
Abstract
As an integral lysosomal transmembrane protein, transmembrane protein 106B (TMEM106B) regulates several aspects of lysosomal function and is associated with neurodegenerative diseases. The TMEM106B gene mutations lead to lysosomal dysfunction and accelerate the pathological progression of Neurodegenerative diseases. Yet, the precise mechanism of TMEM106B in Neurodegenerative diseases remains unclear. Recently, different research teams discovered that TMEM106B is an amyloid protein and the C-terminal domain of TMEM106B forms amyloid fibrils in various Neurodegenerative diseases and normally elderly individuals. In this review, we discussed the physiological functions of TMEM106B. We also included TMEM106B gene mutations that cause neurodegenerative diseases. Finally, we summarized the identification and cryo-electronic microscopic structure of TMEM106B fibrils, and discussed the promising therapeutic strategies aimed at TMEM106B fibrils and the future directions for TMEM106B research in neurodegenerative diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Transmembrane protein 106B (TMEM106B), composed of 274 amino acids, is a type II transmembrane lysosomal protein with its subcellular location being in the late endosome and lysosomal membranes [1, 2]. It is found primarily within neurons and oligodendrocytes in the central nervous system [3]. It has been reported that TMEM106B is an integral lysosomal protein and has crucial effects on lysosome morphology, localization, trafficking, and functions [2, 4, 5]. Although its exact role in the pathogenesis of neurodegenerative diseases remains unclear, studies have shown that TMEM106B appears to affect the pathological burden of TAR DNA binding protein-43 (TDP-43) pathology [5]. Previous studies found that the expression of TMEM106B (including mRNA and protein) in the brain of Alzheimer’s Disease (AD) patients is significantly reduced [6]. However, mutations in TMEM106B increase its expression level, result in lysosome dysfunction, and promote its aggregation [2, 4]. Genome-wide association studies (GWAS) identified mutations of the TMEM106B gene as a major risk factor for frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP) [7, 8], which was mostly associated with the risk of FTLD-TDP in patients with progranulin (GRN) mutations [7, 9, 10]. Mutations and single nucleotide polymorphisms (SNPs) of the TMEM106B gene lead to lysosomal deficits in the clearance of misfolded proteins, which are the main pathological changes in multiple Neurodegenerative diseases [11,12,13]. Recently, studies revealed that the luminal domain of TMEM106B forms amyloid fibrils in various Neurodegenerative diseases and neurologically normal older adults [14]. However, the precise mechanism of TMEM106B at the lysosomal membrane is undetermined and it remains unknown how TMEM106B contributes to the development of Neurodegenerative diseases. In this review, we will introduce current knowledge of TMEM106B in physiological and pathological function and its potential association with Neurodegenerative diseases. Then, we elucidate the identification and cryo-electronic microscopic (cryo-EM) structure of TMEM106B fibrils and analyze the factors that contribute to the polymorphisms of TMEM106B fibrils. Finally, we discussed the potential pathogenic role of TMEM106B fibrils and the future directions for TMEM106B research in Neurodegenerative diseases.
Structure of TMEM106B in the native state
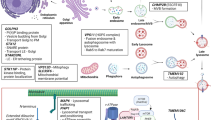
TMEM106B is mainly expressed on the lysosomal membranes of neurons and oligodendrocytes in the central nervous system [15]. This protein consists of three parts, an N-terminal domain (NTD, 1–96 aa) facing the cytosol, a transmembrane domain (TMD, 97-117aa), and a C-terminal domain (CTD, 118–274 aa) facing the lysosome lumen [1]. TMEM106B is likely to be processed by lysosomal proteases at a position close to G127 [2]. The resulting C-terminal fragment contains five highly glycosylated sites at N145, N151, N164, N183 and N256 [1]. Following shedding of the ectodomain, the residual N-terminal fragment (NTF) is anchored to the lysosomal membrane, which is further cleaved by signal peptide peptidase-like 2A (SPPL2A) through intramembrane proteolysis to release intracellular cytosolic domain [16] (Fig. 1). However, many questions remain on (i) which protease(s) is responsible for cleaving the luminal domain, (ii) the precise cleavage site, (iii) the factors that contribute to enzyme digestion of TMEM106B, and (iv) the potential functions of the generated peptides. The mechanisms behind the proteolysis of TMEM106B are important because this contributes to the understanding of what gives rise to TMEM106B fibrils formation.

Structure, physiological and pathologic function of TMEM106B. ① TMEM106B is a type II transmembrane protein located on late endosome and lysosome, with its N-terminus facing the cytosol and C-terminus facing the lysosome lumen. TMEM106B consists of 274 residues and three structural domains. This C-terminal fragment (118–274 residue) contains five important N-glycosylation sites (N145, N151, N164, N183, N256). TMEM106B is processed into a N-terminal fragment (NTF) by lysosomal proteases, which is further cleaved by SPPL2a through intramembrane proteolysis to generate intracellular cytosolic domain. ② TMEM106B SNPs and mutations lead to TMEM106B abnormal expression and dysfunction in morphology, transportation, acidification and maturation of lysosomes. TMEM106B fragment may form fibrils after cleavage. ③Lysosomal activity is modulated by intraluminal pH. The interaction between TMEM106B and v-ATPase controls the acidification of lysosomes. TMEM106B knockdown results in less efficient fusion with autophagosome, poor protein degradation efficiency, and insufficient acidification. TMEM106B also regulates lysosomal trafficking in neurons. In dendrites, TMEM106B interacts with MAP6 to restrict retrograde transport of lysosomes. Loss of TMEM106B leads to the formation of LAMP1-positive lysosomal vacuoles at the axon initial segment region. This is partly due to increased retrograde transport of lysosomes along axons. TMEM106B further activates TFEB-dependent lysosome biogenesis. Endosome and lysosome fusion are also mediated by TMEM106B
Recent studies found that TMEM106B is made up of a single rod-like structure or a doublet of filaments forming a twisted ribbon, of which several polymorphisms have been identified: 4 singlets and 2 doublets [12]. The structural domains are thought to be related but have little to do with the N-terminal domain, as the TMEM106B gene polymorphisms are conserved at the N-terminus domain, and the differences in structure are primarily within the C-terminal region [12]. Misfolding and aggregation of ND-related proteins (amyloid-β, phosphorylated tau, α-synuclein, and TDP-43) result in the formation of filamentous cellular inclusions. These abnormal amyloid fibrils are resistant to sarkosyl and found to be pathogenic hallmarks of Neurodegenerative diseases. A better observation of the molecular structures of these pathogenetic proteins strengthens our understanding of neuropathogenesis. Recently, the cryo-EM results revealed that TMEM106B protein belongs to amyloid proteins and has the potential to form amyloid fibrils. A new amyloid fibril composed of a C-terminal fragment of TMEM106B was found in many Neurodegenerative diseases and normal elderly [17,18,19,20]. Previous studies found that the TDP-43 protein is the pathological protein in FTLD-TDP disease, but recent studies indicate that the disease is associated with amyloid fibrils that are formed by TMEM106B [19]. This makes the TMEM106B protein a recent research hotspot.
