
Abstract
The genetic basis of chronic traumatic encephalopathy (CTE) is poorly understood. Variation in transmembrane protein 106B (TMEM106B) has been associated with enhanced neuroinflammation during aging and with TDP-43-related neurodegenerative disease, and rs3173615, a missense coding SNP in TMEM106B, has been implicated as a functional variant in these processes. Neuroinflammation and TDP-43 pathology are prominent features in CTE. The purpose of this study was to determine whether genetic variation in TMEM106B is associated with CTE risk, pathological features, and ante-mortem dementia. Eighty-six deceased male athletes with a history of participation in American football, informant-reported Caucasian, and a positive postmortem diagnosis of CTE without comorbid neurodegenerative disease were genotyped for rs3173615. The minor allele frequency (MAF = 0.42) in participants with CTE did not differ from previously reported neurologically normal controls (MAF = 0.43). However, in a case-only analysis among CTE cases, the minor allele was associated with reduced phosphorylated tau (ptau) pathology in the dorsolateral frontal cortex (DLFC) (AT8 density, odds ratio [OR] of increasing one quartile = 0.42, 95% confidence interval [CI] 0.22–0.79, p = 0.008), reduced neuroinflammation in the DLFC (CD68 density, OR of increasing one quartile = 0.53, 95% CI 0.29–0.98, p = 0.043), and increased synaptic protein density (β = 0.306, 95% CI 0.065–0.546, p = 0.014). Among CTE cases, TMEM106B minor allele was also associated with reduced ante-mortem dementia (OR = 0.40, 95% CI 0.16–0.99, p = 0.048), but was not associated with TDP-43 pathology. All case-only models were adjusted for age at death and duration of football play. Taken together, variation in TMEM106B may have a protective effect on CTE-related outcomes.
Similar content being viewed by others

Introduction
Chronic traumatic encephalopathy (CTE) is a progressive neurodegenerative disease that has been neuropathologically diagnosed in individuals with a history of repetitive head impacts (RHI) [22], including contact and collision sport athletes who participated in American football, ice hockey, rugby, mixed martial arts, soccer, and boxing [20]. Currently, CTE can only be diagnosed post-mortem. In a recent report describing a convenience sample of 202 former American football players, 99% of former National Football League (NFL) players were neuropathologically diagnosed with CTE at autopsy. Although the frequency of CTE in individuals with less football exposure was substantial, it was nonetheless lower (highest level of play - college: 91%; highest level of play – high school: 21%) [24]. Further, among those with CTE, former college and professional players had both mild and severe CTE pathology. It is unclear why among players with comparable RHI exposure, only some develop CTE or why disease severity varies. This variation may be due to individual differences in genetic, demographic, athletic or comorbid pathologic factors [1, 17, 39]. Previous small studies have identified suggestive relationships between CTE and candidate genetic factors such as apolipoprotein E4 (APOE ϵ4), microtubule associated protein tau (MAPT), and transmembrane protein 106b (TMEM106B) [3]. However, these relationships were not statistically significant and/or have not been replicated [21, 38].
Several converging lines of evidence suggest that TMEM106B may be involved in the development of CTE. In CTE, previous studies have implicated neuroinflammation as a potential disease mechanism, with chronic microglial activation triggering a positive feedback loop with the hyperphosphorylation and aggregation of tau [10]. TMEM106B may have a protective role with respect to neuroinflammation. A recent genome wide associate study (GWAS) of age-associated transcription changes implicated TMEM106B and found that the effect size was amplified when limited to a microglia-associated gene expression cluster [34]. The top-ranked single nucleotide polymorphism (SNP), rs1990622, was associated with a transcription pattern in the frontal cortex linked to younger chronological age. Among participants greater than age 65, the rs1990622 minor allele was associated with improved cognitive status. In human monocyte-derived dendritic cells, rs1990622 was associated with a reduced inflammatory state. Additionally, variation in TMEM106B is associated with TDP-43 pathology [43], a feature found in many cases of CTE [21], as well as several other neurodegenerative diseases. In a GWAS of frontotemporal lobar degeneration (FTLD)-TDP, rs1990622 was the top SNP and achieved genome wide significance [43]. Rs1990622 also has been associated with cognitive impairment in amyotrophic lateral sclerosis (ALS) [44], hippocampal sclerosis in normal aging [28], and incidence of Alzheimer’s disease (AD) in APOE ϵ4 negative individuals [14, 36].
Here, we assessed rs3173615, the only coding SNP in high linkage disequilibrium (LD) with rs1990622, for association with CTE-related outcomes. Rs3173615 encodes a change from threonine to serine at position 185 (p.T185S), may regulate TMEM106B protein levels, and is a suggested functional variant underlying the association with FTLD-TDP [30]. We hypothesized that the minor allele of rs3173615 is associated with CTE risk and, among those with CTE, with reduced tau aggregation, TDP-43 burden, neuroinflammation, synaptic loss and ante-mortem dementia.
