Shu L, Chin DN, Shen Y, et al. Discovery of small molecule splicing modulators of survival motor neuron-2 (SMN2) for the treatment of spinal muscular atrophy (SMA). J Med Chem. 2018;61:11021–36. https://doi.org/10.1021/acs.jmedchem.8b01291
Axford J, Sung MJ, Manchester J, Chin D, Jain M, Shin Y, et al. Use of intramolecular 1,5-sulfur-oxygen and 1,5-sulfur-halogen interactions in the design of N-methyl-5-aryl-N-(2,2,6,6-tetramethylpiperidin-4-yl)-1,3,4-thiadiazol-2-amine SMN2 splicing modulators. J Med Chem. 2021;64:4744–61. https://doi.org/10.1021/acs.jmedchem.0c02173
Zhang Z, Shi J. Preparationof 4-[(5-aryl-1,3,4-oxadiazol-2-yl)amino]-2,2,6,6-tetramethylpiperidine 1-oxylderivatives.Chin J Org Chem.1991;11:404–7. http://sioc-journal.cn/Jwk_yjhx/EN/Y1991/V11/I4/404
Sosnovskikh VY, Usachev BI. Vorontsov II. Unusual reaction of 2-(trifluoromethyl)-1,2-dihydro-3λ6-thieno-[2,3-c]chromen-3,3,4-triones with hydrazine as a new route to 3-hydrazinopyridazine derivatives. J Org Chem. 2002;67:6738–42. https://doi.org/10.1021/jo0258406
Beno BR, Yeung K-S, Bartberger MD, Pennington LD, Meanwell NA. A survey of the role of noncovalent sulfur interactions in drug design. J Med Chem. 2015;58:4383–438. https://doi.org/10.1021/jm501853m
Chein RJ, Corey EJ. Strong conformational preferences of heteroaromatic ethers and electron pair repulsion. Org Lett. 2010;12:132–5. https://doi.org/10.1021/ol9025364
Krach F, Stemick J, Boerstler T, Weiss A, Lingos I, Reischl S, et al. An alternative splicing modulator decreases mutant HTT and improves the molecular fingerprint in Huntington’s disease patient neurons. Nat Commun. 2022;13:6797. https://doi.org/10.1038/s41467-022-34419-x
Ahamad S, Bhat SA. The emerging landscape of small-molecule therapeutics for the treatment of Huntington’s disease. J Med Chem. 2022;65:15993–16032. https://doi.org/10.1021/acs.jmedchem.2c00799
Taylor AM, Bailey C, Belmont LD, Campbell R, Cantone N, Cote A, et al. GNE-064: a potent, selective, and orally bioavailable chemical probe for the bromodomains of SMARCA2 and SMARCA4 and the fifth bromodomain of PBRM1. J Med Chem. 2022;65:11177–86. https://doi.org/10.1021/acs.jmedchem.2c00662
Wanio M, Preuss F, Ni X, Kraemer A, Mathea S, Goebel T, et al. Pan-SMARCA/PB1 bromodomain inhibitors and their role in regulating adipogenesis. J Med Chem. 2020;63:14680–99. https://doi.org/10.1021/acs.jmedchem.0c01242
Clegg MA, Tomkinson NCO, Prinjha RK, Humphreys PG. Advancements in the development of non-BET bromodomain chemical probes. ChemMedChem. 2019;14:362–85. https://doi.org/10.1002/cmdc.201800738
Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–31. https://doi.org/10.1016/j.cell.2012.02.013
Sharp PP, Garnier J-M, Huang DCS, Burns CJ. Evaluation of functional groups as acetyl-lysine mimetics for BET bromodomain inhibition. Med Chem Commun. 2014;5:1834–42. https://doi.org/10.1039/C4MD00182F
Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol. 2019;15:672–80. https://doi.org/10.1038/s41589-019-0294-6
Crew AP, Wang J, Berlin M, Dragovich P, Chen H, Staben L. Preparation of bifunctional PROTAC compounds and methods for degradation of targeted BRM proteins. US Pat Appl. US 20190300521 A1 20191003. April 1, 2019. https://image-ppubs.uspto.gov/dirsearch-public/print/downloadPdf/20190300521
Gentles RG, Ding M, Zheng X, Chupak L, Poss MA, Beno BR, et al. SAR studies on a series of N-benzyl-4-heteroaryl-1-(phenylsulfonyl)piperazine-2-carboxamides: potent inhibitors of the polymerase enzyme (NS5B) of the hepatitis C virus. Bioorg Med Chem Lett. 2011;21:3142–7. https://doi.org/10.1016/j.bmcl.2011.03.008
Komkov AV, Komendantova AS, Menchikov LG, Chernoburova EI, Volkova YA. Zavarzin IV. A straightforward approach toward multifunctionalized pyridazines via imination/electrocyclization. Org Lett. 2015;17:3734–7. https://doi.org/10.1021/acs.orglett.5b01718
Gentles RG, Sheriff S, Beno BR, Wan C, Kish K, Ding M, et al. Investigation of the mode of binding of a novel series of 1-benzyl-4-heteroaryl-1-(phenylsulfonyl)piperazine-2-carboxamides to the hepatitis C virus polymerase. Bioorg Med Chem Lett. 2011;21:2212–5. https://doi.org/10.1016/j.bmcl.2011.03.011
Zhang Z, Dales NA, Winther MD. Opportunities and challenges in developing stearoyl-coenzyme A desaturase-1 inhibitors as novel therapeutics for human disease. J Med Chem. 2014;57:5039–56. https://doi.org/10.1021/jm401516c
William NS, Gonzales S, Naidoo J, Rivera-Cancel G, Voruganti S, Mallipeddi P, et al. Tumor-activated benzothiazole inhibitors of stearoyl-CoA desaturase. J Med Chem. 2020;63:9773–86. https://doi.org/10.1021/acs.jmedchem.0c00899
Zhang Z, Sun S, Kodumuru V, Hou D, Liu S, Chakka N, et al. Discovery of piperazin-1-ylpyridazine-based potent and selective stearoyl-CoA desaturase-1 inhibitors for the treatment of obesity and metabolic syndrome. J Med Chem. 2013;56:568–83. https://doi.org/10.1021/jm301661h
Uto Y, Kiyotsuka Y, Ueno Y, Miyazawa Y, Kurata H, Ogata T. et al. Novelspiropiperidine-based stearoyl-CoA desaturase-1 inhibitors: identification of1'-{6-[5-(pyridin-3-ylmethyl)-1,3,4-oxadiazol-2-yl]pyridazin-3-yl}-5-(trifluoromethyl)-3,4-dihydrospiro[chromene-2,4'-piperidine]. BioorgMed Chem Lett. 2020;20:746–54. https://doi.org/10.1016/j.bmcl.2009.11.043
Bai Y, McCoy JG, Levin EJ, Sobrado P, Rajashankar KR, Fox BG, et al. X-ray structure of a mammalian stearoyl-CoA desaturase. Nature. 2015;524:252–6. https://doi.org/10.1038/nature14549
Liu G. Stearoyl-CoA desaturase 1 (SCD1) inhibitors: bench to bedside must only go through liver. RSC Drug Disco Ser. 2012;27:249–69. https://doi.org/10.1039/9781849735322-00249
