
Abstract
Hydrogen sulfide (H2S) has been known for years as a poisoning gas and until recently evoked mostly negative associations. However, the discovery of its gasotransmitter functions suggested its contribution to various physiological and pathological processes. Although H2S has been found to exert cytoprotective effects through modulation of antioxidant, anti-inflammatory, anti-apoptotic, and pro-angiogenic responses in a variety of conditions, its role in the pathophysiology of skeletal muscles has not been broadly elucidated so far. The classical example of muscle-related disorders is Duchenne muscular dystrophy (DMD), the most common and severe type of muscular dystrophy. Mutations in the DMD gene that encodes dystrophin, a cytoskeletal protein that protects muscle fibers from contraction-induced damage, lead to prominent dysfunctions in the structure and functions of the skeletal muscle. However, the main cause of death is associated with cardiorespiratory failure, and DMD remains an incurable disease. Taking into account a wide range of physiological functions of H2S and recent literature data on its possible protective role in DMD, we focused on the description of the ‘old’ and ‘new’ functions of H2S, especially in muscle pathophysiology. Although the number of studies showing its essential regulatory action in dystrophic muscles is still limited, we propose that H2S-based therapy has the potential to attenuate the progression of DMD and other muscle-related disorders.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Duchenne muscular dystrophy (DMD): an overview
Duchenne muscular dystrophy (DMD) represents one of the most common, severe, and lethal types of dystrophinopathies, which are caused by mutations in the X-linked DMD gene that encodes dystrophin, a structural muscle protein. The clinical presentation of DMD was first described in the 1850s–1860s; however, the first fragments of the DMD gene cDNA were identified more than one hundred years later (for references, see: [1]). DMD is characterized by progressive weakness of the skeletal and cardiac muscle due to muscular damage and degeneration. Patients suffer from motor delays, loss of ambulation, cardiomyopathy, and respiratory impairment [2]. The first symptoms of the disease appear around 2–3 years of age, including frequent falls, difficulty getting up from the floor, the need for help with the hands to stand up (Gower’s sign), or problems with running and climbing stairs. Rapid progression of the disease is observed around the age of 7 with later motor disability, which requires wheelchair use around the age of 12. This is followed by progressive heart problems that require pharmacological treatment, mechanical cardiac support and surgical solutions, and respiratory failure that leads to assisted ventilation.
Respiratory dysfunction and cardiological complications are among the most devastating effects of the disease in patients with DMD, contributing to their morbidity and mortality. Progressive sequelae of DMD, such as scoliosis and kyphosis, result in altered lung function, and respiratory muscle weakness can lead to decreased ventilation capacity and pulmonary infections. Improved survival of patients with respiratory failure was achieved thanks to the introduction of noninvasive nocturnal mechanical ventilation; however, enhanced survival and extension of life expectancy of dystrophic patients result in increased DMD-related cardiomyopathy [3, 4]. Notably, heart problems represent an alternative cause of death associated with DMD and do not correlate with the extent of skeletal muscle degeneration [5]. The first incidence of cardiomyopathy occurs at the age of 6 years. Symptoms manifest themselves in dilated cardiomyopathy, which progresses to end-stage heart failure with accompanying supraventricular and ventricular arrhythmias [6]. Unfortunately, until now, DMD is an incurable disease, and even with optimal care, patients die between the second and fourth decade of life [7, 8] (Fig. 1).

Progression of DMD and its complications. Duchenne muscular dystrophy (DMD) is caused by an X-linked mutation in the DMD gene. Dystrophin deficiency leads to membrane instability and cytoplasm leakage, leading to progressive muscle degeneration. The main hallmarks of DMD are calcium imbalance, chronic inflammation, excessive fibrosis, impaired regeneration, altered autophagy, and dysregulated angiogenesis. The first symptoms of DMD occur around age 2–3 and manifest themselves as frequent falls, Gower's sign, and running problems. Progressive muscle weakness results in disability around the age of 12. This is followed by heart failure and respiratory dysfunction that leads to premature death in the second to fourth decades of life
DMD gene, dystrophin, and dystrophin–glycoprotein complex (DGC)
The gene encoding dystrophin represents one of the largest human genes, spanning more than 2200 kb (0.1% of the whole genome) [9]. It contains 79 exons, and the most common mutation causing DMD is the deletion of one or more exons, which constitutes about 60–70% of all DMD cases, while point mutations account for 26% of cases and exonic duplication cause another 10–15%. Missense mutations, splice mutations, and subexonic insertions or deletions are also responsible for some cases. Importantly, mutations that cause DMD disrupt the protein reading frame, causing premature stop codons, resulting in no or insufficient protein production [10].
DMD affects around 1:5000 male newborns; however, it also rarely occurs in women (less than one per million) with Turner syndrome, biallelic DMD mutations, or translocations involving DMD. In most cases, female carriers are usually asymptomatic, however, some manifest symptoms resembling the milder type of dystrophinopathy, Becker muscular dystrophy (BMD) [8]. BMD is also caused by mutations in the gene encoding dystrophin; however, these mutations (in-frame deletions) maintain the correct reading frame. In contrast to DMD, this leads to the production of a shorter, partially functional form of DMD products, which are internally truncated and expressed at lower levels compared to healthy individuals [10].
Due to the complex structure of the DMD gene (for example, two polyadenylation sites and seven independent tissue-specific promoters), alternative splicing gives the number of various DMD isoforms, different in size and tissue distribution [11]. The full-length muscular isoform of dystrophin with a molecular weight of 427 kDa (Dp427m) is composed of four main functional domains, an N-terminal actin-binding domain (ABD), a central coiled-coil segment of 24 spectrin-like repeats, and 4 hinge domains that form so so-called rod domain (ROD), cysteine-rich domain (CRD), and C-terminal domain (CTD) [12]. Almost 50% of the mutations that occur in patients with DMD are located within the ABD fragment, to a lesser extent in the CRD and ROD, and just over 10% of the mutations are associated with the CTD [13].
Dystrophin is located on the cytoplasmic side of the muscle and the cardiac sarcolemma. The important function of the protein is to maintain the integrity of the myofiber plasma membrane during force generation, providing mechanical stabilization. Dystrophin is one of the key proteins that forms the dystrophin-associated protein complex (DAPC), also known as the dystrophin–glycoprotein complex (DGC), and provides linkage via the N-terminus and C-terminus (binding to the transmembrane dystroglycan complex) between the actin cytoskeleton and the extracellular matrix, respectively [10, 14]. Furthermore, DGC plays a signaling role by controlling mechanical force transduction and cell adhesion and functions as a scaffold for signaling proteins [15]. DGC regulates muscle cell NO signaling and maintains optimal activity of neuronal nitric oxide synthase (nNOS) and calcium (Ca2+) homeostasis [4].
Consequences of dystrophin deficiency and pathological hallmarks of DMD
Although dystrophin represents only approximately 0.002% of total muscle protein, its lack causes enormous changes in skeletal and cardiac muscle functions such as inflammation, fibrosis, and degeneration [16] (Fig. 1). Increased permeability of the cell membrane allows larger proteins, including muscle creatine kinase (CK) and lactate dehydrogenase (LDH), to enter the circulation. In fact, serum CK and LDH can be used as biomarkers of DMD [17]. Furthermore, under dystrophin-deficient conditions, an increased Ca2+ influx and Ca2+ overload were demonstrated in both human and animal models of the disease [18, 19]. Consequently, protein degradation and activation of calcium-dependent proteases such as calpains and numerous chemokines and cytokines occur [20]. This induces inflammation by activating the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which triggers transcription of inducible nitric oxide synthase (iNOS) [21]. Calcium imbalance is also closely related to mitochondrial function. Increased intracellular Ca2+ leads to upregulation in mitochondrial Ca2+ uptake, which further results in accelerated mitochondrial production of reactive oxygen species (ROS). This causes depolarization of the mitochondrial membrane and the opening of the mitochondrial permeability transition pore (MPTP) and decreased ATP production. It should be noted that an imbalance in calcium homeostasis has a more detrimental effect on cardiac cells compared to skeletal muscle [21]; however, in both cell types these disturbances result in cell death through necrosis and apoptosis [20].
Necrosis is the first step in stimulating an inflammatory response, and chronic inflammation and infiltration of immune cells are one of the main hallmarks of DMD. Several mechanisms contribute to these processes, but the innate immune system is first activated by membrane instability and cytoplasm leakage. Destroyed fibers release damage-associated molecular patterns (DAMPs), such as nucleic acids, ROS, and heat shock proteins (HSPs), and activate toll-like receptors (TLRs). Activation of TLRs and the interleukin-1 receptor (IL-1R) initiates a pro-inflammatory signaling cascade, including activation of the myeloid differentiation primary response 88/IL-1R-associated kinase (MyD88/IRAK) pathway. This, in turn, leads to stimulation of mitogen-activated protein kinases (MAPKs) and NF-κB [22]. Dystrophin-deficient muscles are infiltrated by immune cells such as macrophages, neutrophils, and mast cells. Furthermore, in DMD muscles, an elevated level of eosinophils is also present in the early stage of the disease. Among all leukocytes, myeloid cells are the most abundant immune cells that infiltrate dystrophin-deficient muscles; however, lymphoid cells, such as cytotoxic T-lymphocytes, are also present and contribute to the cytolytic and immunomodulatory effect [23]. Self-sustaining activation of the innate immune response leads to the constant release of pro-inflammatory cytokines including IL-1β, tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), IL-6, induction of constitutive expression of MHC class I and II in muscle cells, and recruitment of T and B cells, which generates an adaptive immune response [24].
The diverse nature of macrophages contributes to the progression of DMD. In the early stage of the disease, Ly6Cpos pro-inflammatory macrophages promote muscle lesions due to their NO-mediated cytolytic capacity. Later, during the progression of the disease, IL-10-producing cells, such as regulatory T cells (Tregs), induce alternative activation of Ly6Cneg anti-inflammatory macrophages. It leads to the deactivation of the inflammatory response and tissue repair [24]. This shows the complexity of the immune response in the course of DMD with repeated cycles of inflammation, damage, and activation of compensation mechanisms to counteract disease progression. In addition, macrophages also release pro-fibrotic molecules such as transforming growth factor-β (TGF-β), that is crucial in the initiation of the fibrotic process [24, 25]. Fibroblasts activated by TGF-β produce extracellular matrix (ECM) components and ECM-remodeling factors (collagens, fibronectin, metalloproteinases) [26]. As a result of chronic injury, persistent inflammation, and the presence of macrophages, an excessive amount of ECM components accumulate in muscle tissue. This inhibits myogenic repair and contributes to muscle replacement by the fibrotic scar [27]. Notably, a different, more rigid structure of the fibrotic tissue compared to the muscles affects the efficiency of muscle contraction. During this time, fibrotic tissue can also be replaced by adipocytes (fatty degeneration) [27, 28].
Myonecrosis, increased inflammation, oxidative stress, and progressive fibrosis alter the myogenesis and regeneration process in dystrophic muscles, which leads to a subsequent loss of muscle tissues [29]. Impaired regeneration in DMD muscles is associated with an inefficient generation of myogenic progenitors. Notably, dystrophin is expressed in muscle stem cells, satellite cells (SCs), defined as CD45−CD31−α7-integrin+Sca-1− cells expressing Pax7, the canonical marker that drives their survival and proper functioning [30]. In SCs, dystrophin regulates their polarity and asymmetric division [31], and, interestingly, both an increase in their number [32], and their exhaustion due to telomere shortening [33] under dystrophic conditions have been shown. Despite an increased quantity of SCs in DMD muscles, the absence of dystrophin alters their self-renewal and maintenance, due to the decreased expression of asymmetric divisions-regulating microtubule affinity-regulating kinase 2 (MARK2) [31]. Our results, obtained in the mdx mouse model, also indicate a higher number of Pax7+ SCs in dystrophin-deficient mice, suggesting a lack of association between defects in the regenerative potential of SCs and their number [34,35,36]. It should be noted that the functioning of SCs is also regulated by infiltrating macrophages. Pro-inflammatory macrophages promote SCs proliferation, while anti-inflammatory macrophages favor their differentiation and fusion. Under dystrophic conditions, the increased and persistent presence of different macrophage phenotypes may contribute to alterations in SC function [26].
Recent studies have also shown that impaired autophagy is another hallmark of DMD and can be considered a new therapeutic target [37]. Lack of dystrophin leads to increased activation of Akt and mTOR, negative autophagy regulators [37, 38]. Furthermore, in dystrophin-deficient SCs enhanced phosphorylation and decreased nuclear translocation of coactivator-associated arginine methyltransferase 1 (CARM1) are observed. This not only leads to reduced transcription of Myf5 and other Pax7 target genes, resulting in impaired function of satellite cells, but may also affect the autophagy process [39]. CARM1 regulates autophagy in the adenosine 5′-monophosphate-activated protein kinase (AMPK)-dependent way [40]; therefore, its increased phosphorylation in DMD may be responsible for the inability to properly activate autophagy. Insufficient autophagy causes the accumulation of damaged organelles and protein aggregates that affects proper muscle repair [41]. Under dystrophic conditions, the gene expression of many proteins that participate in the autophagy process is dysregulated. We have found a decrease in the mRNA level of beclin-1 (Becn1), autophagy-related genes 5 and 7 (Atg5, Atg7), and lysosomal-associated membrane protein 1 (Lamp1) in muscles from mdx mice [42]. The conversion of the soluble form of the microtubule-associated protein 1 light chain 3 (LC3-I) to lipid-bound LC3-II contributes to the formation of an autophagosome and is necessary, but not sufficient, to trigger cell autophagy [43]. A decrease in the level of the autophagy marker LC3-II was also observed in the DMD muscles [37]. There are also some studies suggesting activation of autophagy under dystrophic conditions, but this can be affected by age and progression of the disease [44, 45].