TMEM106B modulates the physiological function of lysosomes
The lysosome is a degradative organelle that acts as the waste disposal system by clearing out debris and unnecessary proteins. A typical signature of TMEM106B is its post-translational glycosylation. TMEM106B contains five putative consensus sequence motifs for N-glycosylation and additional modifications that are correlated with the location of lysosome [1]. The effects of glycosylation at different sites of TMEM106B are diverse. Glycosylation is partially required for the transport of TMEM106B beyond the endoplasmic reticulum to late cellular compartments. Mutations of the noncomplex glycosylation sites (N145, N151 and N164) do not affect the localization of TMEM106B. However, glycosylation of residues N183 and N256 may regulate its lysosomal localization. Moreover, glycosylation at N183 appears to be required for the anterograde trafficking and guides the normal transport of TMEM106B to late endosomes/lysosomes, the deficiency of which leads to the accumulation of TMEM106B in the endoplasmic reticulum [1, 21, 22]. These data indicate that N183 glycosylation is responsible for the anterograde transport of TMEM106B to late endosomes/lysosomes. N256 glycosylation causes TMEM106B localization to the cell surface, suggesting a direct effect of N256 glycosylation on the sorting of TMEM106B into endosomes [1].
Ectopic expression of TMEM106B induces morphologic changes in lysosome compartments and delays the degradation of endocytic cargoes by the endolysosomal pathway [2]. TMEM106B deficiency reduces the number of lysosomes, and the remaining lysosomes cluster at the axon initial segment or perinuclear space with a vacuole-like morphology [22, 23]. Low-expression of TMEM106B results in a disruption of lysosome maturation, presented as less efficient fusion with autophagosome, poor protein degradation efficiency, and insufficient acidification [24]. Intriguingly, cells over-expressing TMEM106B result in the loss of vacuolar phenotype and exhibit impaired lysosomal acidification and degradative function [21]. Furthermore, TMEM106B overexpression enhances oxidative stress-induced cytotoxicity [11, 25, 26], induces lysosomal enlargement, and results in cell death [1, 2, 27]. Neuronal TMEM106B overexpression enlarges LAMP1-positive structures and alters lysosomal stress signaling, causing a translocation of transcription factor EB to neuronal nuclei and increasing the expression of Coordinated Lysosomal Expression and Regulation (CLEAR) genes [22]. Similarly, TMEM106B moves from cytosolic and lysosomal compartments into the nucleus to activate lysosomal genes [28]. Reduction of TMEM106B increases axonal transported lysosomes, while TMEM106B elevation inhibits neuronal lysosomal transport, redistributes from the cell periphery to the perinuclear region, and yields large lysosomes in the soma [22].
Overexpression of TMEM106B results in abnormalities in endosomal-lysosomal dysfunction, such as endosome-lysosome morphology, acidification, and trafficking [29]. Since lysosome morphology and size are intricately regulated by fission events and fusion with other organelles [30], and may be partially dependent on pH and successful trafficking. Disorders of either fission or fusion events lead to clustering lysosomes and the formation of large swollen vacuoles [13]. This indicates that TMEM106B may regulate many aspects of lysosomal function, including lysosomal pH, lysosome movement, and lysosome exocytosis [11]. Usually, lysosomes are transported along microtubules with the help of the motor proteins. TMEM106B was demonstrated to interact with microtubule-associated protein 6 (MAP6). This may inhibit the retrograde transport of lysosomes, facilitate appropriate transport of lysosomes, and prevent transport along microtubules through motor proteins [31]. By analyzing the interaction between TMEM106B and MAP6, researchers found that overexpression of TMEM106B affects bidirectional transport, in particular favoring retrograde transport [2]. Furthermore, the lysosome maintains its acidic environment by pumping in protons (H+ ions) from the cytosol via the vacuolar-ATPases (vATPase). TMEM106B overexpression interacts with accessory proteins of vATPase, resulting in a reduction in vATPase activity and the destruction of an acidic environment in lysosomes [11]. Interestingly, the inhibition of vATPases significantly increases the levels of TMEM106B [1]. A diagram is presented to illustrate the physiological and pathological roles of TMEM106B on lysosomes (Fig. 1).