Materials and methods
Participant selection
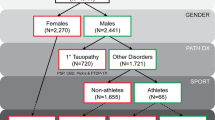
All CTE cases were ascertained from the Veterans Affairs-Boston University-Concussion Legacy Foundation (VA-BU-CLF) Brain Bank. Details on inclusion criteria have been described previously [26]. To be eligible for the brain bank, participants needed to have exposure to RHI, either from contact sports, military service or domestic violence. Clinical symptoms were not considered in the inclusion criteria. Participants of the current study were restricted to those who played American football, who were reported to be Caucasian, and who were neuropathologically diagnosed with CTE without significant co-morbid neurodegenerative disease. Because TMEM106B has been implicated in several other neurodegenerations many of which can occur comorbid with CTE [39, 46], we excluded participants with other significant co-morbid neurodegenerative disease. A total of 86 of 261 brain bank participants met these criteria (Fig. 1). An authorized legal representative provided written consent for participation and brain donation. IRB approval for the brain donation program was obtained through the Boston University Alzheimer’s Disease & CTE Center and the Edith Nourse Rogers Memorial Veterans Hospital. Neurologically normal controls were ascertained from a previous study of frontotemporal dementia [42]. These 376 participants had undergone genotyping at rs3173615 and were found to have no evidence of cognitive impairment or motor neuron disease on clinical assessment.

Inclusion flow chart for CTE Cases. Red curved arrows indicate participants that were excluded. Tissue was not available for genotyping for the following reasons: 1) consults with tissue returned to consulting neuropathologist; 2) only small fragments received; 3) tissue was significantly degraded and did not pass quality control for genotyping
Neuropathological assessment
VA-BU-CLF Brain Bank methods for pathologic processing of tissue and neuropathologic evaluation have been detailed elsewhere [45]. Briefly, the following stains were used: luxol fast blue, hematoxylin and eosin, Bielschowsky’s silver, hyperphosphorylated tau (p-tau) (AT8), alpha synuclein, amyloid beta (Aβ), and phosphorylated TDP-43 using previously published methods [18, 19, 22]. TDP-43 pathology was assessed in paraffin sections from the dorsolateral frontal cortex (DLFC), hippocampus, amygdala, entorhinal cortex, and midbrain. The neuropathological diagnosis of CTE was made using the National Institute of Neurological Disorders and Stroke (NINDS)/ National Institute of Biomedical Imaging and Bioengineering (NIBIB) consensus criteria based on the presence of abnormal perivascular accumulations of p-tau in neurons, astrocytes, and cell processes in an irregular and patchy distribution concentrated at the depths of cortical sulci [18]. Other neurodegenerative diseases were diagnosed using well-established criteria for AD, dementia with Lewy bodies (DLB), FTLD, and motor neuron disease (MND) [4, 6, 7, 12, 15, 16, 23, 27, 29]. Neuropathologists were blinded to all clinical data at the time of diagnosis.
Clinical assessment
VA-BU-CLF Brain Bank clinical assessment was conducted as previously described utilizing telephone clinical interviews with informants, online questionnaires, and medical record review [26, 38, 40]. Briefly, each of the following was assessed: type and amount (in years) of contact sport play, military service, whether mood, behavioral or cognitive symptoms were present and progressed over time, presence of functional impairment, and cause of death. Clinical history was presented at a multidisciplinary clinical consensus conference where it was determined whether the participant met criteria for dementia [26]. All interviews were conducted blinded to the results of the neuropathological examination.
Genotyping
For CTE cases, genomic DNA was extracted from cerebellum using the Qiagen DNeasy Blood & Tissue Kit. Genotyping of rs3173615 was performed using iPLEX Assay and MassARRAY System as per manufactures protocols. Controls were also genotyped using an iPLEX Assay and MassARRAY System, but not as part of the same batch.
Enzyme-linked immunosorbent assay (ELISA)
Flash frozen brain tissue was obtained from 37 of the 86 CTE cases as previously described [9]. Frozen tissue was collected from identical regions in Broadman area 8/9. Briefly, freshly prepared, ice cold 5 M Guanidine Hydrochloride in Tris-buffered saline (20 mM Tris-HCl, 150 mM NaCl, pH 7.4 TBS) containing 1:100 Halt protease inhibitor cocktail (Thermo Scientific) and 1:100 Phosphatase inhibitor cocktail 2 & 3 (Sigma) was added to the brain tissue at 5:1 and homogenized with Qiagen Tissue Lyser LT, at 50 Hz for 5 min. Lysate was diluted according to manufacture protocol and spun down at 17,000 g, 4 °C, for 15 min. Supernatant was investigated using a PSD-95 ELISA (MSD #K250QND) and run according to manufactures protocols. Plates were analyzed with an MSD SECTOR S 600 Imager, and results were reported as arbitrary values. Values appeared normally distributed on visual inspection and then were converted to z-scores with a mean of zero and standard deviation of one.