Furthermore, defective mitochondria-specific autophagy, mitophagy was found in dystrophic muscles. The mechanism may involve dysregulation of the PINK1 (PTEN-induced kinase-1)/PARKIN (Parkinson juvenile disease protein-2) pathway [42, 46]. The decrease in critical mitophagy-related factors, such as PINK1, PARK2, and BNIP3, has been demonstrated in dystrophic patients and various animal models of DMD (mice and worms) [42, 47]. On the other hand, the use of the mitophagy activator urolithin A alleviated the symptoms of DMD [47]. Kuno et al. demonstrated the beneficial cardioprotective effects of resveratrol in mdx mice, which was the consequence of the reactivation of defective mitophagy [48]. Similarly, activation of AMPK, the regulator of the autophagy-mitophagy pathway, improved the contractile functions of the dystrophic diaphragm by improving mitochondrial integrity [49]. Taken together, many studies have shown dysregulated autophagy and mitophagy in DMD; however, due to the complex nature of this process, more studies are warranted.
Angiogenesis, a process of the formation of new blood vessels, has also been suggested to be affected under dystrophic conditions (reviewed in: [50]). Importantly, dystrophin is expressed in vascular smooth muscle cells [51, 52] and, as mentioned above, in SCs that are close to the capillaries and secrete the key angiogenic factor, vascular endothelial growth factor (VEGF). Furthermore, the presence of dystrophin was suggested in endothelial cells [53, 54]; however, these results need further confirmation, as endothelial cell fractions instead of pure endothelial cells have been used in the above studies. Nevertheless, the absence of dystrophin can cause aberrations in the structure and functions of blood vessels and impaired angiogenesis in an age-dependent manner [50, 55, 56]. In DMD-affected muscles, a decrease in VEGF expression was observed in our studies [34,35,36, 56] and an increase in its level was proposed as a strategy that could exert a beneficial effect on the pathology of DMD (reviewed in: [50]).
Treatment of Duchenne muscular dystrophy—possibilities and limitations
Despite extensive research on the molecular mechanisms of DMD, it remains an incurable and fatal disease. However, pharmacological, gene, and cell therapies, aimed at counteracting the processes that contribute to disease progression (described above) or the restoration of functional dystrophin, are currently being investigated (reviewed in [4]) (Fig. 2). Although some approaches are extremely promising, their widespread clinical application may be a matter of the distant future, also because of the drug-specific limitations. Combinatory therapies that simultaneously use strategies to target the cause of the disease and mitigate the secondary effects of the absence of dystrophin can have additive therapeutic benefits [57].

Examples of different therapeutic approaches for DMD treatment. Among the main therapeutic approaches for DMD are genetic and pharmacological therapies. Genetic therapies focus on restoring the expression and synthesis of functional dystrophin through the mini/micro-dystrophin approach, the exon skipping strategy, readthrough therapy, and the CRISPR/Cas9 technology. On the other hand, pharmacological therapies use factors such as glucocorticoids, modulators of utrophin level, histone deacetylase inhibitors, and compounds targeting angiotensin activity that mitigate the downstream effects of dystrophin deficiency
Gene therapies
There are several possibilities to restore the expression and synthesis of functional dystrophin through gene delivery; unfortunately, most viral vectors, apart from adeno-associated virus (AAV) vectors, do not infect myocytes with satisfactory efficiency. Although mature muscle cells can be targeted by AAV, discrepant data has been published on the ability of these vectors to transduce myogenic stem cells. Arnett et al. demonstrated that quiescent SCs are resistant to transduction in vivo in adult mice [58]. In contrast, Tabebordbar et al. were able to show that dystrophic SCs are transduced after systemic delivery of AAV9-Dmd CRISPR (AAV serotype 9) that leads to the restoration of the dystrophin reading frame [59]. Furthermore, further studies confirmed the ability of transduction and gene editing in SCs after AAV9 delivery of CRISPR-Cas9 components, albeit with rather a low efficiency [60].
More studies are needed to analyze in-depth, for example, the role of the satellite cell niche on SCs transduction and to increase efficiency by testing other muscle-specific promoters instead of the commonly used muscle creatine kinase 8 (CK8e) promoter, which is not highly active in resident SCs [60]. It should be noted that the AAV capacity is not sufficient to accommodate a complete dystrophin construct, and these vectors also cause an inflammatory response. However, much effort has been made to improve genetic methods for DMD treatment. Current approaches can be classified into four main groups, including (1) mini/micro-dystrophin (truncated forms of dystrophin that contain only the domains necessary for their function) through AAVs; (2) exon skipping strategy using antisense oligonucleotides (AONs) sequences to restore the open reading frame of dystrophin mRNA; (3) readthrough therapy related to the presence of nonsense mutations, and (4) the CRISPR/Cas9 gene-editing system to repair mutations in the DMD gene (summarized in: [4]).
It should be emphasized that all strategies aimed at restoring the expression and synthesis of functional dystrophin are challenging both from a technical point of view (e.g., the optimal route of administration must be selected to ensure the production of dystrophin not only in skeletal muscles but also in the heart) and from a biological point of view (e.g., to limit the risk of an immune response to dystrophin and/or AAV vectors). Another important issue is the estimation of the minimal level of dystrophin required to achieve a therapeutic effect [61].
Gene transfer of the full-length dystrophin coding sequence is technically difficult, as AAV vectors have a small packaging capacity (around 5 kb). Due to this limitation, truncated forms of dystrophin, the so-called mini/micro-dystrophin, have been generated, containing only the domains necessary for their function (ABD, CRD, and CTD, with the versatile length of the ROD structure) [62,63,64]. Despite positive effects in animal studies, the clinical efficacy of this strategy was not sufficient and problems with a very low level of micro-dystrophin in the muscles and an unsatisfactory transduction efficiency of the diaphragm and particularly the heart were evident [65]. Positive effects, observed in some clinical studies [66], can also be related to concomitant treatment with a high dose of corticosteroids, necessary to halt the immune response against the viral vector.
To overcome immune reactions generated by the newly produced dystrophin protein, therapy with utrophin, structural, and functional dystrophin paralog has been proposed. Like dystrophin, utrophin interacts with the dystrophin-associated protein complex to link the actin cytoskeleton to the extracellular matrix [67]. However, its expression in the extrajunctional sarcolemma declines after birth and, despite high expression during fetal development, it is replaced by dystrophin in adults [68, 69]. Therefore, stimulation of utrophin expression appeared to be an attractive strategy for treating dystrophic patients. However, although utrophin and dystrophin share many similarities, they also differ in some properties and functions, mostly related to the lack of nNOS binding sites and some parts of the central coiled-coil segment of the ROD structure in the utrophin gene [70, 71]. Therefore, utrophin is not fully capable of complementing the function of dystrophin [72], and combinatorial therapies, such as upregulation of utrophin and restoration of dystrophin, can be proposed as an approach with better efficacy. Nevertheless, utrophin alone therapy may be plausible in patients with BMD who still express a low level of dystrophin [73].
The exon skipping strategy with predesigned AONs leads to the conversion of frameshift to in-frame deletions, resulting in the expression of a shortened but functional protein [74]. Typically, AONs are 20–30 nucleotide-long fragments of DNA or RNA, which, by binding to the exon/intron boundary or targeting intraexonic regions, can skip the particular exon(s), ‘hide’ it from the splicing machinery, and restore the reading frame [75]. As this method is a mutation-specific approach (e.g., eteplirsen skips exon 51 of the DMD gene, golodirsen and viltolarsen may be used in patients having confirmed exon 53 amenable mutations, while casimersen allows skipping exon 45), different AONs must be used in a form of personalized medicine. Although all four AONs are approved by the FDA [76], several issues regarding their efficacy, applicability, delivery, and cytotoxicity remain questionable [77]. The main drawbacks of AONs include low uptake into heart tissue, with only a temporal effect, and the need for repeated administration, which also affects the cost of the treatment. Nevertheless, multiple exon skipping (multi-exon skipping) or the cocktail of various AONs could theoretically be used to restore the open reading frame of dystrophin mRNA in 80–90% of patients with DMD in total, regardless of the type of mutation [78, 79].
Readthrough therapy can also be applied to only a limited number of patients, who have nonsense mutations in the DMD gene (approximately 13% of boys). The mechanism of action of the compounds used in this strategy is based on the ribosomal readthrough of the stop codon on dystrophin mRNA and the expression of functional protein. The exemplary drug, ataluren [80], is conditionally approved in the European Union [2, 81]. Although the main objectives of some clinical trials have not been met, many data indicate that this drug delays the progression of DMD (reviewed in: [82]).
Finally, the application of the CRISPR/Cas9 system, a highly effective genetic editing technology, has great potential to be used for the treatment of DMD. Functional restoration of the dystrophin gene was obtained not only in vitro, in dystrophic myoblasts derived from induced pluripotent stem cells (iPSCs) [83, 84] but also in various animal models of DMD—mice [59, 85, 86], dogs [87], and finally pigs [88]. Although CRISPR/Cas9-mediated dystrophin correction may represent a one-time therapy with long-term results [89], its clinical application is still under debate due to the possibility of off-target gene editing [90, 91], including the risk of unspecific mutations. More details on the latest findings and modifications of this approach could be found in recent reviews [92, 93].
Cell therapies
The most controversial and least studied and effective in clinical trials are strategies aimed at the transplantation of cells expressing functional dystrophin. Recently, much attention has been paid to the possible use of meso-angioblasts. These vessel-associated progenitors have been shown to cross the blood-vessel wall after intra-arterial delivery [94] and differentiate into muscle fibers [95]. Despite these valuable abilities, a phase 1/2a clinical study did not elicit a significant benefit [96]. Although other cell populations have also been suggested to be used, including bone marrow-derived mesenchymal stem cells (BM-MSCs) and CD133+ progenitors [97,98,99], only muscle SCs are proven bona fide muscle-derived stem cells able to form new muscle fibers. Unfortunately, the success of their clinical application in humans is so far very limited [100, 101].
Pharmacological therapies
Pharmacological compounds, acting through inhibition of NF-κB (corticosteroids–glucocorticoids), downregulation of histone deacetylase activity (HDAC inhibitors), modulation of the utrophin level, or angiotensin activity (angiotensin-converting enzyme inhibitors; ACEIs and angiotensin II receptor blockers; ARB), mostly used as factors mitigating the downstream effects of dystrophin deficiency, have been tested in animal models, as well as clinical trials (reviewed in [4]).
Glucocorticoids: prednisone, prednisolone, and deflazacort have been used as the gold standard for the treatment of DMD for more than 30 years [102, 103]. Their protective effects are mediated mostly through inhibition of NF-κB signaling and reducing the inflammatory response [104, 105]. However, new mechanisms responsible for their beneficial impact have also been identified. St-Pierre et al. [106] observed that in a mouse model, the use of deflazacort leads to increased expression of nuclear factor of activated T cells 1 (NFATc1)-dependent genes, including utrophin. However, upregulation of utrophin was not evident in prednisolone-treated myotubes derived from fibroblasts of patients with DMD [107]. Prednisone and deflazacort can activate the expression of genes, such as Anxa1 and Anxa6, which encode proteins involved in the repair of the sarcolemma after injury (annexin A1 and annexin A6, respectively) [108], while Kameyama et al. also found that prednisolone can inhibit matrix metalloproteinase-2 (MMP-2) mRNA and consequently increase the level of laminin, the main component of the basement membrane of muscle fibers [107]. All these mechanisms may be responsible not only for the glucocorticoid-triggered reduction of muscle damage, but also for their cardioprotective outcomes [109,110,111]. Although glucocorticoids delay loss of ambulation in DMD patients, they are accompanied by prominent adverse effects, including excessive weight gain, growth inhibition, adrenal insufficiency, bone weakness, cataract development, and behavioral changes [103]. To reduce some of these negative consequences, the use of new and safer steroid analogs such as vamorolone (also known as VBP15) [112,113,114,115] or optimized, less frequent, administration of conventional drugs [108] was suggested. However, a recent comparison of the effectiveness of daily prednisone or deflazacort with intermittent prednisone (10 days on and then 10 days off) found the superiority of the daily corticosteroid regimen over the intermittent treatment [116].
Although the field of pharmacological, gene, and cell therapies for the treatment of DMD continues to advance, new promising therapeutic strategies are being evaluated. An example of such an approach also includes the use of hydrogen sulfide (H2S).
Why H2S? How can we increase the level of H2S in vitro and in vivo?