Multiple single nucleotide polymorphisms (SNPs) of TMEM106B
Aberrant TMEM106B expression and deposition were detected in neurodegenerative diseases. GWAS showed that variants of the human TMEM106B gene are risk factors for FTLD-TDP, especially in patients with granulin (GRN) mutation [7]. Six SNPs of TMEM106B were found to modify the disease risk for several Neurodegenerative diseases and are associated with their clinical and pathological phenotypes [32,33,34,35,36]. Five out of these SNPs are located in the non-coding regions of TMEM106B and regulate its expression by influencing the alternative splicing of TMEM106B mRNA. Experimental evidence suggests that variants on TMEM106B haplotypes increase its expression, and are strongly correlated with pathological phenotypes and disease risk of FTLD-TDP. Genotyping data revealed that the risk allele of rs1990622, located in the 3'-untranslated region of TMEM106B, was a risk factor for FTLD-TDP, and carriers of rs1990622 risk allele increase TMEM106B protein levels in the hippocampus with physiological aging [37]. The T allele at this position is considered the major isoform, linked to higher risks of developing Neurodegenerative diseases or exacerbated cognitive decline. In contrast, the minor C allele is associated with a protective phenotype [9, 10, 34]. The levels of TMEM106B mRNA and protein were significantly increased in GRN mutation carriers [27, 38]. The major allele of rs1990622 in TMEM106B is associated with later age of onset and death of FTLD patients with C9orf72 mutation [32]. Still, this effect was not observed in FTLD-TDP patients without GRN or C9orf72 mutation, indicating different roles of TMEM106B in FTLD. Neuroimaging studies have shown reduced left hemispheric grey matter volume in those carrying the A allele of rs1990622 within a healthy population cohort [39], but it is still unclear whether it affects the volume of key brain areas in AD patients. Genotyping data revealed that the frequency of homozygous minor alleles was significantly reduced in GRN mutation carriers [40]. This result was confirmed in the C9orf72 expansion carriers [36, 41, 42]. In amyotrophic lateral sclerosis (ALS), the minor allele of rs1990622 shows more severe cognitive impairments, and poor motor functional status [43,44,45]. However, another study found the contrary results that ALS patients with the major allele of rs1990622 showed better cognition but worse motor functions than patients homozygous for the minor allele [46]. Research conclusions on the effect of TMEM106B SNPs on AD are inconsistent. Yang and colleagues conducted an expression quantitative trait loci (eQTLs) analysis of rs1990622 variant and found that rs1990622 variant T allele could increase TMEM106B expression [47]. However, Satoh et al. demonstrated both the mRNA and protein levels of TMEM106B were significantly reduced in AD brains compared to controls [48, 49]. Hu and colleagues analyzed the GWAS datasets and reported rs1990622 variant T allele only contributes to increased AD risk in females, but not in males. In addition, rs1990622 variant could regulate the expression of TMEM106B in human brain tissues, which vary considerably in different disease status [35]. Moreover, case–control association studies found that the risk allele of rs1990622 confers increased susceptibility to late-onset AD in the apolipoprotein E (APOE) ε4 allele carriers [50]. Furthermore, GWAS analyses identified a novel genome-wide significant association with genetic markers in DNA variants of TMEM106B and neurofilament light chain (NfL) protein concentrations in the cerebrospinal fluid of AD patients [51], but the relationship between TMEM106B and Aβ/tau proteins in cerebrospinal fluid (CSF) of AD patients remains to be explored. Intriguingly, recent two in vivo studies investigated the role of TMEM106B in tau P301S transgenic mouse model. Feng et al. show that loss of TMEM106B enhances the accumulation of pathological tau and results in severe neuronal loss, especially in the neuronal soma in the hippocampus [52]. The other found similar results that TMEM106B deletion accelerates cognitive decline and tau pathology in tau P301S mice. In contrast, the T186S (equivalent to the human T185S variant) knock-in mutation protected against tau-associated cognitive decline and synaptic impairment [53]. In PD, the rs1990622 risk allele was linked to a greater and faster cognitive deterioration [44]. The variant rs1990622 was also reported to correlate with reduced neuronal degeneration during aging, independently of disease [54].
Moreover, a noncoding variant, rs1990620, was shown to preferentially recruit the chromatin-organizing protein CCTC-binding factor (CTCF) to modulate TMEM106B expression through transcriptional activation due to CTCF-mediated long-range chromatin-looping interactions [25]. It was also shown that one coding variant rs3173615 encoding a threonine to serine change at amino acid position 185 (p.T185S) contributes to the disease-modifying effect [34, 41]. The change of T185 to serine was found to protect against FTLD-TDP [55], possibly because the protein with a serine is more rapidly degraded [56].TMEM106B carrying the risk isoform T185 leads to a higher level of glycosylation at N183, affecting the protein stability and degradation [10, 56]. Alternatively, the coding p.T185S variant might influence disease risk by altering either TMEM106B biology (cleavage, dimerization, etc.) or by affecting lysosomal dysfunction. The T185 variant seems to enhance the binding ability of S185 variant to the charged multivesicular body protein 2B (CHMP2B), which led to a decrease in autophagic flux [57]. Furthermore, overexpression of TMEM106B T185 delayed the degradation of the endogenous epidermal growth factor receptor, suggesting that TMEM106B may drive a defect in late endosome/lysosome fusion or lysosomal degradation [2]. A study revealed that AD risk is significantly influenced by the interaction of APOE with rs1595014 in TMEM106B [58]. The rs1990621, another SNP of TMEM106B, was identified as a protective variant against general aging, independent of disease status [54]. In chronic traumatic encephalopathy (CTE) patients, the homozygous carriers of the major allele of rs3173615 in TMEM106B appear to be more severe in tau pathology than those with the minor allele [59]. This finding suggests that TMEM106B variants modify tau pathology in CTE patients. However, another study reported a negative role of the genetic variations of rs3173615 in TMEM106B in CTE patients, compared to controls [60]. TMEM106B knockdown restores endolysosomal trafficking and branching defects [61]. Furthermore, a novel dominant D252N mutation in TMEM106B causing an amino acid substitution has been recently associated with several cases of hypomyelinating leukodystrophy (HLD) [62, 63]. The SNPs D252N was shown to abolish lysosome enlargement and lysosome acidification induced by wild-type TMEM106B overexpression in oligodendrocytes [15], suggesting that D252N mutation may impair lysosomal function by altering the autophagy process. In addition to chromatin structure alterations, the microRNA-132/miRNA-212 cluster binds to the 3′UTR of TMEM106B gene and inhibits its expression [27]. The microRNA is significantly decreased in FTLD-TDP, thus suggesting an upregulation of TMEM106B expression [64]. Table 1 summarizes the Cohort studies about TMEM106B SNPs and its potential phenotypes.