Oballa RM, Belair L, Black WC, Bleasby K, Chan CC, Desroches C, et al. Development of a liver-targeted stearoyl-CoA desaturase (SCD) inhibitor (MK-8245) to establish a therapeutic window for the treatment of diabetes and dyslipidemia. J Med Chem. 2011;54:5082–96. https://doi.org/10.1021/jm200319u
Llona-Minguez S, Ghassemian A, Baranczewski P, Desroses M, Koolmeister T, Artursson P, et al. Structure-metabolism-relationships in the microsomal clearance of piperazin-1-ylpyridazines. Med Chem Commun. 2017;8:1553–60. https://doi.org/10.1039/C7MD00230K
Llona-Minguez S, Hoeglund A, Ghassemian A, Desroses M, Calderon-Montano JM, Burgos Moron E, et al. Piperazin-1-ylpyridazine derivatives are a novel class of human dCTP pyrophosphatase 1 inhibitors. J Med Chem. 2017;60:4279–92. https://doi.org/10.1021/acs.jmedchem.7b00182
Van Emelen K, Arts J, Backx LJJ, De Winter HJ, Van Brandt SFA, Verdonck MGC, et al. Preparation of sulfonyl-derivatives as novel inhibitors of histone deacetylase. World Patent Appl. 2003;WO-2003076422 A1. September 18th, 2003. https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2003076422
Betts MJ, Darbyshire CJ. Preparation of substituted piperazinyl-phenyl-oxazolidinone derivatives as antibacterial agents. World Patent Appl. 1998;WO-9801446 A1. January 15th, 1998. https://patentscope.wipo.int/search/en/detail.jsf?docId=WO1998001446
Carron CP, Trujillo JI, Olson KL, Huang W, Hamper BC, Dice T, et al. Discovery of an oral potent selective inhibitor of hematopoietic prostaglandin D synthase (HPGDS). ACS Med Chem Lett. 2010;1:59–63. https://doi.org/10.1021/ml900025z
Nguyen T-T-L, Kim JW, Choi H-I, Maeng H-J, Koo T-S. Development of an LC-MS/MS method for ARV-110, a PROTAC molecule, and applications to pharmacokinetic studies. Molecules. 2022;27:1977. https://doi.org/10.3390/molecules27061977
Gristina V, La Mantia M, Iacono F, Galvano A, Russo A, Bazan V. The emerging therapeutic landscape of ALK inhibitors in non-small cell lung cancer. Pharmaceuticals. 2020;13:474. https://doi.org/10.3390/ph13120474
Attwa MW, Kadia AA, Abdelhameed AS. Phase I metabolic profiling and unexpected reactive metabolites in human liver microsome incubations of X-376 using LC-MS/MS: bioactivation pathway elucidation and in silico toxicity studies of its metabolites. RSC Adv. 2020;10:5412. https://doi.org/10.1039/C9RA09115G
Zhao P, Zhuang L, Wang X, Huang S, Wu H, Zhou Y, et al. Discovery of spiro amide SHR902275: A potent, selective, and efficacious RAF inhibitor targeting RAS mutant cancers. Eur J Med Chem. 2022;228:114040. https://doi.org/10.1016/j.ejmech.2021.114040
Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51:5843–55. https://doi.org/10.1021/jm800245z
Moslin R, Zhang Y, Wrobleski ST, Lin S, Mertzman M, Spergel S, et al. Identification of N-methyl nicotinamide and N-methyl pyridazine-3-carboxamide pseudokinase domain ligands as highly selective allosteric inhibitors of tyrosine kinase 2 (TYK2). J Med Chem. 2019;62:8953–72. https://doi.org/10.1021/acs.jmedchem.9b00443
Wrobleski ST, Moslin R, Lin S, Zhang Y, Spergel S, Kempson J, et al. Highly selective inhibition of tyrosine kinase 2 (TYK2) for the treatment of autoimmune diseases: discovery of the allosteric inhibitor BMS-986165. J Med Chem. 2019;62:8973–95. https://doi.org/10.1021/acs.jmedchem.9b00444
Weinstein DS, Moslin RM. Advances in the discovery and development of selective tyrosine kinase 2 (TYK2) inhibitors. Med. Chem Rev. 2018;53:177–200. https://doi.org/10.29200/acsmedchemrev-v53.ch10
Gonzalez Lopez de Turiso F, Guckian K. Selective TYK2 inhibitors as potential therapeutic agents: a patent review (2019-2021). Expert Opin Ther Pat. 2022;32:365–79. https://doi.org/10.1080/13543776.2022.2026927
Goetz GH, Farrell W, Shalaeva M, Sciabola S, Anderson D, Yan J, et al. High throughput method for the indirect detection of intramolecular hydrogen bonding. J Med Chem. 2014;57:2920–29. https://doi.org/10.1021/jm401859b
Wakenhut F, Tran TD, Pickford C, Shaw S, Westby M, Smith-Burchnell C, et al. The discovery of potent nonstructural protein 5A (NS5A) inhibitors with a unique resistance profile - part 2. ChemMedChem. 2014;9:1387–96. https://doi.org/10.1002/cmdc.201400046
Pirali T, Serafini M, Cargnin S, Genazzani AA. Applications of deuterium in medicinal chemistry. J Med Chem. 2019;62:5276–97. https://doi.org/10.1021/acs.jmedchem.8b01808
Kato J-Y, Korenaga S, Iwakura M, Discovery. of a potent and subtype-selective TYK2 degrader based on an allosteric TYK2 inhibitor. Bioorg Med Chem Lett. 2023;79:129083. https://doi.org/10.1016/j.bmcl.2022.129083
Moslin R, Gardner D, Santella J, Zhang Y, Duncia JV, Liu C, et al. Identification of imidazo[1,2-b]pyridazine TYK2 pseudokinase ligands as potent and selective allosteric inhibitors of TYK2 signalling. Med Chem Commun. 2017;8:700–12. https://doi.org/10.1039/C6MD00560H
Liu C, Lin J, Moslin R, Tokarski JS, Muckelbauer J, Chang CY, et al. Identification of imidazo[1,2-b]pyridazine derivatives as potent, selective, and orally active Tyk2 JH2 inhibitors. ACS Med Chem Lett. 2019;10:383–8. https://doi.org/10.1021/acsmedchemlett.9b00035
The structure of NDI-034858 is available at https://drughunter.com/first-disclosures-from-acs-chicago-2022/. Accessed December 1st, 2022
Details can be found at: https://www.nimbustx.com/2022/11/30/nimbus-therapeutics-announces-positive-topline-results-for-phase-2b-clinical-trial-of-allosteric-tyk2-inhibitor-in-psoriasis/. Accessed December 1st, 2022
Zhao L, Zhang Y, Dai C, Guzi T, Wiswell D, Seghezzi W, et al. Design, synthesis and SAR of thienopyridines as potent CHK1 inhibitors. Bioorg Med Chem Lett. 2010;20:7216–21. https://doi.org/10.1016/j.bmcl.2010.10.105.