Gasotransmitters, small gaseous messenger molecules that are freely permeable to cell membranes, produced endogenously in an enzymatically controlled manner, have profound physiological functions and implications in therapeutics. Their discovery has provided new insights into the mechanisms of signaling and cellular interactions. It turned out that signal transduction can occur without the involvement of membrane receptors: the ligand–receptor mechanism is not the only way to modulate cellular activity, and these roles can also be played by endogenously synthesized gas messengers [117]. The first gasotransmitter identified was nitric oxide (NO), followed by carbon monoxide (CO), and, as the third, H2S was classified among gaseous messengers. H2S, like CO, has until recently evoked negative associations. This colorless and flammable gas with the characteristic odor of rotten eggs in higher concentrations has adverse effects on the body, including irritation of the eyes and respiratory system, and can even lead to death through inhibition of mitochondrial respiration [118]. However, all three gases have been shown to act as regulators of many biological functions in animals as a consequence of their anti-inflammatory, cytoprotective, antioxidant, and anti-apoptotic properties [119].
In mammals, H2S biosynthesis can occur through both enzymatic and non-enzymatic pathways [120] (Fig. 3). Most endogenous H2S production is mediated by pyridoxal 5′-phosphate-dependent enzymes: cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST, MPST) using l-cysteine, l-homocysteine or l-methionine as substrates [121]. Although these enzymes can demonstrate organ and tissue-specific distributions, they are all expressed in skeletal muscles, heart, vasculature, and endothelium [122, 123]. An additional biosynthetic pathway was described for the production of H2S from d-cysteine, involving 3-MST and d-amino acid oxidase (DAO). However, this mechanism is not universal and is primarily limited to the brain, kidney, and gastrointestinal system [124,125,126]. Furthermore, Akaike et al. [127] reported that H2S can also be produced from l-cysteine by cysteinyl-tRNA synthetases (CARSs) while Pol et al. [128] found selenium-binding protein 1 (SELENBP1), a methanethiol oxidase (MTO), responsible for the production of H2S (together with H2O2 and formaldehyde) from methanethiol. Regardless of the biosynthesis pathway, subsequently, H2S acts as a gaseous mediator and can finally be metabolized by oxidation in mitochondria, methylation in the cytosol, or can be scavenged by methemoglobin to form sulfhemoglobin [117, 129] (Fig. 3). Oxidation in mitochondria, which occurs in a two-step reaction, is the major catabolic pathway. First, H2S is oxidized by sulfide quinone oxidoreductase (SQR) to a persulfide, which is further oxidized to sulfite by persulfide dioxygenase (ETHE1). Finally, sulfite is converted to sulfate or thiosulfate by sulfite oxidase (SUOX) and thiosulfate transferase, TST (also called rhodanese), respectively. Cytosolic methylation also involves the activity of several enzymes. Thiol S-methyltransferase (TMT) triggers the conversion of H2S to methanethiol and dimethyl sulfide, and then, the latter is oxidized to thiocyanate and sulfate due to the activity of TST. The gasotransmitter can also be removed in an unchanged form through exhalation from the lungs. In the kidney, it is metabolized into final products such as thiosulfate and sulfate, while the liver turns H2S mainly into sulfate (Fig. 3), (for references, see: [130]).

Endogenous and exogenous sources of H2S and its further fate. Endogenous synthesis of H2S occurs primarily through an enzymatic pathway, and the main precursor is l-cysteine derived from the metabolism of homocysteine and cystathionine. l-cysteine can be converted to H2S by cystathionine β-synthase (CBS) or cystathionine γ-lyase (CSE). Other enzymes involved in H2S synthesis may be D-amino acid oxidase (DAO) and cysteine aminotransferase (CAT), which convert d-cysteine and l-cysteine, respectively, to 3-mercaptopyruvate, for further metabolization by 3-mercaptopyruvate sulfurtransferase (3-MST). Exogenously, H2S can be delivered with food and in the form of chemical donors, including inorganic sulfide salts or new-generation synthetic donors. Another approach to increase the level of gasotransmitter through external sources is gene therapy based on the overexpression of H2S-generating enzymes. Subsequently, H2S acts as a gasotransmitter, exerting numerous biological effects or is catabolized through several pathways. It can be scavenged by methemoglobin to form sulfhemoglobin, metabolized into thiosulfate and sulfate in the kidney and liver, or exhaled directly by the lungs. The main catabolic pathway is based on oxidation in the mitochondria. In subsequent reactions catalyzed by sulfide quinone oxidoreductase (SQR) and persulfide dioxygenase (ETHE1), H2S is converted to a persulfide and sulfite, respectively. Sulfite may be turned into sulfate or thiosulfate by sulfite oxidase (SUOX) and thiosulfate transferase, TST (also called rhodanese). In the cytosol, H2S is methylated by thiol S-methyltransferase (TMT) to methanethiol and dimethyl sulfide, which can be further processed by TST to thiocyanate and sulfate. GSH, glutathione; SAM, S-adenosyl-methionine
H2S has been shown to affect different organs and regulate a variety of processes in the organism (Fig. 4). Among others, it acts as a neuromodulator and antioxidant [131], an anti-inflammatory compound [132], a mediator of vasorelaxation [133], a stimulator of angiogenesis [134], a regulator of muscle contractility [135], and a protective factor against heart failure [136]. Furthermore, H2S can modify a wide variety of proteins, including those involved in signal transduction pathways, through post-translational modification of protein cysteine residues in a process known as S-sulfhydration (that is, conversion of cysteine -SH groups to -SSH) [137]. Due to this modification, H2S activity can be found in virtually all physiological processes, and its possible application to treat neurodegenerative, cardiovascular, renal, and other diseases has been widely investigated [130].

Physiological roles and therapeutic targets of H2S. The cytoprotective effects of H2S are exerted by the numerous activities that can be demonstrated in the regulation of various organ functions under physiological and pathological conditions. Among others, H2S has been proposed as a therapeutic in neurodegenerative disorders, acute lung injury, and myocardial infarction. The protective role of H2S has been demonstrated in ischemia–reperfusion injury in the kidney, inflammatory bowel disease and colitis, as well as in reproductive system dysfunctions. Finally, H2S may be a preventive factor in skeletal muscle-related disorders, such as muscle atrophy, sarcopenia, and Duchenne muscular dystrophy
Taking into account the versatile applications of H2S, there are attempts to use this mediator as a therapeutic agent. However, it is important to use a physiologically relevant dose of H2S to avoid its toxic effect. In rat, human, and bovine brain tissues, H2S is present at levels of up to 50–160 μM [138], but there are studies that show its beneficial effects in a wider range of concentrations (10–300 μM) [139]. Several different possibilities of H2S delivery are described. The gaseous nature of the compound does not facilitate its direct administration, as its inhalation raises some concerns about the possible toxicity and the problems in estimating the precise dose administered [140]. However, in a mouse model of Parkinson’s disease (induced by the administration of dopaminergic neurotoxin, MPTP), H2S inhalation for 8 h/day for 7 days, prevented neuronal apoptosis and protected against disease-induced movement dysfunction [141]. A different research group has shown that H2S administered in gaseous form promotes glucose uptake by increasing insulin receptor sensitivity and improves kidney lesions in type II diabetes [142]. For exogenous delivery of H2S, donors in the form of sulfide salts or synthetic molecules are often used (Fig. 3, Table 1). The most common inorganic sulfide salts are sodium hydrosulfide (NaSH) and sodium sulfide (Na2S). They are water-soluble solid analogs of H2S, providing rapid access to biologically relevant forms of sulfide such as H2S and HS− [143]. These compounds have been used in many in vitro studies and have broad applications in vivo. Importantly, short- and long-term treatment was shown to have a favorable outcome. Chen et al. [144] showed that only a single dose of 50 μmol/kg body-weight NaHS improved cardiac function in the mouse model of sepsis-induced myocardial dysfunction. Also long-term (4 weeks) administration of NaHS to C57BL/6J mice by intraperitoneal injection played a protective role in vascular remodeling and inhibited the inflammatory response by activating PPARδ/SOCS3 signaling pathway [145].
Although NaHS has been used as the main tool for increasing H2S concentration in virtually all disease models for years, its main limitation is related to the very rapid release of the gasotransmitter. Upon reaction with water, NaHS and other sulfide salts hydrolyze immediately, leading to a local and transient supra-physiological H2S level followed by a rapid decline [146]. Such poor pharmacokinetic properties limit their use in potential therapies and have led to the discovery of innovative H2S donors, such as GYY4137 and FW1256 [147]. GYY4137 releases H2S significantly slower (10 min) compared to NaHS (10 s) [148] and is the most studied water-soluble compound that releases H2S through hydrolysis. It was used to study the cardioprotective effect of H2S in vitro [149] and in vivo [150]. The use of this donor allowed the demonstration that H2S can have anti-tumor effects [151], stimulate autophagy, and attenuate ferroptosis [152], among other effects.
AP39 is the example of a mitochondria-targeted H2S donor, as it contains a mitochondria-targeting motif, triphenyl phosphonium coupled with an H2S-donating moiety (dithiolethione). This compound was shown to protect against oxidative stress-mediated renal epithelial cell injury in vitro and renal ischemia–reperfusion injury in vivo [153], and oxidative mitochondrial DNA damage in endothelial cells [154]. Furthermore, in a model of cardiac arrest and cardiopulmonary resuscitation-induced neurological injury, treatment with AP39 preserved mitochondrial integrity, reduced oxidative stress, and improved neurological function and long-term survival rates [155]. The cardioprotective effect was also demonstrated [156].
Among different H2S donors, an interesting class of new compounds has been developed that combine traditional nonsteroidal anti‐inflammatory drugs (NSAID) with a chemical moiety that donates H2S. Exemplary compounds, ATB-346, a derivative of naproxen [157, 158] and ATB-352, known as HS-ketoprofen [159], were shown to exert reduced toxic effects in the gastrointestinal track, compared to the parent NSAID. Importantly, their anti-inflammatory and analgesic potential was comparable or even better than traditional NSAIDs. It is noteworthy that the Phase 2 clinical trial (http://ClinicalTrials.gov, NCT03291418) conducted on healthy male and female subjects has demonstrated a superiority of ATB-346 over the naproxen, since less gastroduodenal ulceration was evident in the ATB-346 group [157].
To improve the chemical properties of H2S donors and use those molecules in a more controllable way, new derivatives were recently synthesized. Among them, the pH-controlled factor (ammonium tetrathiomolybdate, ATTM) and the thiol-triggered H2S releaser (SG1002), after the successful results of phase I clinical trials (http://ClinicalTrials.gov, NCT01989208) [160], are suggested for the treatment of cardiovascular diseases and breast cancer. The field of discovery of new small molecules and materials-based approaches for controlled delivery of H2S and related reactive sulfur species (RSS) increases rapidly. Examples of such compounds are N-thiocarboxyanhydrides (NTAs) that release carbonyl sulfide (COS), which is effectively converted to H2S by the action of the enzyme carbonic anhydrase (CA) [161]. A summary of basic information on various H2S donors is provided in Table 1. However, we encourage the readers to seek more details in other articles that focus on donor chemistry, their pros and cons, and possible applications [140, 162, 163].
There are also known natural H2S donors, such as garlic extracts (Allium sativum) and derivatives [164]. The most characterized active component of garlic, allicin (diallyl thiosulfinate), unstable in aqueous media, is quickly converted into several H2S-releasing compounds [e.g., diallyl sulfide (DAS), diallyl disulfide (DADS), diallyl trisulfide (DATS) and allyl methyl sulfide (AMS), allyl methyl disulfide (AMDS), allyl methyl trisulfide (AMTS)] (Fig. 5). Similarly, isothiocyanates such as sulforaphane and allyl isothiocyanate present in cruciferous vegetables have significant H2S-releasing activity. However, the main problem with supplementation with these natural means may be standardization of dosage, as well as bioavailability and absorption of active ingredients.

Garlic-derived natural H2S donors. Allicin (diallyl thiosulfinate) is an organosulfur, active garlic component. Its decomposition results in the formation of several H2S-releasing compounds such as diallyl sulfide (DAS), diallyl disulfide (DADS), diallyl trisulfide (DATS), and methylated forms including allyl methyl sulfide (AMS), allyl methyl disulfide (AMDS), and allyl methyl trisulfide (AMTS). The newly formed compounds are considered natural H2S donors with a broad spectrum of biological activity
Finally, another strategy may be based on the gene therapy approach. Overexpression of H2S-generating enzymes, leading to increased H2S production, may have superior properties over chemical compounds, since the latter may exert functions related not only to H2S signaling. Sen et al. [165] have shown that ex vivo gene therapy with plasmids having CBS, CSE, and 3-MST genes improved the relaxation of hyperhomocysteinemic arterial explants. In D. melanogaster, constitutive ubiquitous overexpression of CBS extended a lifespan, improved locomotor activity, and increased resistance to hyperthermia (35 °C) [166]. On the other hand, genetic overexpression of CBS in the brain caused dysregulation of serotonin and dopamine pathways [167] and contributed to Down syndrome-associated neuronal disturbances [168].