The cryo-EM structure of TMEM106B fibrils
In the past two years, several research groups have reported that TMEM106B forms amyloid fibrils in the brain tissue of different neurodegenerative diseases and elderly normal subjects through cryo-EM [17,18,19,20]. These observations indicate that amyloid fibrils formed by TMEM106B may play a role in the pathogenesis of neurodegenerative diseases. Interestingly, the methods used for the identification of TMEM106B vary among different groups. Chang et al. supplemented cryo-EM with mass spectrometry to identify TMEM106B peptides present in Sarkosyl-insoluble components of FTLD-TDP, progressive supranuclear palsy, and dementia with Lewy bodies [17]. Jiang et al. modeled two query sequences and determined their structures based on cryo-EM density in FTLD-TDP subclasses [19]. Fan et al. found TMEM106B forms amyloid fibrils not only in diseased brains, but also in the brains of normal elders by cryo-EM [18]. Significantly, the burden of TMEM106B fibrillization is much higher in most individuals with Neurodegenerative diseases, when compared with age-matched healthy controls. These data indicate that the TMEM106B fibrils may not be a by-stand in neurodegeneration, but might exert some toxic effects, and contribute to age-dependent neurodegeneration. Schweighauser and colleagues detected TMEM106B filaments based on the unique glycosylation pattern of fibrils [20]. However, all reports observed that amyloid fibrils have ordered cores containing TMEM106B residues S120-G254. These studies suggest that the cleavage between residues 119 and 120 is essential for fibril formation, because the cleaved terminus is buried in the fibril core, leaving no space for other residues. All papers showed that the TMEM106B fibrils stack into protofilaments-single protofilaments form rod-like structures, and pairs of protofilaments form twisted ribbons. The interface in the more prevalent paired fibrils was mediated by residues K178 and R180 of each protofilament interacting with an unresolved density, potentially an anionic cofactor.
Although the TMEM106B fibrils are extracted from the insoluble portion of sarcosine in postmortem tissue, there are some differences in fractionation schemes, including the sarcosine addition stage, the use of ultracentrifugation or low-speed centrifugation, and changes in heating or streptomycin treatment. Jiang et al. extracted and determined the amyloid fibrils from brains of FTLD-TDP patients by cryo-EM [19]. Unexpectedly, all amyloid fibrils were composed of a 135-residue carboxy-terminal fragment of TMEM106B, but not TDP-43 filaments. Jiang et al. used immunogold labeling and identified abundant non-fibrillar aggregated TDP-43 in FTLD-TDP patients [19]. Considering the discrepancy in extraction methods, TDP-43 filaments may be lost during the extraction process and were not obtained in the analyzed process [75]. This emphasizes the importance of sample preparation schemes in cryo-EM research and indicates the need for more samples aiming at analyzing TDP-43 aggregates. Also, different methods should be conducted to verify the amyloid protein properties of TMEM106B in various Neurodegenerative diseases.
Future direction on TMEM106B in neurodegenerative diseases
Several studies identified that TMEM106B may play a role in the pathogenesis of neurodegenerative diseases. However, the exact molecular mechanisms remain unknown. Whether the presence of these fibrils correspond solely to age regardless of whether or not a person has neurodegenerative conditions, or is specifically related to a specific disease state, remains unknown. None of the current studies involves enough autopsy samples to have a statistical power to associate TMEM106B fibrils with FTLD-TDP. TMEM106B fibrils are deposited in many brain regions of patients of various Neurodegenerative diseases, but the mechanism by which TMEM106B fibrils contributes to neurodegenerative pathology is currently unclear. Further research is needed to determine whether there is regional or disease-specific susceptibility for TMEM106B aggregation, and how these aggregates evolve and spread with disease progression. It is possible that increased/reduced TMEM106B expression might affect lysosomal trafficking and neuronal development [76]. However, it remains unclear whether the deposition of TMEM106B fibrils directly leads to neurodegeneration, or does it promote the aggregation and deposition of other pathological proteins, such as Aβ, tau, and α-syn. TMEM106B fibrils are also deposited in neurologically normal older adults, so what is the role of TMEM106B in the elderly, an initiator or a bystander? These issues need to be discussed.
Given that TMEM106B plays an important role in the pathological progression of Neurodegenerative diseases, TMEM106B may be used as a therapeutic target. Finding factors that inhibit the initial aggregation of TMEM106B fibrils will be the focus of future research. The C-terminal fragment of TMEM106B form fibrils. Thus, reducing the generation of the C-terminus of TMEM106B would be an effective way to block the fibrilization process. SPPL2a is a key protease in cleaving TMEM106B, and its antagonist could be a target to block the production of C-terminal fragment [16]. Post-translational modifications could mediate the structural diversity of fibrils by influencing their inter-protofilament interfaces78. Glycosylation is partially required for the transport of TMEM106B beyond the endoplasmic reticulum to late cellular compartments. Complex glycosylation at the N183 site appears to affect anterograde trafficking, whereas N256 site glycosylation appears to influence the direct sorting of TMEM106B to endosomes [1]. Therefore, to regulate the post-translational glycosylation modification of TMEM106B may be a potential therapeutic approach. Third, looking for conformation-specific antibodies and potential small molecules to reduce the formation of TMEM106B β-sheet structure, which is the initial step in inducing amyloid aggregation, may prevent TMEM106B fibrillation. Lysosomes are responsible for the degradation of TMEM106B fibrils and lysosomal dysfunction may alter fibrillation by affecting pH and protease activity. Also, lysosomal dysfunction may result in an increase in intraluminal and lysosomal exocytosis of TMEM106B fibrils due to impaired degradation. Thus, promoting the degradation of TMEM106B fibrils through the lysosome pathway would be promising.