Shen D, Liu H, Qian F, Wang P. Design, synthesis and evaluation of novel thienopyridazine derivatives as Chk1/2 inhibitors. Bioorg Chem. 2022;121:105704. https://doi.org/10.1016/j.bioorg.2022.105704
Johnson F. Allylic strain in six-membered rings. Chem Rev. 1968;68:375–413. https://doi.org/10.1021/cr60254a001
Hu FZ, Zhang M, Song HB, Zou XM, Yang HZ. 3-Benzyloxy-6-fluoropyridazine. Acta Crystallogr E Crystallogr Commun. 2005;61:o2241–2. https://doi.org/10.1107/S1600536805019161
Hu FZ, Zhang M, Song HB, Zou XM, Yang HZ. 3,6-Bis(4-methoxybenzyloxy)pyridazine. Acta Crystallogr Sect E. 2005;61:o2486–8. https://doi.org/10.1107/S1600536805021173
Sharpless KB, Amberg W, Bennani YL, Crispino GA, Hartung J, Jeong KS, et al. The osmium-catalyzed asymmetric dihydroxylation: a new ligand class and a process improvement. J Org Chem. 1992;57:2768–71. https://doi.org/10.1021/jo00036a003
Hartung J, Sharpless KB. New ligands for asymmetric dihydroxylation: multiple cinchona alkaloid units attached to a central heterocyclic core. World Patent Appl. WO-9307142 A1. April 15th, 1993. https://patentscope.wipo.int/search/en/detail.jsf?docId=WO1993007142&_cid=P20-LB6RF4-14977
Amberg W, Bennani YL, Chadha RK, Crispino GA, Davis WD, Hartung J, et al. Syntheses and crystal structures of the cinchona alkaloid derivatives used as ligands in the osmium-catalyzed asymmetric dihydroxylation of olefins. J Org Chem. 1993;58:844–9. https://doi.org/10.1021/jo00056a015
Kolb HC, Andersson PG, Sharpless KB. Toward an understanding of the high enantioselectivity in the osmium-catalyzed asymmetric dihydroxylation (AD). 1. Kinetics. J Am Chem Soc. 1994;116:1278–91. https://doi.org/10.1021/ja00083a014
Corey EJ, Noe MC. Rigid and highly enantioselective catalyst for the dihydroxylation of olefins using osmium tetraoxide clarifies the origin of enantiospecificity. J Am Chem Soc. 1993;115:12579–80. https://doi.org/10.1021/ja00079a045
Corey EJ, Noe MC, Sarshar S. X-Ray crystallographic studies provide additional evidence that an enzyme-like binding pocket is crucial to the enantioselective dihydroxylation of olefins by OsO4-bis-cinchona alkaloid complexes. Tet Lett. 1994;35:2861–4. https://doi.org/10.1016/S0040-4039(00)76644-5
Zhang M, Hu F-Z, Zhao T, Yang L-Q, Yang H-Z. Synthesis and herbicidal evaluation of 3-N-substituted amino-6-benzyloxypyridazine derivatives. J Het Chem. 2014;51:1404–9. https://doi.org/10.1002/jhet.1831
Unlu S, Banoglu E, Ito S, Niiya T, Eren G, Okcelik B, et al. Synthesis, characterization and preliminary screening of regioisomeric 1-(3-pyridazinyl)-3-arylpyrazole and 1-(3-pyridazinyl)-5-arylpyrazole derivatives towards cyclooxygenase inhibition. J Enzym Inhib Med Chem. 2007;22:351–61. https://doi.org/10.1080/14756360601073468
Caliskan B, Luderer S, Ozkan Y, Werz O, Banoglu E. Pyrazol-3-propanoic acid derivatives as novel inhibitors of leukotriene biosynthesis in human neutrophils. Eur J Med Chem. 2011;46:5021–33. https://doi.org/10.1016/j.ejmech.2011.08.009
Levent S, Caliskan B, Ciftci M, Ozkan Y, Yenicesu I, Unver H, et al. Pyrazole derivatives as inhibitors of arachidonic acid-induced platelet aggregation. Eur J Med Chem. 2013;64:42–53. https://doi.org/10.1016/j.ejmech.2013.03.048
Abraham F, Mernari B, Lagrenee M, Sueur S. Crystal structure of 3,6-dihydroxymethylpyridazine. Acta Crystallogr C Struct Chem. 1988;C44:1267–9. https://doi.org/10.1107/S0108270188003610
Brameld KA, Kuhn B, Reuter DC, Stahl M. Small molecule conformational preferences derived from crystal structure data. a medicinal chemistry focused analysis. J Chem Inf Model. 2008;48:1–24. https://doi.org/10.1021/ci7002494
Huang H, Acquaviva L, Berry V, Bregman H, Chakka N, O'Connor A, et al. Structure-based design of potent and selective CK1γ inhibitors. ACS Med Chem Lett. 2012;3:1059–64. https://doi.org/10.1021/ml300278f
Trujillo JI, Kiefer JR, Huang W, Day JE, Moon J, Jerome GM, et al. Investigation of the binding pocket of human hematopoietic prostaglandin (PG) D2 synthase (hH-PGDS): a tale of two waters. Bioorg Med Chem Lett. 2012;22:3795–9. https://doi.org/10.1016/j.bmcl.2012.04.004
Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54:2529–91. https://doi.org/10.1021/jm1013693
Spyrakis F, Ahmed MH, Bayden AS, Cozzini P, Mozzarelli A, Kellogg GE. The roles of water in the protein matrix: a largely untapped resource for drug discovery. J Med Chem. 2017;60:6781–827. https://doi.org/10.1021/acs.jmedchem.7b00057
Chang M-Y, Lu Y-J, Cheng Y-C. In(OTf)3-mediated synthesis of substituted pyridazines. Tetrahedron. 2015;71:6840–5. https://doi.org/10.1016/j.tet.2015.07.025
Ackers B, Blake AJ, Hill SJ, Hubberstey P. 3,6-Di(thiophen-2-yl)pyridazine. Acta Cryst. 2002;C58:o640–1. https://doi.org/10.1107/S0108270102016037
Rahanyan N, Linden A, Baldridge KK, Siegel JS. Diels-Alder reactions of 3,6-disubstituted 1,2,4,5-tetrazines. Synthesis and X-ray crystal structures of diazafluoranthene derivatives. Org Biomol Chem. 2009;7:2082–92. https://doi.org/10.1039/B820551E
Napoletano M, Norcini G, Pellacini F, Marchini F, Morazzoni G, Ferlenga P, et al. Phthalazine PDE4 inhibitors. Part 2: The synthesis and biological evaluation of 6-methoxy-1,4-disubstituted derivatives. Bioorg Med Chem Lett. 2001;11:33–7. https://doi.org/10.1016/S0960-894X(00)00587-4
Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. Comparative molecular field analysis (CoMFA) of phthalazine derivatives as phosphodiesterase IV inhibitors. Bioorg Med Chem Lett. 2003;13:2473–9. https://doi.org/10.1016/S0960-894X(03)00493-1
Haack T, Fattori R, Napoletano M, Pellacini F, Fronza G, Raffaini G, et al. inhibitors: conformational study of some 6-methoxy-1,4-disubstituted derivatives. Bioorg Med Chem. 2005;13:4425–33. https://doi.org/10.1016/j.bmc.2005.04.057
Alessandro C, Antoine D, Perez Marta AS, Olivier M, Vincent Z. SwissBioisostere 2021: updated structural, bioactivity and physicochemical data delivered by a reshaped web interface. Nucleic Acids Res. 2022;50:D1382–90. https://doi.org/10.1093/nar/gkab1047
Chen M, Li Z, Shao X, Maienfisch P. Scaffold-hopping approach to identify new chemotypes of dimpropyridaz. J Agric Food Chem. 2022;70:11109–22. https://doi.org/10.1021/acs.jafc.2c00636
Garofalo AW, Adler M, Aubele DL, Bowers S, Franzini M, Goldbach E, et al. Novel cinnoline-based inhibitors of LRRK2 kinase activity. Bioorg Med Chem Lett. 2013;23:71–4. https://doi.org/10.1016/j.bmcl.2012.11.021