Possible mechanisms of beneficial effects of H2S
H2S exerts anti-inflammatory, anti-apoptotic, and antioxidant activities (Fig. 6). Other known effects are related to neuroprotection (e.g., by mediating the N-methyl-d-aspartate, NMDA) receptor responses) and relaxation of blood vessels (e.g., by enhancing the effects of NO), lowering blood pressure, and regulation of angiogenesis by affecting multiple pathways and interactions with ion channels, enzymes, transcription factors, and receptors [169, 170]. H2S may, similarly to NO, upregulate the activity of soluble guanyl cyclase (sGC) and increase the level of cGMP. This could be at least in part due to direct inhibition of cGMP phosphodiesterase (PDE5) activity, leading to an increase in the half-life of cGMP [170]. As oxidative stress, inflammation, and angiogenesis, disturbances greatly contribute to the progression of DMD and the leading cause of death is related to dilated cardiomyopathy and other cardiac dysfunctions, below we focus on these aspects of H2S-mediated cytoprotection.

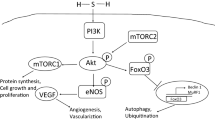
Biochemical functions of H2S. Hydrogen sulfide exerts its cytoprotective properties through several mechanisms. Direct quenching of ROS, enhanced cysteine transport that increases GSH, or post-translational S-sulfhydration of, e.g., p66Shc adaptor protein or SIRT1 contributes to an antioxidant effect. The S-sulfhydration of KEAP1 alters its ability to direct NRF2 to ubiquitylation and proteasomal degradation, thus allowing the accumulation of newly translated NRF2, its nuclear translocation, and increased expression of NRF2-regulated downstream genes such as HMOX1 and NQO1. Activation of cardiac KATP channels mediates the cardioprotective effect of H2S. The anti-inflammatory mechanism of H2S is related to decreased activation and nuclear translocation of NF-κB, resulting in reduced transcription of pro-inflammatory genes, such as TNF-α or IL-6. Furthermore, H2S-mediated cytoprotection can involve activation of AMPK. AMPK also mediates antioxidant and anti-inflammatory effects and contributes to the regulation of autophagy and metabolism. Another mechanism of H2S action is related to its pro-angiogenic activities and may involve VEGFR/PI3K/AKT/eNOS signaling. Neuromodulatory functions are achieved by activating N-methyl-D-aspartate (NMDA) receptors, among other mechanisms. The interaction with NO and CO also contributes to the cytoprotective effect
H2S as an antioxidant
Oxidative stress is caused by the overproduction of ROS relative to cellular antioxidant defense. H2S exerts antioxidant properties through several mechanisms, including direct quenching of ROS [171] and reactive nitrogen species (RNS) [172], increasing antioxidant levels, and regulating the state of certain proteins through S-sulfhydration [137].
An essential pathway associated with maintaining redox balance by H2S is the activation of the nuclear factor-erythroid 2-related factor 2 (NRF2; encoded by the Nfe2l2 gene), which regulates the expression of numerous antioxidant genes [173, 174]. Under normal conditions, NRF2 is located in the cytoplasm and is bound to its negative regulator, the Kelch-like ECH-associated protein 1 (KEAP1) dimer, which targets NRF2 for proteasomal degradation in a ubiquitin-dependent manner. H2S interacts with and modifies KEAP1 through S-sulfhydration, resulting in its conformational change and preventing ubiquitylation of bound NRF2. Newly synthesized NRF2 is not sequestered by KEAP1, but can accumulate in the nucleus, where it binds to the antioxidant response element (ARE) and leads to induction of the expression of NRF2-regulated downstream genes [175,176,177]. One of them is heme oxygenase-1 (HO-1; HMOX1/Hmox1), which exerts anti-inflammatory and antioxidant properties. HO-1 plays a protective role in many pathological conditions, including the cardiovascular, renal, and central nervous systems [178,179,180]. Recently, its role in skeletal muscle differentiation and repair [181,182,183] and the progression of DMD [184] has also been revealed. Other NRF2-regulated genes encode proteins that are strongly involved in the mitigation of oxidative stress with ROS scavenging, such as thioredoxin (TRX), glutathione S-transferase (GST), glutathione peroxidase (GPx), thioredoxin reductase (TRXR), and catalase [185]. The involvement of NRF2 and its downstream effectors in the antioxidative response of H2S has been demonstrated in many studies. For example, the NRF2/HO-1 signaling pathway was activated by NaHS treatment in a rat model of cyclophosphamide-induced toxicity [186]. It should be noted that simultaneously with NRF2 induction, the level of NRF2 targets, such as NAD(P) H quinone oxidoreductase 1 (NQO1), reduced glutathione (GSH), and superoxide dismutase (SOD), also increased. HO-1 was induced by H2S and has cardioprotective effects in rats with volume overload-induced heart failure [187], and in mice with coxsackievirus B3-induced myocarditis [188]. Calvert et al. [189] demonstrated induction of NRF2, HO-1, and Trx-1 in ischemic cardiac tissue after Na2S treatment, leading to a reduction in infarct size, the level of cardiac injury marker, troponin I, and oxidative stress. The protective outcome of H2S-induced HO-1 expression was also shown in pulmonary hypertension [190] and wound healing [191].
Modulation of GSH concentration is another key mechanism in the antioxidant effect of H2S. GSH, a tripeptide, γ-l-glutamyl-l-cysteinylglycine, is synthesized in consecutive reactions catalyzed by two enzymes, glutamate-cysteine ligase (GCL) and glutathione synthetase (GS). Kimura et al. [171] demonstrated that H2S enhances the transport of the sulfur amino acid precursor, cysteine, to increase GSH production. Furthermore, upregulation of the expression of GCL subunits: a catalytic subunit (GCLC) and a modifier subunit (GCLM) by H2S leads to increased intracellular GSH levels [192].
The antioxidant functions of H2S may also involve the regulation of AMPK. The reduction of oxidative stress and apoptosis by NaHS in an experimental model of aging was mediated by activation of the calmodulin-activated protein kinase kinase-β (CaMKKβ)/AMPK pathway [193]. A similar cytoprotective effect of NaHS through activating AMPK was found in a model of dexamethasone-induced osteoblast cell damage [194]. Furthermore, ajoene, a garlic by-product, demonstrated antioxidant properties in hepatic steatosis induced by a high-fat diet (HFD) through stimulation of AMPK signaling [195].
As mentioned above, S-sulfhydration is an important mechanism of H2S-mediated signaling. This post-translational modification of the p66Shc adaptor protein greatly contributes to the antioxidant effect exerted by H2S. p66Shc is involved in mitochondrial redox signaling and its phosphorylation at Ser36 leads to increased ROS production by three different mechanisms in a manner bound to the mitochondria, the plasma membrane, and the nucleus [196, 197]. Oxidative stress associated with p66Shc activity is an important factor in the pathology of many diseases and pathological conditions, including DMD [198], diabetes mellitus, hypercholesterolemia, endothelial dysfunction, and aging (reviewed in [197]). Xie et al. [199] revealed that H2S persulfidates p66Shc at Cys59, which is located near Ser36, preventing the phosphorylation of p66Shc, its translocation to mitochondria, and therefore inhibiting ROS production. These effects were achieved by NaHS treatment and CBS overexpression, which means that exogenous and endogenous H2S reduces oxidative stress. Interestingly, p66Shc is negatively regulated by sirtuin1 (SIRT1) [200]. SIRT1 belongs to the family of nicotine adenine dinucleotide (NAD+)-dependent histone deacetylase proteins, enzymes that catalyze post-translational modifications of many proteins [201]. SIRT1 acts on multiple targets, such as p53, FOXO3, and eNOS, and exhibits diverse biological activities, including negative regulation of oxidative stress (antioxidant properties) [197]. Notably, SIRT1 was suggested to provide beneficial effects in mouse models of DMD through the attenuation of oxidative stress, inflammation, and fibrosis [202]. Increased SIRT1 activity can be induced by direct S-sulfhydration by H2S [203], implying that some of the antioxidant properties of the gasotransmitter can be mediated by regulation of histone deacetylase activity. It is supported by studies in which in vitro application of H2S decreased endoplasmic reticulum stress [204], oxidative stress and senescence [205], while application of SIRT1 inhibitors reversed these effects.
Cardioprotective roles of H2S
The synthesis of endogenous H2S in the heart occurs predominantly through the activity of CSE and substantially less by CBS [206, 207] and the CSE/H2S pathway was shown to be particularly important for the proper functioning of the cardiovascular system. Cardioprotective effects of H2S can result from multiple mechanisms of action, including reduction of oxidative stress, anti-apoptotic and anti-inflammatory effects. Among various possible molecular pathways, H2S-mediated cardioprotection may involve AMPK activation, which was demonstrated in cardiac arrest in mice [208], myocardial ischemia/reperfusion injury [209], cigarette smoking-induced left ventricular dysfunction in rats [210], and high-fat diet-induced diabetic cardiomyopathy [211]. Nevertheless, the influence on ion channels that affect cardiac contractility appears to be the main cardioprotective mechanism of H2S.
ATP-sensitive potassium (KATP) channels are widely distributed and can be found in the heart, muscles, pancreas, and brain [212, 213]. Being highly sensitive to ATP and ADP concentrations, the channels are high-fidelity sensors that match membrane excitability with a given metabolic state of the cell [214]. KATP activation prevents uncontrolled calcium influx, stabilizes membrane potential, and regulates cardiac contractility [215]. Disruption of KATP channel functions has been associated with the progression of many diseases and conditions, including hypertension [216], ischemic heart disease [217], dilated cardiomyopathy [218], or cardiomyopathy in DMD [219]. In myopathic hearts, these channels exhibit abnormal responses to the metabolic state of the cell, particularly to ATP and CK concentrations, resulting in impaired regulation of membrane excitability, which exposes cardiomyocytes to calcium loading and necrosis [220]. Some of these alterations can be mitigated by compounds known to open KATP channels, and interestingly, one of them is H2S [221]. The channels are built from pore-forming subunits, Kir6.x (Kir6.1 or Kir6.2) and a sulfonylurea receptor (SUR1, SUR2, SUR2A, SUR2B), having regulatory activity. H2S was shown to trigger the opening of SUR1 in vascular smooth muscle cells [221] and SUR2B in colonic smooth muscle cells by S-sulfhydration [222]. Mys et al. [223] reported a simultaneous increase in SUR2 and Kir6.1 mRNA levels along with CSE and 3-MST in rat hearts treated with pyridoxal-5-phosphate (PLP) as a cofactor of enzymes that synthesize H2S. This implies that the S-sulfhydration process is also involved in the activation of cardiac channels. The cardioprotective effect of H2S may result from acting through sarcolemmal KATP channels and/or mitochondrial KATP channels. Bian et al. [207] demonstrated the cardioprotective effect of H2S by opening sarcolemmal KATP channels. Increased cell viability and decreased severity and duration of arrhythmias after ischemia/reperfusion in cardiac myocytes treated with NaHS were reported. Blocking sarcolemmal KATP channels reversed the beneficial effects of the H2S donor, whereas inhibiting mitochondrial KATP did not cause any change. On the other hand, Liang et al. [224] showed that the cardioprotective effect of H2S, manifested by increased cell viability, decreased number of apoptotic cells, and reduced oxidative stress, is abolished after inhibition of mitochondrial KATP. Similarly, Testai et al. [225] reported that beneficial effects of the H2S donor in the post-ischemic recovery of the myocardium were antagonized by a mitochondrial KATP channel blocker. Despite such diverse data on the involvement of sarcolemmal and/or mitochondrial KATP channels, H2S-mediated cardioprotective effects are well-documented.
Anti-inflammatory and anti-fibrotic effects of H2S
The cardioprotective properties of H2S may also be related to its anti-inflammatory effect. The inflammatory response induced by tissue damage results in the recruitment of immune cells, followed by a remodeling phase with fibrosis events. Long-term inflammation and defects in the regulatory feedback of inflammatory mediators can facilitate increased deposition of collagen and other matrix proteins, leading to fibrosis and induction of cardiac ventricular dilatation [226]. An important anti-inflammatory mechanism of H2S is related to inhibition of activation and nuclear translocation of NF-κB [227, 228], resulting in reduced transcription of pro-inflammatory genes, such as TNF-α or IL-6. NF-κB is a central mediator of inflammation and immune processes, which was found to be chronically active in many diseases with prolonged inflammation, including DMD [229]. Interestingly, Zhang et al. [230] reported that the reduced recruitment of CD11b+Gr-1+ myeloid cells into the myocardium may be behind one of the mechanisms of the anti-inflammatory effects of H2S with particular cardioprotective importance. Another mechanism involves IL-10/JAK/STAT-3-dependent signaling [231]. Finally, inhibition of inflammation may also be dependent on activation of AMPK [232].
H2S plays a physiological role in the prevention of fibrosis development. This aspect was broadly evaluated in different organs, for example, the kidney. CSE deficiency resulting in decreased H2S and GSH levels caused renal fibrosis with tubular damage, infiltration of inflammatory cells, and deposition of ECM components [233]. On the other hand, exogenous H2S was shown to inhibit the expression of fibrotic cytokines and other mediators and the activation of myofibroblasts that leads to the suppression of renal fibrosis [234]. Other studies evaluated the impact of H2S on cardiac fibrosis. Exogenous delivery of H2S in a form of S-propargyl-cysteine in a special liposomal formulation, which leads to the slow release of the active gasotransmitter, had cardioprotective and anti-fibrotic effects by inhibiting the TGF-β1/SMAD signaling pathway [235]. The same signaling pathway was attenuated by treating spontaneously hypertensive rats with another H2S donor, GYY4137, leading to inhibition of myocardial infarction [236].