Digital histology and analysis
Immunostaining for AT8 and CD68 and analysis using the Aperio ScanScope (Leica) were performed as previously described [10]. Briefly, tissue blocks of cortical samples were taken from Broadman area 8/9 for all cases. Whole stained DLFC sections were scanned and digitized using an Aperio ScanScope AT Turbo. Digital images were viewed and analyzed using Aperio ImageScope (Leica). Analysis of digital images were limited to the depth of the superior frontal sulcus which was denoted as the bottom third of the connecting superior and middle gyri. White matter was excluded. A customized version of the Aperio positive pixel count algorithm (Version 9) was used to determine total AT8 positive staining. Similarly, a modified nuclear count algorithm (Version 9) was used to count total number of CD68 positive cells. Densities in units of count per mm2 were obtained by standardizing quantifications to the area measured. For both AT8-positive pixel density and CD68-positive cell density, participants were stratified into quartiles to account for the rightward skew of the densities.
Statistical analysis
The association of rs3173615 with six dimensions of CTE-related outcomes (presence of positive CTE neuropathological diagnosis, AT8-positive pixel density in the DLFC, CD68-positive cell density in the DLFC, synaptic density as measured by PSD-95 ELISA, presence of TDP-43 pathology in any brain region and ante-mortem dementia) was evaluated using an additive genetic model. Genotype and allele frequencies were compared between cases and controls using the Cochrane-Armitage Trend test and chi-squared test respectively. All other analyses were only conducted among those with CTE (case-only analyses). We used separate ordinal logistic regression models to estimate the relative odds of a one quartile increase in AT8-positive pixel density or CD68-positive cell density for each additional minor allele, linear regression to estimate differences in PSD-95 synaptic density for each additional minor allele, and separate binary logistic regression models to estimate the relative of odds of having anti-mortem dementia or having TDP-43 pathology for each additional minor allele. All case-only analyses were adjusted for age at death and years of football play. Statistical analyses were performed using SPSS (v.24, IBM) and R (v.3.5.0).
Results
TMEM106B genotype is not associated with CTE diagnosis
Clinical and pathological characteristics of the participants with CTE are presented in Table 1. Controls were significantly older than the CTE cases (p = 0.01). Rs3173615 allele and genotype frequencies were not significantly different between cases and controls (p = 0.71 and p = 0.74, respectively).
TMEM106B genotype is associated with CTE-related neuropathology and ante-mortem dementia in persons with CTE
The rs3173615 minor allele (G) was significantly associated with lower p-tau (AT8-positive) pixel density in the DLFC in a dose-dependent manner: for each G allele, the odds of increasing one quartile in p-tau pixel density was 0.42 (Fig. 2, Table 2). The G allele was significantly associated with lower CD68-positive cell density in the DLFC in a dose-dependent manner: for each G allele, the odds of increasing one quartile in CD68-positive cell density was 0.53 (Fig. 2, Table 2). The G allele was significantly associated with higher synaptic density, as measured by PSD-95 ELSIA, in a dose-dependent manner: for each G allele, synaptic density increased by 0.31 standardized units (Table 2). Rs3173615 genotype was not associated with the presence of TDP-43 pathology. However, the presence of each G allele reduced odds of dementia prior to death by 60% (Table 3).

Representative images of ptau (AT8) and neuroinflammation (CD68) staining by TMEM106B genotype. Positive staining for the respective proteins is in red while hematoxylin counterstain is blue. All images are from the DLFC at the depth of the cortical sulcus. Scale bars represent 500 μm (a–c) and 200 μm (d–f)
Discussion
In a series of former American football players with neuropathologically confirmed CTE, we found that rs3173615, a coding SNP in TMEM106B that was previously implicated with risk of FLTD-TDP, was associated with p-tau density, CD68 density, synaptic loss, and dementia status in case-only analyses. However, this variant was not associated with risk of CTE in an analysis that compared allele and genotype frequencies between the CTE cases and a group of neurologically normal controls.