Smith CR, Dougan DR, Komandla M, Kanouni T, Knight B, Lawson JD, et al. Fragment-based discovery of a small molecule inhibitor of Bruton’s tyrosine kinase. J Med Chem. 2015;58:5437–44. https://doi.org/10.1021/acs.jmedchem.5b00734
Yao X, Sun X, Jin S, Yang L, Xu H, Rao Y. Discovery of 4-aminoquinoline-3-carboxamide derivatives as potent reversible Bruton’s tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. J Med Chem. 2019;62:6561–74. https://doi.org/10.1021/acs.jmedchem.9b00329
Wood MR, Noetzel MJ, Poslusney MS, Melancon BJ, Tarr JC, Lamsal A, et al. Challenges in the development of an M4 PAM in vivo tool compound: the discovery of VU0467154 and unexpected DMPK profiles of close analogs. Bioorg Med Chem Lett. 2017;27:171–5. https://doi.org/10.1016/j.bmcl.2016.11.086
Wood MR, Noetzel MJ, Melancon BJ, Poslusney MS, Nance KD, Hurtado MA, et al. Discovery of VU0467485/AZ13713945: an M4 PAM evaluated as a preclinical candidate for the treatment of schizophrenia. ACS Med Chem Lett. 2017;8:233–8. https://doi.org/10.1021/acsmedchemlett.6b00461
Patel L, Chandrasekhar J, Evarts J, Haran AC, Ip C, Kaplan JA, et al. 2,4,6-Triaminopyrimidine as a novel hinge binder in a series of PI3Kδ selective inhibitors. J Med Chem. 2016;59:3532–48. https://doi.org/10.1021/acs.jmedchem.6b00213
Verhoest PR, Chapin DS, Corman M, Fonseca K, Harms JF, Hou X. et al. Discoveryof a novel class of phosphodiesterase 10A inhibitors and identification ofclinical candidate 2-[4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl]quinoline(PF-2545920) for the treatment of schizophrenia. J Med Chem. 2009;52:5188–96. https://doi.org/10.1021/jm900521k
Hobson AD, Judge RA, Aguirre AL, Brown BS, Cui Y, Ding P, et al. Identification of selective dual ROCK1 and ROCK2 inhibitors using structure-based drug design. J Med Chem. 2018;61:11074–100. https://doi.org/10.1021/acs.jmedchem.8b01098
Kunikawa S, Tanaka A, Takasuna Y, Tasaki M, Chida N. Discovery of 2,4-diamino-5-cyanopyrimidine derivatives as protein kinase C theta inhibitors with mitigated time-dependent drug-drug interactions. Bioorg Med Chem. 2019;27:790–9. https://doi.org/10.1016/j.bmc.2019.01.019
Kim MJ, Lee J, Kang SY, Lee S-H, Son E-J, Jung ME, et al. Novel C-aryl glucoside SGLT2 inhibitors as potential antidiabetic agents: pyridazinylmethylphenyl glucoside congeners. Bioorg Med Chem Lett. 2010;20:3420–5. https://doi.org/10.1016/j.bmcl.2010.04.006
Chen M, Li Z, Shao X, Maienfisch P. Bioisosteric-replacement-driven lead optimization of tyclopyrazoflor. J Agric Food Chem. 2022;70:11123–37. https://doi.org/10.1021/acs.jafc.2c00654
Kuduk SD, Di Marco CN, Cofre V, Pitts DR, Ray WJ, Ma L, et al. Pyridine containing M1 positive allosteric modulators with reduced plasma protein binding. Bioorg Med Chem Lett. 2010;20:657–61. https://doi.org/10.1016/j.bmcl.2009.11.059
Kuduk SD, Di Marco CN, Cofre V, Pitts DR, Ray WJ, Ma L, et al. N-Heterocyclic derived M1 positive allosteric modulators. Bioorg Med Chem Lett. 2010;20:1334–7. https://doi.org/10.1016/j.bmcl.2010.01.013
Rover S, Andjelkovic M, Benardeau A, Chaput E, Guba W, Hebeisen P, et al. 6-Alkoxy-5-aryl-3-pyridinecarboxamides, a new series of bioavailable cannabinoid receptor type 1 (CB1) antagonists including peripherally selective compounds. J Med Chem. 2013;56:9874–96. https://doi.org/10.1021/jm4010708
Ahmed S, Ayscough A, Barker GR, Canning HE, Davenport R, Downham R, et al. 1,2,4-Triazolo-[1,5-a]pyridine HIF prolylhydroxylase domain-1 (PHD-1) inhibitors with a novel monodentate binding interaction. J Med Chem. 2017;60:5663–72. https://doi.org/10.1021/acs.jmedchem.7b00352
Anan K, Iso Y, Oguma T, Nakahara K, Suzuki S, Yamamoto T, et al. Trifluoromethyl dihydrothiazine-based β-secretase (BACE1) inhibitors with robust central β-amyloid reduction and minimal covalent binding burden. ChemMedChem. 2019;14:1894–910. https://doi.org/10.1002/cmdc.201900478
Otrubova K, Ezzili C, Boger DL. The discovery and development of inhibitors of fatty acid amide hydrolase (FAAH). Bioorg Med Chem Lett. 2011;21:4674–85. https://doi.org/10.1016/j.bmcl.2011.06.096
Otrubova K, Boger DL. α-Ketoheterocycle-based inhibitors of fatty acid amide hydrolase (FAAH). ACS Chem Neurosci. 2012;3:340–8. https://doi.org/10.1021/cn2001206
Maryanoff BE, Costanzo MJ. Inhibitors of proteases and amide hydrolases that employ α-ketoheterocycles as a key enabling functionality. Bioorg Med Chem. 2008;16:1562–95. https://doi.org/10.1016/j.bmc.2007.11.015
Long JZ, Cravatt BF. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem Rev. 2011;111:6022–63. https://doi.org/10.1021/cr200075y
Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. Binding and inactivation mechanism of a humanized fatty acid amide hydrolase by α-ketoheterocycle inhibitors revealed from cocrystal structures. J Am Chem Soc. 2009;131:10497–506. https://doi.org/10.1021/ja902694n
Boger DL, Miyauchi H, Du W, Hardouin C, Fecik RA, Cheng H, et al. Discovery of a potent, selective, and efficacious class of reversible α-ketoheterocycle inhibitors of fatty acid amide hydrolase effective as analgesics. J Med Chem. 2005;48:1849–56. https://doi.org/10.1021/jm049614v
Garfunkle J, Ezzili C, Rayl TJ, Hochstatter DG, Hwang I, Boger DL. Optimization of the central heterocycle of α-ketoheterocycle inhibitors of fatty acid amide hydrolase. J Med Chem. 2008;51:4392–403. https://doi.org/10.1021/jm800136b
Fleming FF, Yao L, Ravikumar PC, Funk L, Shook BC. Nitrile-containing pharmaceuticals: efficacious roles of the nitrile pharmacophore. J Med Chem. 2010;53:7902–17. https://doi.org/10.1021/jm100762r
Siklos M, BenAissa M, Thatcher GRJ. Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm Sin B 2015;5:506–19. https://doi.org/10.1016/j.apsb.2015.08.001
Brogi S, Ibba R, Rossi S, Butini S, Calderone V, Gemma S, et al. Covalent reversible inhibitors of cysteine proteases containing the nitrile warhead: recent advancement in the field of viral and parasitic diseases. Molecules. 2022;27:2561. https://doi.org/10.3390/molecules27082561
Ehmke V, Quinsaat JEQ, Rivera-Fuentes P, Heindl C, Freymond C, Rottmann M, et al. Tuning and predicting biological affinity: aryl nitriles as cysteine protease inhibitors. Org Biomol Chem. 2012;10:5764–8. https://doi.org/10.1039/C2OB00034B
Quesne MG, de Visser SP, Ward RA. Cysteine protease inhibition by nitrile-based inhibitors: a computational study. Front Chem. 2013;1:39. https://doi.org/10.3389/fchem.2013.00039
Oballa RM, Truchon J-F, Bayly CI, Chauret N, Day S, Crane S, et al. A generally applicable method for assessing the electrophilicity and reactivity of diverse nitrile-containing compounds. Bioorg Med Chem Lett. 2007;17:998–1002. https://doi.org/10.1016/j.bmcl.2006.11.044