Modulation of angiogenesis by H2S
Another protective effect of H2S is related to its pro-angiogenic activities. NaHS treatment increased endothelial cell migration, proliferation, and tube formation in Matrigel in an Akt-dependent way, as well as promoted neovascularization in vivo in the Matrigel plug assay in mice [237]. Similarly, Papapetropoulos et al. demonstrated delayed wound healing and neovascularization in CSE−/− mice, while direct administration of H2S donor to injured skin stimulated wound closure in a rat model. Concomitantly, in vitro studies (using endothelial cells) and ex vivo experiments (done on aortic rings isolated from CSE-deficient mice) underscored the cross talk between H2S synthesis and the pro-angiogenic action of vascular endothelial growth factor (VEGF) [238]. Recent data reveal more details on the molecular mechanisms responsible for the H2S-dependent promotion of angiogenesis. Among them, the involvement of specific microRNAs, for example, upregulation of miR-192 [239] or miR-126-3p [240] has been suggested.
H2S in healthy and diseased skeletal muscles
Initially, mouse skeletal muscles express were shown to express very low levels of H2S-generating enzymes or completely lack these proteins [241]; however, Lu et al. [242] and Ellwood et al. [243] were able to detect the CSE protein by Western blotting in mouse gastrocnemius muscle. Discrepant data were also shown when the level of the third enzyme, 3-MST, was analyzed. Its protein level was very low or even undetectable in mouse skeletal muscles [244], while strong expression has been shown in gastrocnemius muscle of C57BL/10ScSn mice [243]. Zhang et al. [245] found that although CSE, CBS, and 3-MST mRNA were expressed in mouse skeletal muscle, the level of CBS protein was negligible. Although in the work by Zhang et al. [245], both CSE and 3-MST were easily detected at the protein level, additional analysis (for example, with the use of DL-propargylglycine (PPG), a CSE-specific inhibitor) revealed that CSE is the main enzyme in mouse skeletal muscles. In rats, detectable amounts of all three enzymes were evident [246], while in human skeletal muscles, high expression of CBS and CSE has been demonstrated [241]. These variations might be related to several divergences in the applied methodology, e.g., concentration and type of antibodies, but they may also indicate species-dependent regulation. However, an increase in H2S content (by pharmacological treatment or gene delivery of H2S-generating enzymes) may have plausible effects on skeletal muscle biology in various models.
The level of H2S and H2S-generating enzymes declines with age and during various pathological conditions. In the tibialis muscles of 51-week-old mice, CSE expression and H2S production decreased compared to 10-week-old animals. The level of cysteine, the main substrate for H2S production, was also reduced in the muscles of elderly mice [245]. A lower endogenous H2S concentration was detected with a concomitant reduction in NRF2 cytoprotective factor and a significantly increased oxidative stress in the skeletal muscles of patients with critical limb ischemia [247]. H2S levels in serum and muscles and the expression of H2S-generating enzymes were decreased in diabetic mice [242] and rats [248] compared to control animals.
Skeletal muscle-protective effects of H2S
Taking into account the diversity of protective effects exerted by H2S, it is surprising that its role in skeletal muscles and various muscle diseases has not been extensively evaluated. However, recent experiments indicated that H2S can play a role in the regulation of muscle health [123]. Some assumptions on the possible role of H2S in skeletal muscles could be analyzed based on the results of studies in patients with homocystinuria. The decrease in CBS expression/activity in these patients resulted in morphological abnormalities in many organs, including muscles, and represented by fragmented Z disks and disorganized myofilaments with collagen deposits in the basal lamina [249]. Some indications of the importance of H2S in muscles also come from animal models lacking the activity of H2S-generating enzymes. CTH-deficient (Cth−/−) mice on a low cysteine diet were characterized by reduced glutathione levels in skeletal muscles and higher levels of autophagy regulators, LC3 and p62, in skeletal myofibers. Enhanced autophagy was related to acute skeletal muscle atrophy (myopathy) and resulted in severe paralysis of the extremities [250].
Only a few studies describe the molecular mechanism of H2S activity in healthy and diseased skeletal muscles. Bitar et al. [248] concentrated on the evaluation of NaHS treatment on the development of sarcopenia. Such loss of skeletal muscle mass and impaired functions has been described as a complication in diabetic patients; therefore, in this study, Goto Kakizaki (GK) rats (model for type-2 diabetes) with decreased systemic and muscle H2S bioavailability were used. Increased muscle mass and strength and decreased myostatin levels were evident in NaHS-treated diabetic animals compared to controls. In GK rats, an increase in ROS generation was evident, but NaHS delivery resulted in a lower level of superoxide and hydrogen peroxide (H2O2) in the muscle membrane and mitochondrial fractions, as well as better antioxidant capacity measured by the GSH/GSSG ratio [248]. Similar findings were found in vitro in the mouse C2C12 myoblast cell line. After stimulation with NaHS, an increase in GSH level and a reduction in ROS generation were observed [251]. On the other hand, the knockdown of CSE with siRNA resulted in the opposite effects, with increased H2O2 generation and decreased expression of enzymes in the GSH biosynthesis pathway. This indicated that H2S is an important modulator of oxidative balance in myoblasts and skeletal muscles.
Another work, performed in diabetic mice, showed that H2S exerts muscle-protective effects through S-sulfhydration of the muscle RING finger 1 (MuRF1) [242]. MuRF1 (also known as TRIM63) is an E3 ubiquitin ligase, and its increased expression is responsible for muscle mass loss in diabetic conditions. NaHS treatment of db/db mice attenuated skeletal muscle mass atrophy, decreased ROS production, and reduced the degradation of myomesin-1 (MYOM1) and myosin heavy chain 4 (MYH4). Analysis performed in vitro in the C2C12 myoblast cell line revealed that H2S modified MuRF1 by S-sulfhydration at Cys44. This mechanism was suggested to be responsible for the reduction in MYOM1 and MYH4 ubiquitination leading to the attenuation of skeletal muscle atrophy [242].
Zhang et al. [245] performed an interesting study on the evaluation of H2S effect on myogenesis and concluded that this gaseous molecule is capable of inducing regeneration of skeletal muscle. Cardiotoxin-induced injury, as well as age-dependent sarcopenia, were accelerated under conditions of CSE deficiency, while NaHS treatment promoted myogenesis. The mechanism behind this effect was related to the facilitation of heterodimer formation between myogenic factors: myocyte enhancer factor 2 (MEF2c) and myogenic regulatory factor-4 (MRF4) and the promotion of their binding to the myogenin promoter. Therefore, an increase in H2S level might be suggested as a possible treatment of muscle injuries or/and as a preventive factor for age-related sarcopenia.
H2S may also act as an anti-fibrotic agent. Attenuated skeletal muscle fibrosis (decreased mRNA levels of Col1a1, Col1a3, and TGFb, as well as collagen content measured by trichrome staining, and regulation of MMPs) was evident in the mouse contusion model. Furthermore, reduced inflammation (lower levels of pro-inflammatory cytokines and chemokines) and oxidative stress (decreased expression level of a key subunit of NADPH oxidases, gp91phox) were also present in the gastrocnemius muscles [252]. Although this study shows the pleiotropic effect of H2S in reducing skeletal muscle injury, the conclusions are mainly based on the assessment of the level of mRNA and should therefore be interpreted with caution. Nevertheless, the protective effect of H2S against fibrosis was also found in diabetic diaphragms [253]. In the rat model of streptozotocin-induced diabetes, NaHS supplementation reduced collagen deposition, slightly decreased pro-fibrotic Col1a1, Col1a3, and TGFb levels, as well as pro-inflammatory cytokines (IL-1β, IL-6, IL-18, and TNF-α), and finally, suppressed activation of the NLRP3 inflammasome. These effects contributed to the better biomechanical properties of the diaphragm [253].
Another important aspect of the protective effect of H2S on the diaphragm was demonstrated in the ventilator-induced diaphragm dysfunction (VIDD) model [254]. VIDD is a common problem in patients who undergo mechanical ventilation (MV) for the treatment of hypoventilation. The main complications: the decrease in contractile properties of the diaphragm and oxidative stress, which contribute to diaphragm failure, were eliminated by H2S. The H2S donor protected against VIDD by abrogating mitochondrial dysfunction and activation of calpain and caspase-3 protease in diaphragm fibers [254].
Finally, the requirement of H2S for proper muscle vascularization has been underlined [255]. Intramuscular injections with adenoviral vectors-expressing mouse CSE led to higher muscle H2S production and improved muscle functions. The number of CD31-positive blood vessels increased in the gastrocnemius muscle after CSE overexpression, indicating improved vascular density. This study validated CSE as a necessary factor for VEGF-mediated neovascularization in vivo. Also in CBS± mice subjected to hind-limb femoral artery ligation (FAL), treatment with a long-acting H2S donor, GYY4137, resulted in a pro-angiogenic effect and improved muscle-tissue vascularization and blood-vessel function, The H2S donor was also able to counteract the post-FAL-triggered reduction in blood flow and collateral vessel density [256]. Additionally, the downregulation of pro-angiogenic HIF1-α, VEGF, PPAR-γ, and phosphorylated-eNOS was reversed in animals treated with H2S donor. Together, this indicates that H2S can mitigate neoangiogenic defects in skeletal muscles.
The possible roles of H2S in DMD
Hypothetically, many DMD-related complications could potentially be alleviated by H2S due to known mechanisms of gasotransmitter activity. However, until recently, the scientific literature has not broadly addressed the role of H2S in DMD.
The initial study focused on evaluating the cardioprotective potential of H2S. A brief scientific report (published in the form of the conference abstract) by Cain et al. [257] shows that supplementation of ‘humanized’ dystrophic mice (mdx4cv/mTRG2 mouse model with ‘humanized’ telomere lengths) with an orally active slow-release H2S prodrug (SG1002) results in maintenance of the ejection fraction (ET) at a level similar to wild-type mice, indicating preserved cardiac function. Furthermore, a decreasing trend in cardiac fibrosis was observed in the treated animals versus the untreated group. Although these findings need to be confirmed and expanded, we can hypothesize that H2S may support the functionality of dystrophic cardiomyocytes and may be a possible therapeutic option to improve DMD-related cardiomyopathy.
In addition to cardioprotective effects, Cain et al. found attenuated fibrosis in the gastrocnemius and diaphragm after SG1002 treatment [257]. More studies demonstrating the beneficial muscle-related effects of H2S in mitigating the progression of DMD were published in 2021. When the expression of enzymes related to H2S production (CBS, CSE, 3-MST) was analyzed, a significantly decreased level was detected in mdx mice, as well as in human primary myoblasts isolated from DMD donors [258]. Although Ellwood et al. did not observe such a prominent effect in mdx mice, they demonstrated a potent decrease in total sulfide and 3-MST and CSE levels in dystrophin/utrophin double knockout mice (representing a more severe model of DMD than mdx mice) [243]. Additionally, in mdx animals, a reduction in the levels of metabolites associated with the transsulfuration pathway (TSP) such as glycine, glutathione, methionine, glutamate, and taurine was evident [258]. Interestingly, changes were observed in young animals and became more severe with disease progression. Furthermore, it follows that mdx mice exhibit physiological similarities to animal models of cystathioninemia/cystathioninuria (CSE−/− mice), including reduced expression of a major cellular antioxidant, GSH, in skeletal muscles. NaHS treatment, both short-term (2 weeks) and long-term (12 weeks), had a prominent effect in reducing fibrosis, necrosis, and inflammatory-cell infiltration. Furthermore, reactivation of the autophagy process was also evident after H2S delivery. In the quadriceps muscle of mdx mice, normalization of decreased levels of autophagy regulators (Atg3, Atg7, Atg12, and Ulk1), elevated expression of pro-inflammatory cytokines (Il1β, Il6, Tnfα), and pro-fibrotic Tgfb was demonstrated after NaHS. Finally, the effect of H2S on preventing loss of locomotor activity was shown in mdx mice subjected to the rotarod and weight test [258].
Saclier et al. [259] investigated the role of H2S in chronic inflammation in DMD. Using a mouse model (DMDmdx4Cv), they demonstrated that dystrophin-deficient myofibers stimulate a transition of macrophages to a pro-inflammatory phenotype, which is equivalent to induction of muscle fibrosis. NaHS inhibited the development of inflammation, as reflected by a decrease in the number of macrophages expressing pro-inflammatory markers (TNF-α, iNOS), and an increasing percentage of macrophages positive for the anti-inflammatory marker CD206. The mechanism of suppression of inflammation may involve the phosphorylation of the AMPK catalytic α1 subunit, AMPKα1 what leads to the acquisition of the macrophage anti-inflammatory phenotype. Unlike the potent regulation of inflammation, treatment of dystrophic mice with NaHS for 3 weeks caused only a very moderate (approximately 10%) decrease in collagen accumulation in the tibialis anterior, while it did not attenuate fibrosis in the diaphragm [259].