Conclusions
The deposition of TMEM106B fibrils identified by Cryo-EM will undoubtedly highlight the role of TMEM106B in neurodegenerative diseases, but there are many questions to be answered. First, TMEM106B fibrils are present in older adults without neurodegenerative disease, and their pathogenicity needs to be further determined. Second, as TMEM106B fibrils have been found in various Neurodegenerative diseases, it needs to be clarified whether TMEM106B fibrils have synergistic or antagonistic effects on promoting aggregation of pathological proteins in Neurodegenerative diseases. Third, TMEM106B risk haplotype may affect fiber load by modulating TMEM106B expression or processing, or directly by affecting glycosylation at the N183 site. Detailed studies of haplotype contributions are needed. Finally, it would be necessary to elucidate the mechanism of TMEM106B C-terminus generation, the precise steps, the proteases, and cellular conditions required for TMEM106B fibrillization, and to determine the precise cell location of TMEM106B fibrils. Further work will be required to explain these ambiguities and determine whether the fibrils are harmful, irrelevant, or even protective against disease.
Data availability
Not applicable.
References
Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, Capell A, Haass C (2012) Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem 287:19355–19365. https://doi.org/10.1074/jbc.M112.365098
Brady OA, Zheng Y, Murphy K, Huang M, Hu F (2013) The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet 22:685–695. https://doi.org/10.1093/hmg/dds475
Feng T, Mai S, Roscoe JM, Sheng RR, Ullah M, Zhang J, Katz II, Yu H, Xiong W, Hu F (2020) Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep 21:50219
Schwenk BM, Lang CM, Hogl S, Tahirovic S, Orozco D, Rentzsch K, Lichtenthaler SF, Hoogenraad CC, Capell A, Haass C, Edbauer D (2014) The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. Embo j 33:450–467. https://doi.org/10.1002/embj.201385857
Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT, Strittmatter SM (2017) Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron 95:281-296.e286. https://doi.org/10.1016/j.neuron.2017.06.026
Satoh J, Kino Y, Kawana N, Yamamoto Y, Ishida T, Saito Y, Arima K (2014) TMEM106B expression is reduced in Alzheimer’s disease brains. Alzheimers Res Ther 6:17. https://doi.org/10.1186/alzrt247
Van Deerlin VM, Sleiman PMA, Martinez-Lage M, Chen-Plotkin A, Wang L-S, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42:234–239. https://doi.org/10.1038/ng.536
Pottier C, Ren Y, Perkerson RB 3rd, Baker M, Jenkins GD, van Blitterswijk M, DeJesus-Hernandez M, van Rooij JGJ, Murray ME, Christopher E et al (2019) Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD. Acta Neuropathol 137:879–899. https://doi.org/10.1007/s00401-019-01962-9
van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, Santens P, Van den Broeck M, Joris G, Brys J et al (2011) TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 134:808–815. https://doi.org/10.1093/brain/awr007
Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L et al (2011) TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76:467–474. https://doi.org/10.1212/WNL.0b013e31820a0e3b
Feng T, Lacrampe A, Hu F (2021) Physiological and pathological functions of TMEM106B: a gene associated with brain aging and multiple brain disorders. Acta Neuropathol 141:327–339. https://doi.org/10.1007/s00401-020-02246-3
Perneel J, Rademakers R (2022) Identification of TMEM106B amyloid fibrils provides an updated view of TMEM106B biology in health and disease. Acta Neuropathol 144:807–819. https://doi.org/10.1007/s00401-022-02486-5
Nicholson AM, Rademakers R (2016) What we know about TMEM106B in neurodegeneration. Acta Neuropathol 132:639–651. https://doi.org/10.1007/s00401-016-1610-9
Perneel J, Neumann M, Heeman B, Cheung S, Van den Broeck M, Wynants S, Baker M, Vicente CT, Faura J, Rademakers R, Mackenzie IRA (2022) Accumulation of TMEM106B C-terminal fragments in neurodegenerative disease and aging. Acta Neuropathol 145:285–302. https://doi.org/10.1007/s00401-022-02531-3
Feng T, Sheng RR, Solé-Domènech S, Ullah M, Zhou X, Mendoza CS, Enriquez LCM, Katz II, Paushter DH, Sullivan PM et al (2020) A role of the frontotemporal lobar degeneration risk factor TMEM106B in myelination. Brain 143:2255–2271. https://doi.org/10.1093/brain/awaa154
Brady OA, Zhou X, Hu F (2014) Regulated intramembrane proteolysis of the frontotemporal lobar degeneration risk factor, TMEM106B, by signal peptide peptidase-like 2a (SPPL2a). J Biol Chem 289:19670–19680. https://doi.org/10.1074/jbc.M113.515700
Chang A, Xiang X, Wang J, Lee C, Arakhamia T, Simjanoska M, Wang C, Carlomagno Y, Zhang G, Dhingra S et al (2022) Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 185:1346-1355.e1315. https://doi.org/10.1016/j.cell.2022.02.026
Fan Y, Zhao Q, Xia W, Tao Y, Yu W, Chen M, Liu Y, Zhao J, Shen Y, Sun Y et al (2022) Generic amyloid fibrillation of TMEM106B in patient with Parkinson’s disease dementia and normal elders. Cell Res 32:585–588. https://doi.org/10.1038/s41422-022-00665-3