Sinha S, Ahire D, Wagh S, Mullick D, Sistla R, Selvakumar K, et al. Electrophilicity of pyridazine-3-carbonitrile, pyrimidine-2-carbonitrile, and pyridine-carbonitrile derivatives: a chemical model to describe the formation of thiazoline derivatives in human liver microsomes. Chem Res Toxicol. 2014;27:2052–61. https://doi.org/10.1021/tx500256j
Sharma LK, Subramanian C, Yun M-K, Frank MW, White SW, Rock CO, et al. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat Commun. 2018;9:1–15. https://doi.org/10.1038/s41467-018-06703-2
Sharma LK, Yun M-K, Subramanian C, Tangallapally R, Jackowski S, Rock CO, et al. LipE guided discovery of isopropylphenyl pyridazines as pantothenate kinase modulators. Bioorg Med Chem. 2021;52:116504. https://doi.org/10.1016/j.bmc.2021.116504
Subramanian C, Frank MW, Tangallapally R, Yun M-K, Edwards A, White SW, et al. Pantothenate kinase activation relieves coenzyme A sequestration and improves mitochondrial function in mice with propionic acidemia. Sci Transl Med. 2021;13:eabf5965. https://doi.org/10.1126/scitranslmed.abf596
Subramanian C, Yun M-K, Yao J, Sharma LK, Lee RE, White SW, et al. Allosteric regulation of mammalian pantothenate kinase. J Biol Chem. 2016;291:22302–14. https://doi.org/10.1074/jbc.M116.748061
Lin H, Luengoa JI. Exploiting binding-site arginines in drug design: recent examples. Bioorg Med Chem Lett. 2020;30:127442. https://doi.org/10.1016/j.bmcl.2020.127442
Regueiro-Ren A, Swidorski JJ, Liu Z, Chen Y, Sin N, Sit S-Y, et al. Design, synthesis, and SAR of C‑3 benzoic acid, C‑17 triterpenoid derivatives. Identification of the HIV‑1 maturation inhibitor 4‑((1R,3aS,5aR,5bR,7aR,11aS,11bR,13aR,13bR)‑3a-((2-(1,1-dioxidothiomorpholino)ethyl)amino)-5a,5b,8,8,11a-pentamethyl-1-(prop-1-en-2-yl) 2,3,3a,4,5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a,13b-octadecahydro‑1H‑cyclopenta[a]chrysen-9-yl)benzoic acid (GSK3532795, BMS-955176). J Med Chem. 2018;61:7289–313. https://doi.org/10.1021/acs.jmedchem.8b00854
Marcoux D, Duan JJ-W, Shi Q, Cherney RJ, Srivastava AS, Cornelius L, et al. Rationally designed, conformationally constrained inverse agonists of RORγt - identification of a potent, selective series with biologic-like in vivo efficacy. J Med Chem. 2019;62:9931–46. https://doi.org/10.1021/acs.jmedchem.9b01369
Paulin R, Müller K, Diederich F. Orthogonal multipolar interactions in structural chemistry and biology. Angew Chem Int Ed. 2005;44:1788–805. https://doi.org/10.1002/anie.200462213
Parrish RM, Sitkoff DF, Cheney DL, Sherrill CD. The surprising importance of peptide bond contacts in drug–protein interactions. Chem Eur J 2017;23:7887–90. https://doi.org/10.1002/chem.201701031
Chen JM, Xu SL, Wawrzak Z, Basarab GS, Jordan DB. Structure-based design of potent inhibitors of scytalone dehydratase: displacement of a water molecule from the active site. Biochemistry. 1998;37:17735–44. https://doi.org/10.1021/bi981848r
Ladbury JE. Just add water! The effect of water on the specificity of protein ligand binding sites and its potential application to drug design. Chem Biol. 1996;3:973–80. https://doi.org/10.1016/S1074-5521(96)90164-7
Ellermann M, Jakob-Roetne R, Lerner C, Borroni E, Schlatter D, Roth D, et al. Molecular recognition at the active site of catechol-O-methyltransferase: Energetically favorable replacement of a water molecule imported by a bisubstrate inhibitor. Angew Chem Int Ed. 2009;48:9092–6. https://doi.org/10.1002/anie.200904410
Varnes JG, Gero T, Huang S, Diebold RB, Ogoe C, Grover PT, et al. Towards the next generation of dual Bcl-2/Bcl-xL inhibitors. Bioorg Med Chem Lett 2014;24:3026–33. https://doi.org/10.1016/j.bmcl.2014.05.036
Gleave RJ, Beswick PJ, Brown AJ, Giblin GMP, Goldsmith P, Haslam CP, et al. Synthesis and evaluation of 3-amino-6-aryl-pyridazines as selective CB2 agonists for the treatment of inflammatory pain. Bioorg Med Chem Lett. 2010;20:465–8. https://doi.org/10.1016/j.bmcl.2009.11.117
Ohno H, Minamiguchi D, Nakamura S, Shu K, Okazaki S, Honda M, et al. Structure-activity relationship study of 4-(thiazol-5-yl)benzoic acid derivatives as potent protein kinase CK2 inhibitors. Bioorg Med Chem. 2016;24:1136–41. https://doi.org/10.1016/j.bmc.2016.01.043
Barsanti PA, Pan Y, Lu Y, Jain R, Cox M, Aversa RJ, et al. Structure-based drug design of novel, potent, and selective azabenzimidazoles (ABI) as ATR inhibitors. ACS Med Chem Lett. 2015;6:42–6. https://doi.org/10.1021/ml500352s
Debenham JS, Madsen-Duggan C, Clements MJ, Walsh TF, Kuethe JT, Reibarkh M, et al. Discovery of N-[bis(4-methoxyphenyl)methyl]-4-hydroxy-2-(pyridazin-3-yl)pyrimidine-5-carboxamide (MK-8617), an orally active pan-inhibitor of hypoxia-inducible factor prolyl hydroxylase 1-3 (HIF PHD1-3) for the treatment of anemia. J Med Chem. 2016;59:11039–49. https://doi.org/10.1021/acs.jmedchem.6b01242
Cohen SM. Bioinorganic approach to fragment-based drug discovery targeting metalloenzymes. Acc Chem Res. 2017;50:2007–16. https://doi.org/10.1021/acs.accounts.7b00242
Vachal P, Miao S, Pierce JM, Guiadeen D, Colandrea VJ, Wyvratt MJ, et al. 1,3,8-Triazaspiro[4.5]decane-2,4-diones as efficacious pan-inhibitors of hypoxia-inducible factor prolyl hydroxylase 1–3 (HIF PHD1–3) for the treatment of anemia. J Med Chem. 2012;55:2945–59. https://doi.org/10.1021/jm201542d
Berlin M, Boyce CW, de Lera Ruiz M. Histamine H3 receptor as a drug discovery target. J Med Chem. 2011;54:26–53. https://doi.org/10.1021/jm100064d
Berlin M, Lee YJ, Boyce CW, Wang Y, Aslanian R, McCormick KD, et al. Reduction of hERG inhibitory activity in the 4-piperidinyl urea series of H3 antagonists. Bioorg Med Chem Lett. 2010;20:2359–64. https://doi.org/10.1016/j.bmcl.2010.01.121
Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, Donatelli CA, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med Chem Lett. 2010;1:39–43. https://doi.org/10.1021/ml900028r
Gunzner-Toste J, Zhao G, Bauer P, Baumeister T, Buckmelter AJ, Caligiuri M, et al. Yuen P-w, Zak M, Zhang L, Zheng X, Bair KW, Dragovich, PS. Discovery of potent and efficacious urea-containing nicotinamide phosphoribosyltransferase (NAMPT) inhibitors with reduced CYP2C9 inhibition properties. Bioorg Med Chem Lett. 2013;23:3531–8. https://doi.org/10.1016/j.bmcl.2013.04.040