Not only were mouse models applied to check whether H2S donors may have therapeutic potential in DMD. In the C. elegans DMD model with a nonsense mutation at position 3,287 of the DYS-1 dystrophin ortholog, supplementation with H2S donors improved motor skills, contractile force, and muscle mitochondrial structure [243]. Although the overall lifespan of mutated worms was not affected by H2S supplementation, the delay in muscle cell death was evident, suggesting an improvement in healthspan. An interesting observation was found when two different H2S donors were compared: a slow-release sodium GYY4137 (NaGYY) was effective at a considerably higher dose (100 μM) than the mitochondria-targeted donor—AP39 (100 pM). It should be noted that the positive effect with AP39 indicates that the direct action of H2S in the mitochondria is sufficient to exert DMD protective effects. In addition, similar results were obtained when C. elegans was treated with prednisone, a corticosteroid drug approved for use in patients with DMD.
Although data on the influence of H2S donors on the progression of DMD are limited, initial published results strongly support the hypothesis that this gasotransmitter can delay disease progression, by attenuating inflammation and fibrosis and improving muscle strength. Future studies should carefully consider the dose and route of administration of various H2S donors, as well as analyze different DMD models. Despite the fact that already published studies utilized discrepant protocols in terms of dose and duration of treatment with H2S donors, as well as their route of administration and animal age, they suggest that H2S is a beneficial factor in mitigating the dystrophic phenotype (Fig. 7).

Potential roles of H2S in counteracting DMD. Several mechanisms of the cytoprotective effect of H2S on the dystrophic phenotype are suggested. The anti-fibrotic effect is the result of reduced fibrosis-related cytokine production, decreased collagen accumulation, the main component of the ECM, and inhibition of the TGF-β1/SMAD signaling pathway. H2S prevents loss of locomotor activity by enhancing muscle strength. The improvement in the DMD phenotype is also caused by the anti-inflammatory properties of H2S, as indicated not only by an increase in the number of cells expressing anti-inflammatory markers such as CD206, but also by a decrease in the production of pro-inflammatory cytokines and infiltration of inflammatory cells. H2S increases the expression of autophagy regulators and antioxidant enzymes while simultaneously preventing ROS production. Furthermore, it exerts a cardioprotective effect by opening KATP channels, thus reducing calcium loading and preserving cell viability
Limitations and future directions
Despite enormous work in the field of the discovery of new therapeutic strategies as well as optimization of the currently available possibilities to treat DMD, the disease is incurable. Although muscle wasting is one of the main hallmarks of DMD, it is not only a skeletal muscle disorder. Therefore, the ideal drug/approach should regulate many symptoms and be able to deal with all the harmful consequences of dystrophin deficiency in other organs, including the diaphragm and heart.
H2S, due to its anti-inflammatory, antioxidant, anti-fibrotic, pro-angiogenic, and cardioprotective properties, seems to be an attractive candidate. As mentioned above, this gasotransmitter affects many signaling pathways, including AMPK activity. AMPK serves as a metabolic sensor and regulates lipid and glucose metabolism, and metabolic alterations contribute to the progression of DMD [260] and were also recapitulated in mdx mice [261]. Therefore, H2S, in addition to alleviating muscle-related symptoms of DMD, can hypothetically affect dysregulated metabolism. Future studies may help to better understand this possible role of H2S in DMD.
Until now, knowledge of the effects of H2S on skeletal muscles is limited even in animal models of DMD. Although various factors that liberate H2S after in vivo delivery have been described, their effectiveness in patients with DMD has not yet been evaluated. Unfortunately, systematic treatment with such compounds can be accompanied by the generation of reactive by-products, which can counteract the effect of H2S itself [163]. The cooperation of chemists and biologists is required to design and deeply characterize the new donors that will release this cytoprotective gas in a controlled way at a concentration comparable to endogenous H2S production.
Availability of data and material
Not applicable.
Abbreviations
- AMPK:
-
Adenosine 5′-monophosphate-activated protein kinase
- BMD:
-
Becker muscular dystrophy
- CBS:
-
Cystathionine β-synthase
- CK:
-
Creatine kinase
- CSE:
-
Cystathionine γ-lyase
- DAPC:
-
Dystrophin-associated protein complex
- DGC:
-
Dystrophin–glycoprotein complex
- DMD:
-
Duchenne muscular dystrophy
- ECM:
-
Extracellular matrix
- HO-1:
-
Heme oxygenase-1
- LDH:
-
Lactate dehydrogenase
- 3-MST:
-
MPST: 3-mercaptopyruvate sulfurtransferase
- NOS:
-
Nitric oxide synthase
- NRF2:
-
Nuclear factor-erythroid 2-related factor 2
- ROS:
-
Reactive oxygen species
- SCs:
-
Satellite cells
References
Hoffman EP (2020) The discovery of dystrophin, the protein product of the Duchenne muscular dystrophy gene. FEBS J 287:3879–3887. https://doi.org/10.1111/febs.15466
Birnkrant DJ, Bushby K, Bann CM et al (2018) Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 17:251–267. https://doi.org/10.1016/S1474-4422(18)30024-3
Kamdar F, Garry DJ (2016) Dystrophin-deficient cardiomyopathy. J Am Coll Cardiol 67:2533–2546. https://doi.org/10.1016/j.jacc.2016.02.081
Łoboda A, Dulak J (2020) Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: past, present, and future. Pharmacol Rep 72:1227–1263. https://doi.org/10.1007/s43440-020-00134-x
Kieny P, Chollet S, Delalande P et al (2013) Evolution of life expectancy of patients with Duchenne muscular dystrophy at AFM Yolaine de Kepper centre between 1981 and 2011. Ann Phys Rehabil Med 56:443–454. https://doi.org/10.1016/j.rehab.2013.06.002
Florczyk-Soluch U, Polak K, Dulak J (2021) The multifaceted view of heart problem in Duchenne muscular dystrophy. Cell Mol Life Sci 78:5447–5468. https://doi.org/10.1007/s00018-021-03862-2
Mercuri E, Bönnemann CG, Muntoni F (2019) Muscular dystrophies. Lancet 394:2025–2038. https://doi.org/10.1016/S0140-6736(19)32910-1
Duan D, Goemans N, Takeda S et al (2021) Duchenne muscular dystrophy. Nat Rev Dis Prim 7:13. https://doi.org/10.1038/s41572-021-00248-3
Koenig M, Hoffman EP, Bertelson CJ et al (1987) Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50:509–517. https://doi.org/10.1016/0092-8674(87)90504-6
Gao QQ, McNally EM (2015) The dystrophin complex: structure, function, and implications for therapy. Compr Physiol 5:1223–1239. https://doi.org/10.1002/cphy.c140048
Sironi M, Cagliani R, Pozzoli U et al (2002) The dystrophin gene is alternatively spliced throughout its coding sequence. FEBS Lett 517:163–166. https://doi.org/10.1016/S0014-5793(02)02613-3
Ervasti JM (2007) Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta 1772:108–117. https://doi.org/10.1016/j.bbadis.2006.05.010
Bagchi A (2015) Domain wise distribution of mutations in dystrophin protein and Duchenne muscular dystrophy. Gene Technol. https://doi.org/10.4172/2329-6682.1000128
Waldrop MA, Flanigan KM (2019) Update in Duchenne and Becker muscular dystrophy. Curr Opin Neurol 32:722–727. https://doi.org/10.1097/WCO.0000000000000739
Constantin B (2014) Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta 1838:635–642. https://doi.org/10.1016/j.bbamem.2013.08.023
Hoffman EP, Brown RH, Kunkel LM (1987) Dystrophin: the protein product of the duchenne muscular dystrophy locus. Cell 51:919–928. https://doi.org/10.1016/0092-8674(87)90579-4
Hathout Y, Brody E, Clemens PR et al (2015) Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc Natl Acad Sci USA 112:7153–7158. https://doi.org/10.1073/pnas.1507719112
Jackson MJ, Jones DA, Edwards RH (1985) Measurements of calcium and other elements in muscle biopsy samples from patients with Duchenne muscular dystrophy. Clin Chim Acta 147:215–221. https://doi.org/10.1016/0009-8981(85)90202-5
Robert V, Massimino ML, Tosello V et al (2001) Alteration in calcium handling at the subcellular level in mdx myotubes. J Biol Chem 276:4647–4651. https://doi.org/10.1074/jbc.M006337200
Law ML, Cohen H, Martin AA et al (2020) Dysregulation of calcium handling in duchenne muscular dystrophy-associated dilated cardiomyopathy: mechanisms and experimental therapeutic strategies. J Clin Med 9:520. https://doi.org/10.3390/jcm9020520
van Westering T, Betts C, Wood M (2015) Current understanding of molecular pathology and treatment of cardiomyopathy in Duchenne muscular dystrophy. Molecules 20:8823–8855. https://doi.org/10.3390/molecules20058823
Starosta A, Konieczny P (2021) Therapeutic aspects of cell signaling and communication in Duchenne muscular dystrophy. Cell Mol Life Sci 78:4867–4891. https://doi.org/10.1007/s00018-021-03821-x
Tidball JG, Welc SS, Wehling-Henricks M (2018) Immunobiology of inherited muscular dystrophies. Compr Physiol 8:1313–1356. https://doi.org/10.1002/cphy.c170052
Rosenberg AS, Puig M, Nagaraju K et al (2015) Immune-mediated pathology in Duchenne muscular dystrophy. Science Transl Med 7:299rv4–299rv4. https://doi.org/10.1126/scitranslmed.aaa7322
Juban G, Saclier M, Yacoub-Youssef H et al (2018) AMPK activation regulates LTBP4-dependent TGF-β1 secretion by pro-inflammatory macrophages and controls fibrosis in Duchenne muscular dystrophy. Cell Rep 25:2163-2176.e6. https://doi.org/10.1016/j.celrep.2018.10.077
Mann CJ, Perdiguero E, Kharraz Y et al (2011) Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle 1:21. https://doi.org/10.1186/2044-5040-1-21
Magrath P, Maforo N, Renella P et al (2018) Cardiac MRI biomarkers for Duchenne muscular dystrophy. Biomark Med 12:1271–1289. https://doi.org/10.2217/bmm-2018-0125
Natarajan A, Lemos DR, Rossi FMV (2010) Fibro/adipogenic progenitors: a double-edged sword in skeletal muscle regeneration. Cell Cycle 9:2045–2046. https://doi.org/10.4161/cc.9.11.11854
Grounds MD, Terrill JR, Al-Mshhdani BA et al (2020) Biomarkers for Duchenne muscular dystrophy: myonecrosis, inflammation and oxidative stress. Dis Model Mech 13:dmm043638. https://doi.org/10.1242/dmm.043638
von Maltzahn J, Jones AE, Parks RJ, Rudnicki MA (2013) Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc Natl Acad Sci USA 110:16474–16479. https://doi.org/10.1073/pnas.1307680110
Dumont NA, Wang YX, von Maltzahn J et al (2015) Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med 21:1455–1463. https://doi.org/10.1038/nm.3990
Kottlors M, Kirschner J (2010) Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res 340:541–548. https://doi.org/10.1007/s00441-010-0976-6
Sacco A, Mourkioti F, Tran R et al (2010) Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 143:1059–1071. https://doi.org/10.1016/j.cell.2010.11.039
Bronisz-Budzyńska I, Chwalenia K, Mucha O et al (2019) miR-146a deficiency does not aggravate muscular dystrophy in mdx mice. Skelet Muscle 9:22. https://doi.org/10.1186/s13395-019-0207-0
Bronisz-Budzyńska I, Kozakowska M, Podkalicka P et al (2020) The role of Nrf2 in acute and chronic muscle injury. Skelet Muscle 10:35. https://doi.org/10.1186/s13395-020-00255-0
Mucha O, Podkalicka P, Kaziród K et al (2021) Simvastatin does not alleviate muscle pathology in a mouse model of Duchenne muscular dystrophy. Skelet Muscle 11:21. https://doi.org/10.1186/s13395-021-00276-3
De Palma C, Morisi F, Cheli S et al (2014) Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis 5:e1363. https://doi.org/10.1038/cddis.2014.312
Spitali P, Grumati P, Hiller M et al (2013) Autophagy is impaired in the tibialis anterior of dystrophin null mice. PLoS Curr 5:ecurrents.md.e1226cefa851a2f079bbc406c0a21e80. https://doi.org/10.1371/currents.md.e1226cefa851a2f079bbc406c0a21e80
Chang NC, Sincennes M-C, Chevalier FP et al (2018) The dystrophin glycoprotein complex regulates the epigenetic activation of muscle stem cell commitment. Cell Stem Cell 22:755-768.e6. https://doi.org/10.1016/j.stem.2018.03.022