Jiang YX, Cao Q, Sawaya MR, Abskharon R, Ge P, DeTure M, Dickson DW, Fu JY, Ogorzalek Loo RR, Loo JA, Eisenberg DS (2022) Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 605:304–309. https://doi.org/10.1038/s41586-022-04670-9
Schweighauser M, Arseni D, Bacioglu M, Huang M, Lövestam S, Shi Y, Yang Y, Zhang W, Kotecha A, Garringer HJ et al (2022) Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature 605:310–314. https://doi.org/10.1038/s41586-022-04650-z
Busch JI, Unger TL, Jain N, Tyler Skrinak R, Charan RA, Chen-Plotkin AS (2016) Increased expression of the frontotemporal dementia risk factor TMEM106B causes C9orf72-dependent alterations in lysosomes. Hum Mol Genet 25:2681–2697. https://doi.org/10.1093/hmg/ddw127
Stagi M, Klein ZA, Gould TJ, Bewersdorf J, Strittmatter SM (2014) Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol Cell Neurosci 61:226–240. https://doi.org/10.1016/j.mcn.2014.07.006
Stroobants S, D’Hooge R, Damme M (2021) Aged Tmem106b knockout mice display gait deficits in coincidence with Purkinje cell loss and only limited signs of non-motor dysfunction. Brain Pathol 31:223–238. https://doi.org/10.1111/bpa.12903
Klein ZA, Takahashi H, Ma M, Stagi M, Zhou M, Lam TT, Strittmatter SM (2017) Loss of TMEM106B Ameliorates Lysosomal and Frontotemporal Dementia-Related Phenotypes in Progranulin-Deficient Mice. Neuron. https://doi.org/10.1016/j.neuron.2017.06.026
Gallagher MD, Posavi M, Huang P, Unger TL, Berlyand Y, Gruenewald AL, Chesi A, Manduchi E, Wells AD, Grant SFA et al (2017) A Dementia-Associated Risk Variant near TMEM106B Alters Chromatin Architecture and Gene Expression. Am J Hum Genet 101:643–663. https://doi.org/10.1016/j.ajhg.2017.09.004
Suzuki H, Matsuoka M (2016) The Lysosomal Trafficking Transmembrane Protein 106B Is Linked to Cell Death. J Biol Chem 291:21448–21460. https://doi.org/10.1074/jbc.M116.737171
Chen-Plotkin AS, Unger TL, Gallagher MD, Bill E, Kwong LK, Volpicelli-Daley L, Busch JI, Akle S, Grossman M, Van Deerlin V et al (2012) TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci 32:11213–11227. https://doi.org/10.1523/jneurosci.0521-12.2012
Perez-Canamas A, Takahashi H, Lindborg JA, Strittmatter SM (2021) Fronto-temporal dementia risk gene TMEM106B has opposing effects in different lysosomal storage disorders. Brain Commun. https://doi.org/10.1093/braincomms/fcaa200
de Araujo MEG, Liebscher G, Hess MW, Huber LA (2020) Lysosomal size matters. Traffic 21:60–75. https://doi.org/10.1111/tra.12714
Root J, Merino P, Nuckols A, Johnson M, Kukar T (2021) Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis. https://doi.org/10.1016/j.nbd.2021.105360
Gallagher MD, Suh E, Grossman M, Elman L, McCluskey L, Van Swieten JC, Al-Sarraj S, Neumann M, Gelpi E, Ghetti B et al (2014) TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol 127:407–418. https://doi.org/10.1007/s00401-013-1239-x
Harding SR, Bocchetta M, Gordon E, Cash DM, Cardoso MJ, Druyeh R, Ourselin S, Warren JD, Mead S, Rohrer JD (2017) The TMEM106B risk allele is associated with lower cortical volumes in a clinically diagnosed frontotemporal dementia cohort. J Neurol Neurosurg Psychiatry 88:997–998. https://doi.org/10.1136/jnnp-2017-315641
Pottier C, Zhou X, Perkerson RB 3rd, Baker M, Jenkins GD, Serie DJ, Ghidoni R, Benussi L, Binetti G, Lopez de Munain A et al (2018) Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: a genome-wide association study. Lancet Neurol 17:548–558. https://doi.org/10.1016/S1474-4422(18)30126-1
Hu Y, Sun JY, Zhang Y, Zhang H, Gao S, Wang T, Han Z, Wang L, Sun BL, Liu G (2021) rs1990622 variant associates with Alzheimer’s disease and regulates TMEM106B expression in human brain tissues. BMC Med 19:11. https://doi.org/10.1186/s12916-020-01883-5
Mao F, Robinson JL, Unger T, Posavi M, Amado DA, Elman L, Grossman M, Wolk DA, Lee EB, Van Deerlin VM et al (2021) TMEM106B modifies TDP-43 pathology in human ALS brain and cell-based models of TDP-43 proteinopathy. Acta Neuropathol 142:629–642. https://doi.org/10.1007/s00401-021-02330-2
Katsumata Y, Nelson PT, Ellingson SR, Fardo DW (2017) Gene-based association study of genes linked to hippocampal sclerosis of aging neuropathology: GRN, TMEM106B, ABCC9, and KCNMB2. Neurobiol Aging 53:193.e117-193.e125. https://doi.org/10.1016/j.neurobiolaging.2017.01.003
Rothhammer V, Muschaweckh A, Gasteiger G, Petermann F, Heink S, Busch DH, Heikenwälder M, Hemmer B, Drexler I, Korn T (2014) α4-integrins control viral meningoencephalitis through differential recruitment of T helper cell subsets. Acta Neuropathol Commun 2:27. https://doi.org/10.1186/2051-5960-2-27
Adams HH, Verhaaren BF, Vrooman HA, Uitterlinden AG, Hofman A, van Duijn CM, van der Lugt A, Niessen WJ, Vernooij MW, Ikram MA (2014) TMEM106B influences volume of left-sided temporal lobe and interhemispheric structures in the general population. Biol Psychiatry 76:503–508. https://doi.org/10.1016/j.biopsych.2014.03.006
Premi E, Formenti A, Gazzina S, Archetti S, Gasparotti R, Padovani A, Borroni B (2014) Effect of TMEM106B polymorphism on functional network connectivity in asymptomatic GRN mutation carriers. JAMA Neurol 71:216–221. https://doi.org/10.1001/jamaneurol.2013.4835