Although several genes have been proposed as potential CTE risk factors, this is the first study to demonstrate that a variant in TMEM106B is associated with CTE-related outcomes. The findings in this study provide further evidence that variation in TMEM106B is linked with neurodegeneration. Interestingly, the effects of TMEM106B are heterogeneous across all diseases with which it has been associated. In FTLD, hippocampal sclerosis, and APOE ϵ4-negative AD, variation in TMEM106B is associated with disease risk [14, 28, 35, 43]. However, variation in TMEM106B does not change the risk for ALS, but among those with ALS, it is associated with the presentation of cognitive impairment [44]. Similarly, in the present study, the TMEM106B SNP rs3173615 was not associated with CTE risk, but among those with CTE, it modified the neuropathological and clinical presentation, influencing p-tau density, CD68 density, synaptic loss, and odds of dementia.
Rs3173615 underlies the production of two different isoforms of the TMEM106B protein. The more common C allele codes for a highly conserved threonine (T185), whereas the less frequent G allele codes for a substitution of serine (S185) at this location [30]. Functionally, the protein product with S185 is more rapidly degraded than the protein product with T185 [30]. This results in more TMEM106B protein present in C allele carriers. Although the full mechanistic action of the protein is unclear, TMEM106B has been implicated in controlling the size, shape, and acidification of the lysosome [5]. Overexpression of TMEM106B can result in larger lysosomes that do not properly acidify and have impaired function. Enhanced TMEM106B expression has also been demonstrated to result in cell oxidative stress and cytotoxicity [41]. These changes present in rs3173615 G allele carriers with CTE could help explain the increase in microglial activation and p-tau when compared to those lacking this allele. A properly functioning lysosome is critical for the phagocytosis and elimination of toxic proteins. Microglia as well as neurons have been shown to participate in the removal of proteins such as Aβ [8] and tau [2, 33]. An impaired lysosome could result in the buildup of pathogenic proteins as seen in CTE. Future studies will be needed to investigate impaired lysosomal function and how that might prevent proper elimination of ptau.
The association of TMEM106B with neuroinflammation as indicated by increased CD68-positive cell density in participants with CTE is consistent with a recent GWAS of age-associated transcriptional changes that identified a genome-wide significant finding with TMEM106B. The TMEM106B signal was amplified when the outcome was limited to a microglia-associated gene expression cluster [34]. Furthermore, ex vivo analysis in monocyte-derived dendritic cells showed an enhanced stimulated inflammatory response in participants with the risk allele [34]. This increased sensitization is similar to the priming response observed in microglia [31]. Microglia that have been exposed to a previous inflammatory stimulus can exhibit enhanced reactive markers and a more severe immune response when stimulated for a second time [32]. Microglia from individuals with the risk allele might exist in a primed-like state. Exposure to RHI may thus elicit an increased glial reactivity. Our previous work on CTE suggests that microglia and p-tau exist in a positive feedback loop where each component can enhance the other [10]. However, the full spectrum of microglial-mediated inflammatory changes will not be fully captured using only one marker (CD68). Future studies should utilize a combination of histologic markers and biochemical techniques to further explore the effect TMEM106B on neuroinflammation.
Finally, TMEM106B genotype was related to dementia status, adjusting for age at death and duration of football play. For each protective minor allele, odds of dementia decreased by 60%. This finding provides insight into why certain individuals with CTE may progress to dementia while others have slower progression and do not become functionally impaired, even though the exposure to contact sports might be similar. One possible pathologic mechanism for the TMEM106B-dementia relationship comes from the relationship between TMEM106B and PSD-95 protein levels. PSD proteins are post-synaptic density proteins that are often used as markers of synaptic loss and dysfunction [13]. Loss of synaptic proteins can impair neuronal signaling and recruitment of essential neuronal proteins, leading to loss of long-term potentiation and cognitive dysfunction. Synapse loss can occur through a variety of ways [37]. Two common pathways for synaptic loss that may occur in CTE are neuronal death and microglia-mediated synaptic pruning [11]. These pathways may be mediated by tau-induced synaptic dysfunction and neuroinflammation respectively. Future studies should further tease apart the mechanisms leading to synaptic loss and its relationship to clinical impairment.