Mammoliti O, Palisse A, Joannesse C, El Bkassiny S, Allart B, Jaunet A. et al. Discovery of the S1P2 antagonist GLPG2938(1-[2-ethoxy-6-(trifluoromethyl)-4-pyridyl]-3-[[5-methyl-6-[1-methyl-3-(trifluoromethyl)pyrazol-4-yl]pyridazin-3-yl]methyl]urea), a preclinical candidate for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2021;64:6037–58. https://doi.org/10.1021/acs.jmedchem.1c00138
Ahn K, Johnson DS, Cravatt BF. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin Drug Disco. 2009;4:763–84. https://doi.org/10.1517/17460440903018857
Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, et al. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry. 2007;46:13019–30. https://doi.org/10.1021/bi701378g
Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16:411–20. https://doi.org/10.1016/j.chembiol.2009.02.013
Johnson DS, Ahn K, Kesten S, Lazerwith SE, Song Y, Morris M, et al. Benzothiophene piperazine and piperidine urea inhibitors of fatty acid amide hydrolase (FAAH). Bioorg Med Chem Lett. 2009;19:2865–9. https://doi.org/10.1016/j.bmcl.2009.03.080
Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, et al. Discovery of PF-04457845: a highly potent, orally bioavailable, and selective urea FAAH inhibitor. ACS Med Chem Lett. 2011;2:91–6. https://doi.org/10.1021/ml100190t
Li B-F, Moree WJ, Yu J, Coon T, Zamani-Kord S, Malany S, et al. Selectivity profiling of novel indene H1-antihistamines for the treatment of insomnia. Bioorg Med Chem Lett. 2010;20:2629–33. https://doi.org/10.1016/j.bmcl.2010.02.055
Moree WJ, Li B-F, Zamani-Kord S, Yu J, Coon T, Huang C, et al. Identification of a novel selective H1-antihistamine with optimized pharmacokinetic properties for clinical evaluation in the treatment of insomnia. Bioorg Med Chem Lett. 2010;20:5874–8. https://doi.org/10.1016/j.bmcl.2010.07.117
Luo H, Hawes EM, McKay G, Korchinski ED, Midha K. N+-glucuronidation of aliphatic tertiary amines, a general phenomenon in the metabolism of H1-antihistamines in humans. Xenobiotica. 1991;21:1281–8. https://doi.org/10.3109/00498259109043203
Brooker S. Some copper and cobalt complexes of Schiff-base macrocycles containing pyridazine head units. Eur J Inorg Chem. 2002;2535-47. https://doi.org/10.1002/1099-0682(200210)2002:10<2535::AID-EJIC2535>3.0.CO;2-Z
Horeau M, Lautrette G, Wicher B, Blot V, Lebreton J, Pipelier M, et al. Metal-coordination-assisted folding and guest binding in helical aromatic oligoamide molecular capsules. Angew Chem Int Ed. 2017;56:6823–7. https://doi.org/10.1002/anie.201701693
de Sahb C, Watson LA, Nadas J, Hay BP. Design criteria for polyazine extractants to separate AnIII from LnIII. Inorg Chem. 2013;52:10632–42. https://doi.org/10.1021/ic401666m
Onggo D, Rae AD, Goodwin HA. Coordination of the strong field di-imine 3,3'-bipyridazine. Structural, magnetic and spectroscopic properties of the iron(II), cobalt(II) and nickel(II) complexes. Inorg Chim Acta. 1990;178:151–63. https://doi.org/10.1016/S0020-1693(00)86776-6
Lincker F, Kreher D, Attias A-J, Do J, Kim E, Hapiot P, et al. Rodlike fluorescent π-conjugated 3,3'-bipyridazine ligand: optical, electronic, and complexation properties. Inorg Chem. 2010;49:3991–4001. https://doi.org/10.1021/ic901925w
Bembenek SD, Venkatesan H, Peltier HM, Rosen MD, Barrett TD, Kanelakis KC, et al. Beyond traditional structure-based drug design: the role of iron complexation, strain, and water in the binding of inhibitors for hypoxia-inducible factor prolyl hydroxylase 2. ACS Omega. 2019;4:6703–8. https://doi.org/10.1021/acsomega.9b00199
Franklin TJ, Hales NJ, Johnstone D, Morris WB, Cunliffe CJ, Millest AJ, et al. Approaches to the design of anti-fibrotic drugs. Biochem Soc Trans. 1991;19:812–5. https://doi.org/10.1042/bst0190812
Joharapurkar AA, Pandya VB, Patel VJ, Desai RC, Jain MR. Prolyl hydroxylase inhibitors: a breakthrough in the therapy of anemia associated with chronic diseases. J Med Chem. 2018;61:6964–82. https://doi.org/10.1021/acs.jmedchem.7b01686
Vasta JD, Raines RT. Collagen prolyl 4-hydroxylase as a therapeutic target. J Med Chem. 2018;61:10403–11. https://doi.org/10.1021/acs.jmedchem.8b00822
Meanwell NA. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J Med Chem. 2018;61:5822–80. https://doi.org/10.1021/acs.jmedchem.7b01788
Maramai S, Benchekroun M, Ward SE, Atack JR. Subtype selective γ-aminobutyric acid type A Receptor (GABAAR) modulators acting at the benzodiazepine binding site: an update. J Med Chem. 2020;63:3425–46. https://doi.org/10.1021/acs.jmedchem.9b01312
McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nat Neurosci. 2000;3:587–92. https://doi.org/10.1038/75761
Carling RW, Madin A, Guiblin A, Russell MGN, Moore KW, Mitchinson A, et al. 7-(1,1-Dimethylethyl)-6-(2-ethyl-2H-1,2,4-triazol-3-ylmethoxy)-3-(2-fluorophenyl)-1,2,4-triazolo[4,3-b]pyridazine: a functionally selective γ-aminobutyric acidA (GABAA) α2/α3-subtype selective agonist that exhibits potent anxiolytic activity but is not sedating in animal models. J Med Chem 2005;48:7089–92. https://doi.org/10.1021/jm058034a
Russell MGN, Carling RW, Street LJ, Hallett DJ, Goodacre S, Mezzogori E, et al. Discovery of imidazo[1,2-b][1,2,4]triazines as GABAA α2/3 subtype selective agonists for the treatment of anxiety. J Med Chem. 2006;49:1235–8. https://doi.org/10.1021/jm051200u
Jennings ASR, Lewis RT, Russell MGN, Hallett DJ, Street LJ, Castro JL, et al. Imidazo[1,2-b][1,2,4]triazines as α2/α3 subtype selective GABAA agonists for the treatment of anxiety. Bioorg Med Chem Lett. 2006;16:1477–80. https://doi.org/10.1016/j.bmcl.2005.12.044
Owen RM, Blakemore D, Cao L, Flanagan N, Fish R, Gibson KR, et al. Design and identification of a novel, functionally subtype selective GABAA positive allosteric modulator (PF-06372865). J Med Chem. 2019;62:5773–96. https://doi.org/10.1021/acs.jmedchem.9b00322
Nickolls SA, Gurrell R, van Amerongen G, Kammonen J, Cao L, Brown AR, et al. Pharmacology in translation: the preclinical and early clinical profile of the novel alpha2/3 functionally selective GABAA receptor positive allosteric modulator PF-06372865. Br J Pharm. 2018;175:708–25. https://doi.org/10.1111/bph.14119
Cerne R, Lippa A, Poe MM, Smith JL, Jin X, Ping X, et al. GABAkines - advances in the discovery, development, and commercialization of positive allosteric modulators of GABAA receptors. Pharm Ther. 2022;234:108035 https://doi.org/10.1016/j.pharmthera.2021.108035
Goodacre SC, Street LJ, Hallett DJ, Crawforth JM, Kelly S, Owens AP, et al. Imidazo[1,2-a]pyrimidines as functionally selective and orally bioavailable GABAAα2/α3 binding site agonists for the treatment of anxiety disorders. J Med Chem. 2006;49:35–8. https://doi.org/10.1021/jm051065l