van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Murray ME et al (2014) TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol 127:397–406. https://doi.org/10.1007/s00401-013-1240-4
Deming Y, Cruchaga C (2014) TMEM106B: a strong FTLD disease modifier. Acta Neuropathol 127:419–422. https://doi.org/10.1007/s00401-014-1249-3
Rollinson S, Mead S, Snowden J, Richardson A, Rohrer J, Halliwell N, Usher S, Neary D, Mann D, Hardy J, Pickering-Brown S (2011) Frontotemporal lobar degeneration genome wide association study replication confirms a risk locus shared with amyotrophic lateral sclerosis. Neurobiol Aging 32(758):e751-757. https://doi.org/10.1016/j.neurobiolaging.2010.12.005
Tropea TF, Mak J, Guo MH, Xie SX, Suh E, Rick J, Siderowf A, Weintraub D, Grossman M, Irwin D et al (2019) TMEM106B Effect on cognition in Parkinson disease and frontotemporal dementia. Ann Neurol 85:801–811. https://doi.org/10.1002/ana.25486
Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, Elman L, McCluskey L, Lee VM, Van Deerlin VM et al (2011) Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol 121:373–380. https://doi.org/10.1007/s00401-010-0782-y
Manini A, Ratti A, Brusati A, Maranzano A, Fogh I, Peverelli S, Messina S, Gentilini D, Verde F, Poletti B et al (2022) TMEM106B Acts as a Modifier of Cognitive and Motor Functions in Amyotrophic Lateral Sclerosis. Int J Mol Sci. https://doi.org/10.3390/ijms23169276
Yang HS, White CC, Klein HU, Yu L, Gaiteri C, Ma Y, Felsky D, Mostafavi S, Petyuk VA, Sperling RA et al (2020) Genetics of Gene Expression in the Aging Human Brain Reveal TDP-43 Proteinopathy Pathophysiology. Neuron. https://doi.org/10.1016/j.neuron.2020.05.010
Chang A, Xiang X, Wang J, Lee C, Arakhamia T, Simjanoska M, Wang C, Carlomagno Y, Zhang G, Dhingra S et al (2022) Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 185(1346–1355):e1315. https://doi.org/10.1016/j.cell.2022.02.026
Ren Y, van Blitterswijk M, Allen M, Carrasquillo MM, Reddy JS, Wang X, Beach TG, Dickson DW, Ertekin-Taner N, Asmann YW, Rademakers R (2018) TMEM106B haplotypes have distinct gene expression patterns in aged brain. Mol Neurodegener 13:35. https://doi.org/10.1186/s13024-018-0268-2
Lu RC, Wang H, Tan MS, Yu JT, Tan L (2014) TMEM106B and APOE polymorphisms interact to confer risk for late-onset Alzheimer’s disease in Han Chinese. J Neural Transm (Vienna) 121:283–287. https://doi.org/10.1007/s00702-013-1106-x
Hong S, Dobricic V, Ohlei O, Bos I, Vos SJB, Prokopenko D, Tijms BM, Andreasson U, Blennow K, Vandenberghe R et al (2021) TMEM106B and CPOX are genetic determinants of cerebrospinal fluid Alzheimer’s disease biomarker levels. Alzheimers Dement 17:1628–1640. https://doi.org/10.1002/alz.12330
Feng T, Du H, Yang C, Wang Y, Hu F (2024) Loss of TMEM106B exacerbates Tau pathology and neurodegeneration in PS19 mice. Acta Neuropathol 147:62. https://doi.org/10.1007/s00401-024-02702-4
Edwards GA 3rd, Wood CA, He Y, Nguyen Q, Kim PJ, Gomez-Gutierrez R, Park KW, Xu Y, Zurhellen C, Al-Ramahi I, Jankowsky JL (2024) TMEM106B coding variant is protective and deletion detrimental in a mouse model of tauopathy. Acta Neuropathol 147:61. https://doi.org/10.1007/s00401-024-02701-5
Li Z, Farias FHG, Dube U, Del-Aguila JL, Mihindukulasuriya KA, Fernandez MV, Ibanez L, Budde JP, Wang F, Lake AM et al (2020) The TMEM106B FTLD-protective variant, rs1990621, is also associated with increased neuronal proportion. Acta Neuropathol 139:45–61. https://doi.org/10.1007/s00401-019-02066-0
Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, Bertelsen S, Mayo K, Norton JB, Morris JC, Goate A (2011) Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 68:581–586. https://doi.org/10.1001/archneurol.2010.350
Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB 3rd, Castanedes-Casey M, Rousseau L, Benussi L, Binetti G, Ghidoni R et al (2013) TMEM106B p. T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem 126:781–791. https://doi.org/10.1111/jnc.12329
Jun MH, Han JH, Lee YK, Jang DJ, Kaang BK, Lee JA (2015) TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD-3-linked CHMP2B, a complex of ESCRT-III. Mol Brain 8:85. https://doi.org/10.1186/s13041-015-0177-z
Jun G, Ibrahim-Verbaas CA, Vronskaya M, Lambert JC, Chung J, Naj AC, Kunkle BW, Wang LS, Bis JC, Bellenguez C et al (2016) A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 21:108–117. https://doi.org/10.1038/mp.2015.23
Bieniek KF, Ross OA, Cormier KA, Walton RL, Soto-Ortolaza A, Johnston AE, DeSaro P, Boylan KB, Graff-Radford NR, Wszolek ZK et al (2015) Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol 130:877–889. https://doi.org/10.1007/s00401-015-1502-4
Cherry JD, Mez J, Crary JF, Tripodis Y, Alvarez VE, Mahar I, Huber BR, Alosco ML, Nicks R, Abdolmohammadi B et al (2018) Variation in TMEM106B in chronic traumatic encephalopathy. Acta Neuropathol Commun 6:115. https://doi.org/10.1186/s40478-018-0619-9
Clayton EL, Milioto C, Muralidharan B, Norona FE, Edgar JR, Soriano A, Jafar-Nejad P, Rigo F, Collinge J, Isaacs AM (2018) Frontotemporal dementia causative CHMP2B impairs neuronal endolysosomal traffic-rescue by TMEM106B knockdown. Brain 141:3428–3442. https://doi.org/10.1093/brain/awy284