There are several limitations to this study. CTE cases were largely self-selected or referred by the next-of-kin after death and are not necessarily representative of all individuals who play football. However, selection should only bias a genetic relationship if there are pleiotropic effects that influence selection into the study [25]. Additionally, methods for determination of RHI exposure and clinical and medical history depended on retrospective review and inaccuracies associated with informant-report may introduce measurement error. Another limitation comes from the inclusion of a separately genotyped control group for the case-control analysis. As the control group was clinically, but not neuropathologically assessed, it is possible they may have underlying sub-clinical pathology. Additionally, although the same genotyping platform was used for cases and controls, they were genotyped in separate batches, potentially introducing bias. Ideal controls would have played football and would not have evidence of CTE or other neurodegenerative pathology. Unfortunately, most football players from the VA-BU-CLF brain bank have evidence of CTE pathology; therefore, we relied on controls from another study who may have developed CTE if they were exposed to football. This misclassification may have biased our case-control analysis toward the null, but would not affect our case-only analyses. Future studies should include controls with a complete athletic history and neuropathological evaluation and should not genotype cases and controls separately. An additional limitation is the small sample size by genetic standards. However, studies have only recently ascertained contact sport history or conducted neuropathological examinations for CTE. The current study was conducted in the largest group of CTE cases available to date. Additionally, to maximize statistical power, these cases were densely phenotyped using a quantitative measure of tau pathology. Nonetheless, the findings should be interpreted with caution until they can be independently replicated. Lastly, sufficient genetic data was not available to account for population substructure, which could confound a genetic relationship. However, the analysis was limited to informant reported Caucasian participants to grossly account for population differences. Future studies will be needed to better understand the effects of rs3173615 in non-Caucasian ethnicities.
Conclusions
In conclusion, this study reports one of the first genetic associations for CTE-related outcomes. Although TMEM106B was not associated with CTE case-control status, in case-only analyses, the minor allele had a protective effect for multiple CTE-related neuropathological outcomes including neuroinflammation, p-tau density and synaptic dysfunction. Similarly, in case-only analyses, the minor allele had a protective effect for dementia. Future work is required to replicate these findings in an independent sample and to determine the mechanism by which TMEM106B interacts with RHI and other genetic risk factors to modify CTE-related outcomes. Overall, TMEM106B genotype may partially explain why some individuals experience more severe CTE- related outcomes while others are spared despite similar exposure to contact sports.
References
Alosco ML, Mez J, Tripodis Y, Kiernan PT, Abdolmohammadi B, Murphy L, Kowall NW, Stein TD, Huber BR, Goldstein LE, Cantu RC, Katz DI, Chaisson CE, Martin B, Solomon TM, McClean MD, Daneshvar DH, Nowinski CJ, Stern RA, McKee AC (2018) Age of first exposure to tackle football and chronic traumatic encephalopathy. Ann Neurol 83:886–901. https://doi.org/10.1002/ana.25245
Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593. https://doi.org/10.1038/nn.4132
Bieniek KF, Ross OA, Cormier KA, Walton RL, Soto-Ortolaza A, Johnston AE, DeSaro P, Boylan KB, Graff-Radford NR, Wszolek ZK, Rademakers R, Boeve BF, McKee AC, Dickson DW (2015) Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol 130:877–889. https://doi.org/10.1007/s00401-015-1502-4
Bigio EH (2008) Update on recent molecular and genetic advances in frontotemporal lobar degeneration. J Neuropathol Exp Neurol 67:635–648. https://doi.org/10.1097/NEN.0b013e31817d751c
Brady OA, Zheng Y, Murphy K, Huang M, Hu F (2013) The frontotemporal lobar degeneration risk factor, TMEM106B, regulates lysosomal morphology and function. Hum Mol Genet 22:685–695. https://doi.org/10.1093/hmg/dds475
Brownell B, Oppenheimer DR, Hughes JT (1970) The central nervous system in motor neurone disease. J Neurol Neurosurg Psychiatry 33:338–357