Shin H-JR, Kim H, Oh S et al (2016) AMPK–SKP2–CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534:553–557. https://doi.org/10.1038/nature18014
Sandri M, Coletto L, Grumati P, Bonaldo P (2013) Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci 126:5325–5333. https://doi.org/10.1242/jcs.114041
Mucha O, Kaziród K, Podkalicka P et al (2021) Dysregulated autophagy and mitophagy in a mouse model of duchenne muscular dystrophy remain unchanged following heme oxygenase-1 knockout. Int J Mol Sci 23:470. https://doi.org/10.3390/ijms23010470
Giménez-Xavier P, Francisco R, Platini F et al (2008) LC3-I conversion to LC3-II does not necessarily result in complete autophagy. Int J Mol Med 22:781–785
Moore TM, Lin AJ, Strumwasser AR et al (2020) Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front Physiol 11:690. https://doi.org/10.3389/fphys.2020.00690
Vasconcellos LR, Siqueira MS, Moraes R et al (2018) Heme oxygenase-1 and autophagy linked for cytoprotection. Curr Pharm Des 24:2311–2316. https://doi.org/10.2174/1381612824666180727100909
Kang C, Badr MA, Kyrychenko V et al (2018) Deficit in PINK1/PARKIN-mediated mitochondrial autophagy at late stages of dystrophic cardiomyopathy. Cardiovasc Res 114:90–102. https://doi.org/10.1093/cvr/cvx201
Luan P, D’Amico D, Andreux PA et al (2021) Urolithin A improves muscle function by inducing mitophagy in muscular dystrophy. Sci Transl Med 13:eabb0319. https://doi.org/10.1126/scitranslmed.abb0319
Kuno A, Hosoda R, Sebori R et al (2018) Resveratrol ameliorates mitophagy disturbance and improves cardiac pathophysiology of dystrophin-deficient mdx mice. Sci Rep 8:15555. https://doi.org/10.1038/s41598-018-33930-w
Pauly M, Daussin F, Burelle Y et al (2012) AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol 181:583–592. https://doi.org/10.1016/j.ajpath.2012.04.004
Podkalicka P, Mucha O, Dulak J, Loboda A (2019) Targeting angiogenesis in Duchenne muscular dystrophy. Cell Mol Life Sci 76:1507–1528. https://doi.org/10.1007/s00018-019-03006-7
Christov C, Chrétien F, Abou-Khalil R et al (2007) Muscle satellite cells and endothelial cells: close neighbors and privileged partners. MBoC 18:1397–1409. https://doi.org/10.1091/mbc.e06-08-0693
Harricane M-C, Febris E, Lees D et al (1994) Dystrophin does not influence regular cytoskeletal architecture but is required for contractile performance in smooth muscle aortic cells. Cell Biol Int 18:947–958. https://doi.org/10.1006/cbir.1994.1015
Loufrani L, Matrougui K, Gorny D et al (2001) Flow (shear stress)–induced endothelium-dependent dilation is altered in mice lacking the gene encoding for dystrophin. Circulation 103:864–870. https://doi.org/10.1161/01.CIR.103.6.864
Kodippili K, Thorne PK, Laughlin MH, Duan D (2021) Dystrophin deficiency impairs vascular structure and function in the canine model of Duchenne muscular dystrophy. J Pathol 254:589–605. https://doi.org/10.1002/path.5704
Latroche C, Matot B, Martins-Bach A et al (2015) Structural and functional alterations of skeletal muscle microvasculature in dystrophin-deficient mdx mice. Am J Pathol 185:2482–2494. https://doi.org/10.1016/j.ajpath.2015.05.009
Podkalicka P, Mucha O, Kaziród K et al (2021) Age-dependent dysregulation of muscle vasculature and blood flow recovery after hindlimb ischemia in the mdx model of duchenne muscular dystrophy. Biomedicines 9:481. https://doi.org/10.3390/biomedicines9050481
Cordova G, Negroni E, Cabello-Verrugio C et al (2018) Combined therapies for Duchenne muscular dystrophy to optimize treatment efficacy. Front Genet 9:114. https://doi.org/10.3389/fgene.2018.00114
Arnett AL, Konieczny P, Ramos JN et al (2014) Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol Ther Methods Clin Dev 1:14038. https://doi.org/10.1038/mtm.2014.38
Tabebordbar M, Zhu K, Cheng JKW et al (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351:407–411. https://doi.org/10.1126/science.aad5177
Kwon JB, Ettyreddy AR, Vankara A et al (2020) In vivo gene editing of muscle stem cells with adeno-associated viral vectors in a mouse model of duchenne muscular dystrophy. Mol Ther Methods Clin Dev 19:320–329. https://doi.org/10.1016/j.omtm.2020.09.016
Aartsma-Rus A, Morgan J, Lonkar P et al (2019) Report of a TREAT-NMD/World Duchenne organisation meeting on dystrophin quantification methodology. J Neuromuscul Dis 6:147–159. https://doi.org/10.3233/JND-180357
Harper SQ, Hauser MA, DelloRusso C et al (2002) Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med 8:253–261. https://doi.org/10.1038/nm0302-253
Sakamoto M, Yuasa K, Yoshimura M et al (2002) Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem Biophys Res Commun 293:1265–1272. https://doi.org/10.1016/S0006-291X(02)00362-5
Duan D (2018) Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther 26:2337–2356. https://doi.org/10.1016/j.ymthe.2018.07.011
Mendell JR, Campbell K, Rodino-Klapac L et al (2010) Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med 363:1429–1437. https://doi.org/10.1056/NEJMoa1000228
Mendell JR, Sahenk Z, Lehman K, et al (2020) Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2020.1484
Love DR, Hill DF, Dickson G et al (1989) An autosomal transcript in skeletal muscle with homology to dystrophin. Nature 339:55–58. https://doi.org/10.1038/339055a0
Clerk A, Morris GE, Dubowitz V et al (1993) Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem J 25:554–561
Schofield J, Houzelstein D, Davies K et al (1993) Expression of the dystrophin-related protein (utrophin) gene during mouse embryogenesis. Dev Dyn 198:254–264. https://doi.org/10.1002/aja.1001980403
Matsumura K, Ervasti JM, Ohlendieck K et al (1992) Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature 360:588–591. https://doi.org/10.1038/360588a0
Pearce M, Blake DJ, Tinsley JM et al (1993) The utrophin and dystrophin genes share similarities in genomic structure. Hum Mol Genet 2:1765–1772. https://doi.org/10.1093/hmg/2.11.1765
Li D, Bareja A, Judge L et al (2010) Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J Cell Sci 123:2008–2013. https://doi.org/10.1242/jcs.064808
Guiraud S, Edwards B, Babbs A et al (2019) The potential of utrophin and dystrophin combination therapies for Duchenne muscular dystrophy. Hum Mol Genet 28:2189–2200. https://doi.org/10.1093/hmg/ddz049
Dowling JJ (2016) Eteplirsen therapy for Duchenne muscular dystrophy: skipping to the front of the line. Nat Rev Neurol 12:675–676. https://doi.org/10.1038/nrneurol.2016.180
Ferlini A, Goyenvalle A, Muntoni F (2021) RNA-targeted drugs for neuromuscular diseases. Science 371:29–31. https://doi.org/10.1126/science.aba4515
Dulak J (2021) Gene therapy. The legacy of Wacław Szybalski. Acta Biochim Pol 68:359–375. https://doi.org/10.18388/abp.2020_5805
Dzierlega K, Yokota T (2020) Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther 27:407–416. https://doi.org/10.1038/s41434-020-0156-6
Echigoya Y, Yokota T (2014) Skipping multiple exons of dystrophin transcripts using cocktail antisense oligonucleotides. Nucleic Acid Ther 24:57–68. https://doi.org/10.1089/nat.2013.0451
Aslesh T, Maruyama R, Yokota T (2018) Skipping multiple exons to treat DMD-promises and challenges. Biomedicines. https://doi.org/10.3390/biomedicines6010001
Bushby K, Finkel R, Wong B et al (2014) Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 50:477–487. https://doi.org/10.1002/mus.24332
Nakamura A (2019) Mutation-based therapeutic strategies for Duchenne muscular dystrophy: from genetic diagnosis to therapy. J Pers Med. https://doi.org/10.3390/jpm9010016
Michorowska S (2021) Ataluren-promising therapeutic premature termination codon readthrough frontrunner. Pharmaceuticals (Basel) 14:785. https://doi.org/10.3390/ph14080785
Young CS, Hicks MR, Ermolova NV et al (2016) A Single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 18:533–540. https://doi.org/10.1016/j.stem.2016.01.021
Min Y-L, Bassel-Duby R, Olson EN (2019) CRISPR correction of Duchenne muscular dystrophy. Annu Rev Med 70:239–255. https://doi.org/10.1146/annurev-med-081117-010451
Nelson CE, Hakim CH, Ousterout DG et al (2016) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351:403–407. https://doi.org/10.1126/science.aad5143
Long C, Amoasii L, Mireault AA et al (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351:400–403. https://doi.org/10.1126/science.aad5725
Amoasii L, Hildyard JCW, Li H et al (2018) Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362:86–91. https://doi.org/10.1126/science.aau1549
Moretti A, Fonteyne L, Giesert F et al (2020) Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat Med 26:207–214. https://doi.org/10.1038/s41591-019-0738-2
Karri DR, Zhang Y, Chemello F et al (2022) Long-term maintenance of dystrophin expression and resistance to injury of skeletal muscle in gene edited DMD mice. Mol Ther Nucleic Acids 28:154–167. https://doi.org/10.1016/j.omtn.2022.03.004
Kimberland ML, Hou W, Alfonso-Pecchio A et al (2018) Strategies for controlling CRISPR/Cas9 off-target effects and biological variations in mammalian genome editing experiments. J Biotechnol 284:91–101. https://doi.org/10.1016/j.jbiotec.2018.08.007
Zhang X-H, Tee LY, Wang X-G et al (2015) Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids 4:e264. https://doi.org/10.1038/mtna.2015.37
Olson EN (2021) Toward the correction of muscular dystrophy by gene editing. Proc Natl Acad Sci USA 118:e2004840117. https://doi.org/10.1073/pnas.2004840117
Erkut E, Yokota T (2022) CRISPR therapeutics for Duchenne muscular dystrophy. Int J Mol Sci 23:1832. https://doi.org/10.3390/ijms23031832
Sampaolesi M, Blot S, D’Antona G et al (2006) Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444:574–579. https://doi.org/10.1038/nature05282
Minasi MG, Riminucci M, De Angelis L et al (2002) The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 129:2773–2783. https://doi.org/10.1242/dev.129.11.2773
Cossu G, Previtali SC, Napolitano S et al (2015) Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol Med 7:1513–1528. https://doi.org/10.15252/emmm.201505636
Meng J, Chun S, Asfahani R et al (2014) Human skeletal muscle-derived CD133+ cells form functional satellite cells after intramuscular transplantation in immunodeficient host mice. Mol Ther 22:1008–1017. https://doi.org/10.1038/mt.2014.26
Negroni E, Riederer I, Chaouch S et al (2009) In vivo myogenic potential of human CD133+ muscle-derived stem cells: a quantitative study. Mol Ther 17:1771–1778. https://doi.org/10.1038/mt.2009.167
Torrente Y, Belicchi M, Sampaolesi M et al (2004) Human circulating AC133(+) stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle. J Clin Investig 114:182–195. https://doi.org/10.1172/JCI20325
Blau HM, Daley GQ (2019) Stem cells in the treatment of disease. N Engl J Med 380:1748–1760. https://doi.org/10.1056/NEJMra1716145
Dumont NA, Bentzinger CF, Sincennes M-C, Rudnicki MA (2015) Satellite cells and skeletal muscle regeneration. Compr Physiol 5:1027–1059. https://doi.org/10.1002/cphy.c140068
Mendell JR, Moxley RT, Griggs RC et al (1989) Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med 320:1592–1597. https://doi.org/10.1056/NEJM198906153202405
Kourakis S, Timpani CA, Campelj DG et al (2021) Standard of care versus new-wave corticosteroids in the treatment of Duchenne muscular dystrophy: can we do better? Orphanet J Rare Dis 16:117. https://doi.org/10.1186/s13023-021-01758-9
Biggar WD, Harris VA, Eliasoph L, Alman B (2006) Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord 16:249–255. https://doi.org/10.1016/j.nmd.2006.01.010
Miyatake S, Shimizu-Motohashi Y, Takeda S, Aoki Y (2016) Anti-inflammatory drugs for Duchenne muscular dystrophy: focus on skeletal muscle-releasing factors. Drug Des Dev Ther 10:2745–2758. https://doi.org/10.2147/DDDT.S110163
St-Pierre SJG, Chakkalakal JV, Kolodziejczyk SM et al (2004) Glucocorticoid treatment alleviates dystrophic myofiber pathology by activation of the calcineurin/NF-AT pathway. FASEB J 18:1937–1939. https://doi.org/10.1096/fj.04-1859fje
Kameyama T, Ohuchi K, Funato M et al (2018) Efficacy of prednisolone in generated myotubes derived from fibroblasts of Duchenne muscular dystrophy patients. Front Pharmacol 9:1402. https://doi.org/10.3389/fphar.2018.01402