Humphries AC, Gancia E, Gilligan MT, Goodacre S, Hallett D, Merchant KJ, et al. 8-Fluoroimidazo[1,2-a]pyridine: synthesis, physicochemical properties and evaluation as a bioisosteric replacement for imidazo[1,2-a]pyrimidine in an allosteric modulator ligand of the GABAA receptor. Bioorg Med Chem Lett. 2006;16:1518–22. https://doi.org/10.1016/j.bmcl.2005.12.037
Sternfield F, Carling RW, Jelley RA, Ladduwahetty T, Merchant KJ, Moore KW, et al. Selective, orally active γ-aminobutyric acid-A α5 receptor inverse agonists as cognition enhancers. J Med Chem. 2004;47:2176–9. https://doi.org/10.1021/jm031076j
Dawson GR, Maubach KA, Collinson N, Cobain M, Everitt BJ, MacLeod AM, et al. An inverse agonist selective for α5 subunit-containing GABAA receptors enhances cognition. J Pharm Exp Ther. 2006;316:1335–45. https://doi.org/10.1124/jpet.105.092320
Street LJ, Sternfeld F, Jelley RA, Reeve AJ, Carling RW, Moore KW, et al. Synthesis and biological evaluation of 3-heterocyclyl-7,8,9,10-tetrahydro-(7,10-ethano)-1,2,4-triazolo-[3,4-a]phthalazines and analogues as subtype-selective inverse agonists for the GABAA α5 benzodiazepine binding site. J Med Chem. 2004;47:3642–57. https://doi.org/10.1021/jm0407613
Chambers MS, Atack JR, Carling RW, Collinson N, Cook SM, Dawson GR, et al. An orally bioavailable, functionally selective inverse agonist at the benzodiazepine site of GABAA α5 receptors with cognition enhancing properties. J Med Chem. 2004;47:5829–32. https://doi.org/10.1021/jm040863t
Atack JR, Maubach KA, Wafford KA, O’Connor D, Rodrigues AD, Evans DC, et al. In vitro and in vivo properties of 3-tert-butyl-7-(5-methylisoxazol-3-yl)-2-(1-methyl-1H-1,2,4-triazol-5-ylmethoxy)-pyrazolo[1,5-d]-[1,2,4]triazine (MRK-016), a GABAA Receptor α5 subtype-selective inverse agonist. J Pharm Exp Ther. 2009;331:470–84. https://doi.org/10.1124/jpet.109.157636
Van Niel MB, Wilson K, Adkins CH, Atack JR, Castro JL, Clarke DE, et al. A new pyridazine series of GABAA α5 ligands. J Med Chem. 2005;48:6004–11. https://doi.org/10.1021/jm050249x
Lewis RT, Blackaby WP, Blackburn T, Jennings ASR, Pike A, Wilson RA, et al. A pyridazine series of α2/α3 subtype selective GABAA agonists for the treatment of anxiety. J Med Chem. 2006;49:2600–10. https://doi.org/10.1021/jm051144x
Conn PJ, Lindsley CW, Meiler J, Niswender CM. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for treating CNS disorders. Nat Rev Drug Disco. 2014;13:692–708. https://doi.org/10.1038/nrd4308
Barbaro L, Rodriguez AL, Blevins AN, Dickerson JW, Billard N, Boutaud O, et al. Discovery of "molecular switches" within a series of mGlu5 allosteric ligands driven by a “magic methyl” effect affording both PAMs and NAMs with in vivo activity, derived from an M1 PAM chemotype. ACS Bio Med Chem Au. 2021;1:21–30. https://doi.org/10.1021/acsbiomedchemau.1c00024
Orgovan Z, Ferenczy GG, Keseru GM. Allosteric molecular switches in metabotropic glutamate receptors. ChemMedChem. 2021;16:81–93. https://doi.org/10.1002/cmdc.202000444
Yogeeswari P, Sriram D, Bal TR, Thirumurugan R. Epibatidine and its analogues as nicotinic acetylcholine receptor agonist: an update. Nat Prod Res. 2006;20:497–505. https://doi.org/10.1080/14786410600604583
Garraffo HM, Spande TF, Williams M. Epibatidine: from frog alkaloid to analgesic clinical candidates. A testimonial to “true grit”! Heterocycles. 2009;79:207–17. https://doi.org/10.3987/REV-08-SR(D)5
Salehi B, Sestito S, Rapposelli S, Peron G, Calina D, Sharifi-Rad M, et al. Epibatidine: a promising natural alkaloid in health. Biomolecules. 2019;9:6 https://doi.org/10.3390/biom9010006
Che D, Wegge T, Stubbs MT, Seitz G, Meier H, Methfessel C. exo-2-(Pyridazin-4-yl)-7-azabicyclo[2.2.1]heptanes: syntheses and nicotinic acetylcholine receptor. agonist activity of potent pyridazine analogues of (±)-epibatidine. J Med Chem. 2001;44:47–57. https://doi.org/10.1021/jm000949w
Gohlke H, Gündisch D, Schwarz S, Seitz G, Tilotta MC, Wegge T. Synthesis and nicotinic binding studies on enantiopure diazine analogues of the novel (2-chloro-5-pyridyl)-9-azabicyclo[4.2.1]non-2-ene UB-165. J Med Chem. 2002;45:1064–72. https://doi.org/10.1021/jm010936y
Porter JD, Vivas O, Weaver CD, Alsafran A, DiMilo E, Arnold LA, et al. An anthrone-based Kv7.2/7.3 channel blocker with improved properties for the investigation of psychiatric and neurodegenerative disorders. Bioorg Med Chem Lett. 2019;29:126681. https://doi.org/10.1016/j.bmcl.2019.126681
Claesson A, Minidis A. Systematic approach to organizing structural alerts for reactive metabolite formation from potential drugs. Chem Res Toxicol. 2018;31:389–411. https://doi.org/10.1021/acs.chemrestox.8b00046
Kalgutkar AS. Designing around structural alerts in drug discovery. J Med Chem. 2020;63:6276–302. https://doi.org/10.1021/acs.jmedchem.9b00917
Koppel N, Rekdal VM, Balskus EP. Chemical transformation of xenobiotics by the human gut microbiota. Science. 2017;356:1246. https://doi.org/10.1126/science.aag2770
Guo Y, Lee H, Jeong H. Gut microbiota in reductive drug metabolism. Prog Mol Biol Transl Sci. 2020;171:61–93. https://doi.org/10.1016/bs.pmbts.2020.04.002
Xie Y, Hu F, Xiang D, Lu H, Li W, Zhao A, et al. The metabolic effect of gut microbiota on drugs. Drug Metab Rev. 2020;52:139–56. https://doi.org/10.1080/03602532.2020.1718691
Altuntas TG, Gorrod JW. Factors involved in the N-oxidation of isomeric aromatic diazines by microsomal preparations. Drug Metab Drug Interact. 1995;12:117–30. https://doi.org/10.1515/dmdi.1995.12.2.117
Bambury RE, Feeley DT, Lawton GC, Weaver JM, Wemple J. Mesoionic pyridazine and pyridine nucleosides. An unusual biologically active nucleoside metabolite. J Chem Soc Chem Commun. 1984;422-3. https://doi.org/10.1039/C39840000422
Polsky-Fisher SL, Vickers S, Cui D, Subramanian R, Arison BH, Agrawal NGB, et al. Metabolism and disposition of a potent and selective GABA-Aα2/3 receptor agonist in healthy male volunteers. Drug Metab Disp. 2006;34:1004–11. https://doi.org/10.1124/dmd.105.008193
Ma B, Polsky-Fisher SL, Vickers S, Cui D, Rodrigues AD. Cytochrome P450 3A-dependent metabolism of a potent and selective γ-aminobutyric acidAα2/3 receptor agonist in vitro: involvement of cytochrome P450 3A5 displaying biphasic kinetics. Drug Metab Disp. 2007;35:1301–7. https://doi.org/10.1124/dmd.107.014753
Nakagawa M, Ando M. Metabolism of credazine, 3-(2’methylphenoxy)pyridazine, in rats. Agric Biol Chem. 1977;41:1975–84. https://doi.org/10.1080/00021369.1977.10862794