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM, Consortium for Frontotemporal Lobar D (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the consortium for frontotemporal lobar degeneration. Acta Neuropathol 114:5–22. https://doi.org/10.1007/s00401-007-0237-2
Cherry JD, Olschowka JA, O’Banion MK (2015) Arginase 1+ microglia reduce Aβ plaque deposition during IL-1β-dependent neuroinflammation. J Neuroinflammation 12. https://doi.org/10.1186/s12974-015-0411-8
Cherry JD, Stein TD, Tripodis Y, Alvarez VE, Huber BR, Au R, Kiernan PT, Daneshvar DH, Mez J, Solomon TM, Alosco ML, McKee AC (2017) CCL11 is increased in the CNS in chronic traumatic encephalopathy but not in Alzheimer’s disease. PLoS One 12:e0185541. https://doi.org/10.1371/journal.pone.0185541
Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, Mez J, Montenigro PH, Solomon TM, Alosco ML, Stern RA, McKee AC, Stein TD (2016) Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 4:112. https://doi.org/10.1186/s40478-016-0382-8
Chung WS, Welsh CA, Barres BA, Stevens B (2015) Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 18:1539–1545. https://doi.org/10.1038/nn.4142
Dickson DW (2009) Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol 3:1–23
Gong Y, Lippa CF (2010) Review: disruption of the postsynaptic density in Alzheimer’s disease and other neurodegenerative dementias. Am J Alzheimers Dis Other Demen 25:547–555. https://doi.org/10.1177/1533317510382893
Jun G, Ibrahim-Verbaas CA, Vronskaya M, Lambert JC, Chung J, Naj AC, Kunkle BW, Wang LS, Bis JC, Bellenguez C, Harold D, Lunetta KL, Destefano AL, Grenier-Boley B, Sims R, Beecham GW, Smith AV, Chouraki V, Hamilton-Nelson KL, Ikram MA, Fievet N, Denning N, Martin ER, Schmidt H, Kamatani Y, Dunstan ML, Valladares O, Laza AR, Zelenika D, Ramirez A, Foroud TM, Choi SH, Boland A, Becker T, Kukull WA, van der Lee SJ, Pasquier F, Cruchaga C, Beekly D, Fitzpatrick AL, Hanon O, Gill M, Barber R, Gudnason V, Campion D, Love S, Bennett DA, Amin N, Berr C, Tsolaki M, Buxbaum JD, Lopez OL, Deramecourt V, Fox NC, Cantwell LB, Tarraga L, Dufouil C, Hardy J, Crane PK, Eiriksdottir G, Hannequin D, Clarke R, Evans D, Mosley TH Jr, Letenneur L, Brayne C, Maier W, De Jager P, Emilsson V, Dartigues JF, Hampel H, Kamboh MI, de Bruijn RF, Tzourio C, Pastor P, Larson EB, Rotter JI, O’Donovan MC, Montine TJ, Nalls MA, Mead S, Reiman EM, Jonsson PV, Holmes C, St George-Hyslop PH, Boada M, Passmore P, Wendland JR, Schmidt R, Morgan K, Winslow AR, Powell JF, Carasquillo M, Younkin SG, Jakobsdottir J, Kauwe JS, Wilhelmsen KC, Rujescu D, Nothen MM, Hofman A, Jones L, Consortium I, Haines JL, Psaty BM, Van Broeckhoven C, Holmans P, Launer LJ, Mayeux R, Lathrop M, Goate AM, Escott-Price V, Seshadri S, Pericak-Vance MA, Amouyel P, Williams J, van Duijn CM, Schellenberg GD, Farrer LA (2016) A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry 21:108–117. https://doi.org/10.1038/mp.2015.23
Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, McKee A, Dickson D, Bancher C, Tabaton M, Jellinger K, Anderson DW (1996) Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 55:97–105
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4. https://doi.org/10.1007/s00401-009-0612-2
Mahar I, Alosco ML, McKee AC (2017) Psychiatric phenotypes in chronic traumatic encephalopathy. Neurosci Biobehav Rev 83:622–630. https://doi.org/10.1016/j.neubiorev.2017.08.023
McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Dirk Keene C, Litvan I, Perl DP, Stein TD, Vonsattel J-P, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O’Connor K, Alvarez VE, Gordon WA (2015) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86. https://doi.org/10.1007/s00401-015-1515-z
McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:709–735. https://doi.org/10.1097/NEN.0b013e3181a9d503
McKee AC, Daneshvar DH, Alvarez VE, Stein TD (2014) The neuropathology of sport. Acta Neuropathol 127:29–51. https://doi.org/10.1007/s00401-013-1230-6
McKee AC, Stein TD, Kiernan PT, Alvarez VE (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25:350–364. https://doi.org/10.1111/bpa.12248
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. https://doi.org/10.1093/brain/aws307
McKeith IG (2006) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis 9:417–423
Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, Alosco ML, Solomon TM, Nowinski CJ, McHale L, Cormier KA, Kubilus CA, Martin BM, Murphy L, Baugh CM, Montenigro PH, Chaisson CE, Tripodis Y, Kowall NW, Weuve J, McClean MD, Cantu RC, Goldstein LE, Katz DI, Stern RA, Stein TD, McKee AC (2017) Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 318:360–370. https://doi.org/10.1001/jama.2017.8334