Ikemoto S, Hamano SI, Kikuchi K, Koichihara R, Hirata Y, Matsuura R, Hiraide T, Nakashima M, Inoue K, Kurosawa K, Saitsu H (2020) A recurrent TMEM106B mutation in hypomyelinating leukodystrophy: A rapid diagnostic assay. Brain Dev 42:603–606. https://doi.org/10.1016/j.braindev.2020.06.002
Simons C, Dyment D, Bent SJ, Crawford J, D’Hooghe M, Kohlschutter A, Venkateswaran S, Helman G, Poll-The BT, Makowski CC et al (2017) A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain 140:3105–3111. https://doi.org/10.1093/brain/awx314
Piscopo P, Albani D, Castellano AE, Forloni G, Confaloni A (2016) Frontotemporal Lobar Degeneration and MicroRNAs. Front Aging Neurosci 8:17. https://doi.org/10.3389/fnagi.2016.00017
Lattante S, Le Ber I, Galimberti D, Serpente M, Rivaud-Pechoux S, Camuzat A, Clot F, Fenoglio C, French research network on, F.T.D., Ftd, A.L.S. et al (2014) Defining the association of TMEM106B variants among frontotemporal lobar degeneration patients with GRN mutations and C9orf72 repeat expansions. Neurobiol Aging. https://doi.org/10.1016/j.neurobiolaging.2014.06.023
Rutherford NJ, Carrasquillo MM, Li M, Bisceglio G, Menke J, Josephs KA, Parisi JE, Petersen RC, Graff-Radford NR, Younkin SG et al (2012) TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology 79:717–718. https://doi.org/10.1212/WNL.0b013e318264e3ac
Milind N, Preuss C, Haber A, Ananda G, Mukherjee S, John C, Shapley S, Logsdon BA, Crane PK, Carter GW (2020) Transcriptomic stratification of late-onset Alzheimer’s cases reveals novel genetic modifiers of disease pathology. PLoS Genet. https://doi.org/10.1371/journal.pgen.1008775
Murray ME, Cannon A, Graff-Radford NR, Liesinger AM, Rutherford NJ, Ross OA, Duara R, Carrasquillo MM, Rademakers R, Dickson DW (2014) Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol 128:411–421. https://doi.org/10.1007/s00401-014-1302-2
Yu L, De Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA (2015) The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 84:927–934. https://doi.org/10.1212/WNL.0000000000001313
Hu T, Chen Y, Ou R, Wei Q, Cao B, Zhao B, Wu Y, Song W, Chen X, Shang HF (2017) Association analysis of polymorphisms in VMAT2 and TMEM106B genes for Parkinson’s disease, amyotrophic lateral sclerosis and multiple system atrophy. J Neurol Sci 377:65–71. https://doi.org/10.1016/j.jns.2017.03.028
Nelson PT, Wang WX, Partch AB, Monsell SE, Valladares O, Ellingson SR, Wilfred BR, Naj AC, Wang LS, Kukull WA, Fardo DW (2015) Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J Neuropathol Exp Neurol 74:75–84. https://doi.org/10.1097/NEN.0000000000000151
Hokkanen SRK, Kero M, Kaivola K, Hunter S, Keage HAD, Kiviharju A, Raunio A, Tienari PJ, Paetau A, Matthews FE et al (2020) Putative risk alleles for LATE-NC with hippocampal sclerosis in population-representative autopsy cohorts. Brain Pathol 30:364–372. https://doi.org/10.1111/bpa.12773
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C et al (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142:1503–1527. https://doi.org/10.1093/brain/awz099
Lee JY, Harney DJ, Teo JD, Kwok JB, Sutherland GT, Larance M, Don AS (2023) The major TMEM106B dementia risk allele affects TMEM106B protein levels, fibril formation, and myelin lipid homeostasis in the ageing human hippocampus. Mol Neurodegener 18:63. https://doi.org/10.1186/s13024-023-00650-3
Takahashi H, Strittmatter SM (2022) An unexpected protein aggregate in diseased and ageing brains. Nature 605:227–228. https://doi.org/10.1038/d41586-022-00873-2
Jiao HS, Yuan P, Yu JT (2023) TMEM106B aggregation in neurodegenerative diseases: linking genetics to function. Mol Neurodegener 18:54. https://doi.org/10.1186/s13024-023-00644-1
Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, Williams D, DeTure M, Dickson DW, Cook CN et al (2020) Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 180:633-644.e612. https://doi.org/10.1016/j.cell.2020.01.027
Funding
This work was supported by grants from the National Key Research and Development Program of China (2019YFE0115900), the Innovative Research Groups of Hubei Province (2022CFA026), the New 20 Terms of Universities in Jinan (No. 202228022), and the Project for Academic and Technical Leaders of Major Disciplines in Jiangxi Province (20213BCJL22049).
Author information
Authors and Affiliations
Contributions
Min Zhu and Guoxin Zhang wrote the original draft. Lanxia Meng and Tingting Xiao helped in literature search. Xin Fang and Zhentao Zhang conceived the project. All authors contributed to the article and approved the submission.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors have consented to the publication of this review.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, M., Zhang, G., Meng, L. et al. Physiological and pathological functions of TMEM106B in neurodegenerative diseases. Cell. Mol. Life Sci. 81, 209 (2024). https://doi.org/10.1007/s00018-024-05241-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-024-05241-z