Borchert HH, Pfeifer S, Helbig B, Franke P, Heinisch G. Pharmacokinetics and biotransformation of drugs. Part 77. Biotransformation of pyridazines. Part 1. Pyridazine 3-methylpyridazine Pharmazie. 1989;44:625–30
Hutzler JM, Obach RS, Dalvie D, Zientek MA. Strategies for a comprehensive understanding of metabolism by aldehyde oxidase. Expert Opin Drug Metab Toxicol. 2013;9:153–68. https://doi.org/10.1517/17425255.2013.738668
Lepri S, Ceccarelli M, Milani N, Tortorella S, Cucco A, Valeri A, et al. Structure-metabolism relationships in human-AOX: Chemical insights from a large database of aza-aromatic and amide compounds. Proc Natl Acad Sci USA. 2017;114:E3178–87. https://doi.org/10.1073/pnas.1618881114
Manevski N, King L, Pitt WR, Lecomte F, Toselli F. Metabolism by aldehyde oxidase: drug design and complementary approaches to challenges in drug discovery. J Med Chem. 2019;62:10955–94. https://doi.org/10.1021/acs.jmedchem.9b00875
Ghafourian T, Rashidi MR. Quantitative study of the structural requirements of phthalazine/quinazoline derivatives for interaction with human liver aldehyde oxidase. Chem Pharm Bull. 2001;49:1066–71. https://doi.org/10.1248/cpb.49.1066
Coelho C, Foti A, Hartmann T, Santos-Silva T, Leimkuhler S, Romao MJ. Structural insights into xenobiotic and inhibitor binding to human aldehyde oxidase. Nat Chem Biol. 2015;11:779–83. https://doi.org/10.1038/nchembio.1895
Torres RA, Korzekwa KR, McMasters DR, Fandozzi CM, Jones JP. Use of density functional calculations to predict the regioselectivity of drugs and molecules metabolized by aldehyde oxidase. J Med Chem. 2007;50:4642–7. https://doi.org/10.1021/jm0703690
Pryde DC, Tran T-D, Jones P, Duckworth J, Howard M, Gardner I, et al. Medicinal chemistry approaches to avoid aldehyde oxidase metabolism. Bioorg Med Chem Lett. 2012;22:2856–60. https://doi.org/10.1016/j.bmcl.2012.02.069
Zhang C, Crawford JJ, Landry ML, Chen H, Kenny JR, Khojasteh SC, et al. Strategies to mitigate the bioactivation of aryl amines. Chem Res Toxicol. 2020;33:1950–9. https://doi.org/10.1021/acs.chemrestox.0c00138
Boldron C, Besse A, Bordes M-F, Tissandie S, Yvon X, Gau B. et al. N-[6-(4-Butanoyl-5-methyl-1H-pyrazol-1-yl)pyridazin-3-yl]-5-chloro-1-[2-(4-methylpiperazin-1-yl)-2-oxoethyl]-1H-indole-3-carboxamide (SAR216471), a novelintravenous and oral, reversible, and directly acting P2Y12 antagonist. J Med Chem. 2014;57:7293–316. https://doi.org/10.1021/jm500588w
Kim D, Guengerich FP. Cytochrome P450 activation of arylamines and heterocyclic amines. Annu Rev Pharm Toxicol. 2005;45:27–49. https://doi.org/10.1146/annurev.pharmtox.45.120403.100010
Delesque-Touchard N, Pflieger AM, Bonnet-Lignon S, Millet L, Salel V, Boldron C, et al. SAR216471, an alternative to the use of currently available P2Y12 receptor inhibitors? Thrombosis Res. 2014;134:693–703. https://doi.org/10.1016/j.thromres.2014.06.034
Sapi J. In memoriam: Professor Camille-Georges Wermuth. Eur J Med Chem. 2016;108:741. https://doi.org/10.1016/j.ejmech.2015.11.017
Graton J, Le Questel J-Y, Maxwell P, Popelier P. Hydrogen-bond accepting properties of new heteroaromatic ring chemical motifs: a theoretical study. J Chem Inf Model. 2016;56:322–34. https://doi.org/10.1021/acs.jcim.5b00574
Mitra K. Acyl glucuronide and coenzyme a thioester metabolites of carboxylic acid-containing drug molecules: layering chemistry with reactive metabolism and toxicology. Chem Res Toxicol. 2022;35:177–88. https://doi.org/10.1021/acs.chemrestox.2c00188
Stachulski AV, Harding JR, Lindon JC, Maggs JL, Park BK, Wilson ID. Acyl glucuronides: biological activity, chemical reactivity, and chemical synthesis. J Med Chem. 2006;49:6931–45. https://doi.org/10.1021/jm060599z
Smith DA, Hammond T, Baillie TA. Safety assessment of acyl glucuronides—a simplified paradigm. Drug Metab Disp. 2018;46:908–12. https://doi.org/10.1124/dmd.118.080515
Walles M, Brown AP, Zimmerlin A, End P. New perspectives on drug-induced liver injury risk assessment of acyl glucuronides. Chem Res Toxicol. 2020;33:1551–60. https://doi.org/10.1021/acs.chemrestox.0c00131
Bradshaw PR, Athersuch TJ, Stachulski AV, Wilson ID. Acyl glucuronide reactivity in perspective. Drug Disco Today. 2020;25:1639–50. https://doi.org/10.1016/j.drudis.2020.07.009
Grillo MP. Drug-S-acyl-glutathione thioesters: synthesis, bioanalytical properties, chemical reactivity, biological formation and degradation. Curr Drug Metab. 2011;12:229–44. https://doi.org/10.2174/138920011795101886
Trub AG, Hirschey MD. Reactive acyl-CoA species modify proteins and induce carbon stress. Trends Biochem Sci. 2018;43:369–79. https://doi.org/10.1016/j.tibs.2018.02.002
Stroebel D, Buhl DL, Knafels JD, Chanda PK, Green M, Sciabola S, et al. A novel binding mode reveals two distinct classes of NMDA receptor GluN2B-selective antagonists. Mol Pharm. 2016;89:541–51. https://doi.org/10.1124/mol.115.103036
Chin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science. 2020;369:993–9. https://doi.org/10.1126/science.abb4255
Goldberg FW, Kettle JG, Lamont GM, Buttar D, Ting AKT, McGuire TM, et al. Discovery of clinical candidate AZD0095, a selective inhibitor of monocarboxylate transporter 4 (MCT4) for oncology. J Med Chem. 2023; https://doi.org/10.1021/acs.jmedchem.2c01342
Acknowledgements
This review article is offered as a contribution to the special issue of Medicinal Chemistry Research devoted to honoring the outstanding career of Professor Thomas A. Baillie. I have had the good fortune to become acquainted with Tom through my work with Fred Guengerich and the Division of Chemical Toxicology of the American Chemical Society. Tom has been a key opinion leader in the arena of drug metabolism and bioactivation for many years and he has imparted his insights and wisdom to inform the medicinal chemistry community of how to reconcile experimental observations and manage risk in drug candidate optimization. I am delighted to be able to present this synopsis of the applications of pyridazines in drug design as a token of my appreciation for, and recognition of, his sustained and significant contributions to chemical toxicology and drug discovery and development.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Meanwell, N.A. The pyridazine heterocycle in molecular recognition and drug discovery. Med Chem Res 32, 1853–1921 (2023). https://doi.org/10.1007/s00044-023-03035-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03035-9