Mez J, Marden JR, Mukherjee S, Walter S, Gibbons LE, Gross AL, Zahodne LB, Gilsanz P, Brewster P, Nho K, Crane PK, Larson EB, Glymour MM (2017) Alzheimer’s disease genetic risk variants beyond APOE epsilon4 predict mortality. Alzheimers Dement (Amst) 8:188–195. https://doi.org/10.1016/j.dadm.2017.07.002
Mez J, Solomon TM, Daneshvar DH, Murphy L, Kiernan PT, Montenigro PH, Kriegel J, Abdolmohammadi B, Fry B, Babcock KJ, Adams JW, Bourlas AP, Papadopoulos Z, McHale L, Ardaugh BM, Martin BR, Dixon D, Nowinski CJ, Chaisson C, Alvarez VE, Tripodis Y, Stein TD, Goldstein LE, Katz DI, Kowall NW, Cantu RC, Stern RA, McKee AC (2015) Assessing clinicopathological correlation in chronic traumatic encephalopathy: rationale and methods for the UNITE study. Alzheimers Res Ther 7:62. https://doi.org/10.1186/s13195-015-0148-8
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on A, Alzheimer’s A (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123:1–11. https://doi.org/10.1007/s00401-011-0910-3
Nelson PT, Wang WX, Partch AB, Monsell SE, Valladares O, Ellingson SR, Wilfred BR, Naj AC, Wang LS, Kukull WA, Fardo DW (2015) Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J Neuropathol Exp Neurol 74:75–84. https://doi.org/10.1097/NEN.0000000000000151
Newell KL, Hyman BT, Growdon JH, Hedley-Whyte ET (1999) Application of the National Institute on Aging (NIA)-Reagan institute criteria for the neuropathological diagnosis of Alzheimer disease. J Neuropathol Exp Neurol 58:1147–1155
Nicholson AM, Finch NA, Wojtas A, Baker MC, Perkerson RB, Castanedes-Casey M, Rousseau L, Benussi L, Binetti G, Ghidoni R, Hsiung G-YR, Mackenzie IR, Finger E, Boeve BF, Ertekin-Taner N, Graff-Radford NR, Dickson DW, Rademakers R (2013) TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem 126:781–791. https://doi.org/10.1111/jnc.12329
Norden DM, Godbout JP (2012) Microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. https://doi.org/10.1111/j.1365-2990.2012.01306.x
Perry VH, Teeling J (2013) Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol 35:601–612. https://doi.org/10.1007/s00281-013-0382-8
Piras A, Collin L, Gruninger F, Graff C, Ronnback A (2016) Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun 4:22. https://doi.org/10.1186/s40478-016-0292-9
Rhinn H, Abeliovich A (2017) Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that regulate aging phenotypes. Cell Syst 4:404–415 e405. https://doi.org/10.1016/j.cels.2017.02.009
Rutherford NJ, Carrasquillo M, Li M, Bisceglio G, Menke J, Josephs KA, Parisim JE, Petersen RC, Graff-Radford N, Younkin SG, Dickson DW, Rademakers R (2012) TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology 79:718–719
Satoh J, Kino Y, Kawana N, Yamamoto Y, Ishida T, Saito Y, Arima K (2014) TMEM106B expression is reduced in Alzheimer’s disease brains. Alzheimers Res Ther 6:17. https://doi.org/10.1186/alzrt247
Sheng M, Kim E (2011) The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol 3. https://doi.org/10.1101/cshperspect.a005678
Stamm JM, Bourlas AP, Baugh CM, Fritts NG, Daneshvar DH, Martin BM, McClean MD, Tripodis Y, Stern RA (2015) Age of first exposure to football and later-life cognitive impairment in former NFL players. Neurology 84:1114–1120. https://doi.org/10.1212/WNL.0000000000001358
Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y, Daneshvar DH, Mez J, Solomon T, Meng G, Kubilus CA, Cormier KA, Meng S, Babcock K, Kiernan P, Murphy L, Nowinski CJ, Martin B, Dixon D, Stern RA, Cantu RC, Kowall NW, McKee AC (2015) Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathol 130:21–34. https://doi.org/10.1007/s00401-015-1435-y
Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, Fritts NG, Stamm JM, Robbins CA, McHale L, Simkin I, Stein TD, Alvarez VE, Goldstein LE, Budson AE, Kowall NW, Nowinski CJ, Cantu RC, McKee AC (2013) Clinical presentation of chronic traumatic encephalopathy. Neurology 81:1122–1129. https://doi.org/10.1212/WNL.0b013e3182a55f7f
Suzuki H, Matsuoka M (2016) The lysosomal trafficking transmembrane protein 106B is linked to cell death. J Biol Chem 291:21448–21460. https://doi.org/10.1074/jbc.M116.737171
Van Blitterswijk M, Mullen B, Nicholson AM, Bieniek KF, Heckman MG, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Murray ME, Hsiung GY, Stewart H, Karydas AM, Finger E, Kertesz A, Bigio EH, Weintraub S, Mesulam M, Hatanpaa KJ, White CL 3rd, Strong MJ, Beach TG, Wszolek ZK, Lippa C, Caselli R, Petrucelli L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Mackenzie IR, Seeley WW, Grinberg LT, Miller BL, Boylan KB, Graff-Radford NR, Boeve BF, Dickson DW, Rademakers R (2014) TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol 127:397–406. https://doi.org/10.1007/s00401-013-1240-4
Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P, van Swieten JC, Murrell JR, Ghetti B, Spina S, Grafman J, Hodges J, Spillantini MG, Gilman S, Lieberman AP, Kaye JA, Woltjer RL, Bigio EH, Mesulam M, Al-Sarraj S, Troakes C, Rosenberg RN, White CL 3rd, Ferrer I, Llado A, Neumann M, Kretzschmar HA, Hulette CM, Welsh-Bohmer KA, Miller BL, Alzualde A, Lopez de Munain A, McKee AC, Gearing M, Levey AI, Lah JJ, Hardy J, Rohrer JD, Lashley T, Mackenzie IR, Feldman HH, Hamilton RL, Dekosky ST, van der Zee J, Kumar-Singh S, Van Broeckhoven C, Mayeux R, Vonsattel JP, Troncoso JC, Kril JJ, Kwok JB, Halliday GM, Bird TD, Ince PG, Shaw PJ, Cairns NJ, Morris JC, McLean CA, DeCarli C, Ellis WG, Freeman SH, Frosch MP, Growdon JH, Perl DP, Sano M, Bennett DA, Schneider JA, Beach TG, Reiman EM, Woodruff BK, Cummings J, Vinters HV, Miller CA, Chui HC, Alafuzoff I, Hartikainen P, Seilhean D, Galasko D, Masliah E, Cotman CW, Tunon MT, Martinez MC, Munoz DG, Carroll SL, Marson D, Riederer PF, Bogdanovic N, Schellenberg GD, Hakonarson H, Trojanowski JQ, Lee VM (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 42:234–239. https://doi.org/10.1038/ng.536
Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, Elman L, McCluskey L, Lee VM, Van Deerlin VM, Trojanowski JQ, Chen-Plotkin AS (2011) Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol 121:373–380. https://doi.org/10.1007/s00401-010-0782-y
Vonsattel JP, Aizawa H, Ge P, DiFiglia M, McKee AC, MacDonald M, Gusella JF, Landwehrmeyer GB, Bird ED, Richardson EP Jr et al (1995) An improved approach to prepare human brains for research. J Neuropathol Exp Neurol 54:42–56
Walt GS, Burris HM, Brady CB, Spencer KR, Alvarez VE, Huber BR, Guilderson L, Abdul Rauf N, Collins D, Singh T, Mathias R, Averill JG, Walker SE, Robey I, McKee AC, Kowall NW, Stein TD (2018) Chronic traumatic encephalopathy within an amyotrophic lateral sclerosis brain bank cohort. J Neuropathol Exp Neurol. https://doi.org/10.1093/jnen/nly092 [Epub ahead of print]
Acknowledgements
We would like to acknowledge all the donors and their families whose participation made this work possible.
Funding
This study received support from National Institute of Neurological Disorders and Stroke (U01NS086659, R01NS078337, R56NS078337, U01NS093334, and K23NS102399), National Institute on Aging (K23AG046377, P30AG13846 and supplement 0572063345, RF1AG057902, RF1AG054156, R56AG057768), US Department of Defense (grant W81XWH-13-2-0064), US Department of Veterans Affairs (I01CX001038), Veterans Affairs Biorepository (BX002466), Veterans Affairs Rehabilitation Research and Development Traumatic Brain Injury Center of Excellence (B6796-C), Department of Defense Peer Reviewed Alzheimer’s Research Program (13267017), Department of Defense, Chronic Effects of Neurotrauma Consortium (CENC) Award W81XWH-13-2-0095, Department of Veterans Affairs CENC Award I01CX001135, National Center for PTSD, National Operating Committee on Standards for Athletic Equipment, Alzheimer’s Association (NIRG-15-362697, NIRG-305779, AARF-17-529888), Nick and Lynn Buoniconti Foundation, Concussion Legacy Foundation, Andlinger Family Foundation, WWE, and NFL. The views, opinions and/or findings contained in this article are those of the authors and should not be construed as an official Veterans Affairs or Department of Defense position, policy or decision, unless so designated by other official documentation.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
Study design and conception: JDC, JM, ACM, TDS. Acquisition and analysis of data: JDC, JM, JFC, YT, VEA, IA, BRH, MLA, RN, BA, PTK, LE, GM, SS, KB, HMG, GM, CJN, BMM, BD, NWK, RCC, LEG, DIK, RAS, LAF, ACM, TDS. Drafting of the manuscript: JDC, JM, TDS. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
IRB approval for the brain donation program was obtained through the Boston University Alzheimer’s Disease & CTE Center and the Edith Nourse Rogers Memorial Veterans Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cherry, J.D., Mez, J., Crary, J.F. et al. Variation in TMEM106B in chronic traumatic encephalopathy. acta neuropathol commun 6, 115 (2018). https://doi.org/10.1186/s40478-018-0619-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-018-0619-9