Quattrocelli M, Barefield DY, Warner JL et al (2017) Intermittent glucocorticoid steroid dosing enhances muscle repair without eliciting muscle atrophy. J Clin Investig 127:2418–2432. https://doi.org/10.1172/JCI91445
Mavrogeni S, Papavasiliou A, Douskou M et al (2009) Effect of deflazacort on cardiac and sternocleidomastoid muscles in Duchenne muscular dystrophy: a magnetic resonance imaging study. Eur J Paediatr Neurol 13:34–40. https://doi.org/10.1016/j.ejpn.2008.02.006
Schram G, Fournier A, Leduc H et al (2013) All-cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol 61:948–954. https://doi.org/10.1016/j.jacc.2012.12.008
Silversides CK, Webb GD, Harris VA, Biggar DW (2003) Effects of deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. Am J Cardiol 91:769–772. https://doi.org/10.1016/s0002-9149(02)03429-x
Heier CR, Damsker JM, Yu Q et al (2013) VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med 5:1569–1585. https://doi.org/10.1002/emmm.201302621
Hoffman EP, Schwartz BD, Mengle-Gaw LJ et al (2019) Vamorolone trial in Duchenne muscular dystrophy shows dose-related improvement of muscle function. Neurology 93:e1312–e1323. https://doi.org/10.1212/WNL.0000000000008168
Smith EC, Conklin LS, Hoffman EP et al (2020) Efficacy and safety of vamorolone in Duchenne muscular dystrophy: An 18-month interim analysis of a non-randomized open-label extension study. PLoS Med 17:e1003222. https://doi.org/10.1371/journal.pmed.1003222
Li X, Conklin LS, van den Anker J et al (2020) Exposure-response analysis of vamorolone (VBP15) in boys with duchenne muscular dystrophy. J Clin Pharmacol 60:1385–1396. https://doi.org/10.1002/jcph.1632
Guglieri M, Bushby K, McDermott MP et al (2022) Effect of different corticosteroid dosing regimens on clinical outcomes in boys with Duchenne muscular dystrophy: a randomized clinical trial. JAMA. https://doi.org/10.1001/jama.2022.4315
Wang R (2002) Two’s company, three’s a crowd: can H 2 S be the third endogenous gaseous transmitter? FASEB j 16:1792–1798. https://doi.org/10.1096/fj.02-0211hyp
Ng PC, Hendry-Hofer TB, Witeof AE et al (2019) Hydrogen sulfide toxicity: mechanism of action, clinical presentation, and countermeasure development. J Med Toxicol 15:287–294. https://doi.org/10.1007/s13181-019-00710-5
Hendriks KD, Maassen H, van Dijk PR et al (2019) Gasotransmitters in health and disease: a mitochondria-centered view. Curr Opin Pharmacol 45:87–93. https://doi.org/10.1016/j.coph.2019.07.001
Wang R (2003) The gasotransmitter role of hydrogen sulfide. Antioxid Redox Signal 5:493–501. https://doi.org/10.1089/152308603768295249
Rose P, Moore PK, Zhu YZ (2017) H2S biosynthesis and catabolism: new insights from molecular studies. Cell Mol Life Sci 74:1391–1412. https://doi.org/10.1007/s00018-016-2406-8
Katsouda A, Bibli S-I, Pyriochou A et al (2016) Regulation and role of endogenously produced hydrogen sulfide in angiogenesis. Pharmacol Res 113:175–185. https://doi.org/10.1016/j.phrs.2016.08.026
Vellecco V, Armogida C, Bucci M (2018) Hydrogen sulfide pathway and skeletal muscle: an introductory review: Hydrogen sulfide and skeletal muscle. Br J Pharmacol 175:3090–3099. https://doi.org/10.1111/bph.14358
Shibuya N, Koike S, Tanaka M et al (2013) A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat Commun 4:1366. https://doi.org/10.1038/ncomms2371
Kimura H (2015) Signaling molecules: hydrogen sulfide and polysulfide. Antioxid Redox Signal 22:362–376. https://doi.org/10.1089/ars.2014.5869
Souza LKM, Araújo TSL, Sousa NA et al (2017) Evidence that d-cysteine protects mice from gastric damage via hydrogen sulfide produced by d-amino acid oxidase. Nitric Oxide 64:1–6. https://doi.org/10.1016/j.niox.2017.01.010
Akaike T, Ida T, Wei F-Y et al (2017) Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun 8:1177. https://doi.org/10.1038/s41467-017-01311-y
Pol A, Renkema GH, Tangerman A et al (2018) Mutations in SELENBP1, encoding a novel human methanethiol oxidase, cause extraoral halitosis. Nat Genet 50:120–129. https://doi.org/10.1038/s41588-017-0006-7
Whiteman M, Moore PK (2009) Hydrogen sulfide and the vasculature: a novel vasculoprotective entity and regulator of nitric oxide bioavailability? J Cell Mol Med 13:488–507. https://doi.org/10.1111/j.1582-4934.2009.00645.x
Cirino G, Szabo C, Papapetropoulos A (2022) Physiological roles of hydrogen sulfide in mammalian cells, tissues and organs. Physiol Rev. https://doi.org/10.1152/physrev.00028.2021
Shefa U, Kim M-S, Jeong NY, Jung J (2018) Antioxidant and cell-signaling functions of hydrogen sulfide in the central nervous system. Oxid Med Cell Longev 2018:1–17. https://doi.org/10.1155/2018/1873962
Bhatia M, Gaddam RR (2021) Hydrogen sulfide in inflammation: a novel mediator and therapeutic target. Antioxid Redox Signal 34:1368–1377. https://doi.org/10.1089/ars.2020.8211
Yan H, Du J, Tang C (2004) The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun 313:22–27. https://doi.org/10.1016/j.bbrc.2003.11.081
Wu D, Hu Q, Zhu D (2018) An update on hydrogen sulfide and nitric oxide interactions in the cardiovascular system. Oxid Med Cell Longev 2018:1–16. https://doi.org/10.1155/2018/4579140
Tang C, Li X, Du J (2006) Hydrogen sulfide as a new endogenous gaseous transmitter in the cardiovascular system. CVP 4:17–22. https://doi.org/10.2174/157016106775203144
Kondo K, Bhushan S, King AL et al (2013) H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 127:1116–1127. https://doi.org/10.1161/CIRCULATIONAHA.112.000855
Zhang D, Du J, Tang C et al (2017) H2S-induced sulfhydration: biological function and detection methodology. Front Pharmacol 8:608. https://doi.org/10.3389/fphar.2017.00608
Panthi S, Chung H-J, Jung J, Jeong NY (2016) Physiological importance of hydrogen sulfide: emerging potent neuroprotector and neuromodulator. Oxid Med Cell Longev 2016:1–11. https://doi.org/10.1155/2016/9049782
Wang R (2012) Physiological implications of hydrogen sulfide: a Whiff exploration that blossomed. Physiol Rev 92:791–896. https://doi.org/10.1152/physrev.00017.2011
Magli E, Perissutti E, Santagada V et al (2021) H2S donors and their use in medicinal chemistry. Biomolecules 11:1899. https://doi.org/10.3390/biom11121899
Kida K, Yamada M, Tokuda K et al (2011) Inhaled hydrogen sulfide prevents neurodegeneration and movement disorder in a mouse model of Parkinson’s disease. Antioxid Redox Signal 15:343–352. https://doi.org/10.1089/ars.2010.3671
Xue R, Hao D-D, Sun J-P et al (2013) Hydrogen sulfide treatment promotes glucose uptake by increasing insulin receptor sensitivity and ameliorates kidney lesions in type 2 diabetes. Antioxid Redox Signal 19:5–23. https://doi.org/10.1089/ars.2012.5024
Powell CR, Dillon KM, Matson JB (2018) A review of hydrogen sulfide (H2S) donors: chemistry and potential therapeutic applications. Biochem Pharmacol 149:110–123. https://doi.org/10.1016/j.bcp.2017.11.014
Chen Y, Teng X, Hu Z et al (2021) Hydrogen sulfide attenuated sepsis-induced myocardial dysfunction through TLR4 pathway and endoplasmic reticulum stress. Front Physiol 12:653601. https://doi.org/10.3389/fphys.2021.653601
Tian D, Teng X, Jin S et al (2021) Endogenous hydrogen sulfide improves vascular remodeling through PPARδ/SOCS3 signaling. J Adv Res 27:115–125. https://doi.org/10.1016/j.jare.2020.06.005
Hughes MN, Centelles MN, Moore KP (2009) Making and working with hydrogen sulfide: the chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: a review. Free Radic Biol Med 47:1346–1353. https://doi.org/10.1016/j.freeradbiomed.2009.09.018
Feng W, Teo X-Y, Novera W et al (2015) Discovery of new H2S releasing phosphordithioates and 2,3-dihydro-2-phenyl-2-sulfanylenebenzo[d][1,3,2]oxazaphospholes with improved antiproliferative activity. J Med Chem 58:6456–6480. https://doi.org/10.1021/acs.jmedchem.5b00848
Li L, Whiteman M, Guan YY et al (2008) Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation 117:2351–2360. https://doi.org/10.1161/CIRCULATIONAHA.107.753467
Wu Z, Peng H, Du Q et al (2015) GYY4137, a hydrogen sulfide-releasing molecule, inhibits the inflammatory response by suppressing the activation of nuclear factor-kappa B and mitogen-activated protein kinases in Coxsackie virus B3-infected rat cardiomyocytes. Mol Med Rep 11:1837–1844. https://doi.org/10.3892/mmr.2014.2901
Ellmers LJ, Templeton EM, Pilbrow AP et al (2020) Hydrogen sulfide treatment improves post-infarct remodeling and long-term cardiac function in CSE knockout and wild-type mice. IJMS 21:4284. https://doi.org/10.3390/ijms21124284
Lee ZW, Zhou J, Chen C-S et al (2011) The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE 6:e21077. https://doi.org/10.1371/journal.pone.0021077
Li J, Li M, Li L et al (2022) Hydrogen sulfide attenuates ferroptosis and stimulates autophagy by blocking mTOR signaling in sepsis-induced acute lung injury. Mol Immunol 141:318–327. https://doi.org/10.1016/j.molimm.2021.12.003
Ahmad A, Olah G, Szczesny B et al (2016) AP39, a mitochondrially- targeted hydrogen sulfide donor, exerts protective effects in renal epithelial cells subjected to oxidative stress in vitro and in acute renal injury in vivo. Shock 45:88–97. https://doi.org/10.1097/SHK.0000000000000478
Szczesny B, Módis K, Yanagi K et al (2014) AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 41:120–130. https://doi.org/10.1016/j.niox.2014.04.008
Ikeda K, Marutani E, Hirai S et al (2015) Mitochondria-targeted hydrogen sulfide donor AP39 improves neurological outcomes after cardiac arrest in mice. Nitric Oxide 49:90–96. https://doi.org/10.1016/j.niox.2015.05.001
Karwi QG, Bornbaum J, Boengler K et al (2017) AP39, a mitochondria-targeting hydrogen sulfide (H2S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br J Pharmacol 174:287–301. https://doi.org/10.1111/bph.13688
Wallace JL, Nagy P, Feener TD et al (2020) A proof-of-concept, phase 2 clinical trial of the gastrointestinal safety of a hydrogen sulfide-releasing anti-inflammatory drug. Br J Pharmacol 177:769–777. https://doi.org/10.1111/bph.14641
Gemici B, Elsheikh W, Feitosa KB et al (2015) H2S-releasing drugs: anti-inflammatory, cytoprotective and chemopreventative potential. Nitric Oxide 46:25–31. https://doi.org/10.1016/j.niox.2014.11.010
Costa SKPF, Muscara MN, Allain T et al (2020) Enhanced analgesic effects and gastrointestinal safety of a novel, hydrogen sulfide-releasing anti-inflammatory drug (ATB-352): a role for endogenous cannabinoids. Antioxid Redox Signal 33:1003–1009. https://doi.org/10.1089/ars.2019.7884
Polhemus DJ, Li Z, Pattillo CB et al (2015) A novel hydrogen sulfide prodrug, SG 1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc Ther 33:216–226. https://doi.org/10.1111/1755-5922.12128
Kaur K, Enders P, Zhu Y et al (2021) Amino acid-based H2S donors: N-thiocarboxyanhydrides that release H2S with innocuous byproducts. Chem Commun 57:5522–5525. https://doi.org/10.1039/D1CC01309B
Levinn CM, Cerda MM, Pluth MD (2020) Activatable small-molecule hydrogen sulfide donors. Antioxid Redox Signal 32:96–109. https://doi.org/10.1089/ars.2019.7841
Corvino A, Frecentese F, Magli E et al (2021) Trends in H2S-donors chemistry and their effects in cardiovascular diseases. Antioxidants (Basel) 10:429. https://doi.org/10.3390/antiox10030429
Benavides GA, Squadrito GL, Mills RW et al (2007) Hydrogen sulfide mediates the vasoactivity of garlic. Proc Natl Acad Sci USA 104:17977–17982. https://doi.org/10.1073/pnas.0705710104
Sen U, Sathnur PB, Kundu S et al (2012) Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am J Physiol Cell Physiol 303:C41–C51. https://doi.org/10.1152/ajpcell.00398.2011
Shaposhnikov M, Proshkina E, Koval L et al (2018) Overexpression of CBS and CSE genes affects lifespan, stress resistance and locomotor activity in Drosophila melanogaster. Aging (Albany NY) 10:3260–3272. https://doi.org/10.18632/aging.101630
London J, Ndiaye FK, Bui LC et al (2019) Alterations in the serotonin and dopamine pathways by cystathionine beta synthase overexpression in murine brain. Mol Neurobiol 56:3958–3971. https://doi.org/10.1007/s12035-018-1323-2
Marechal D, Brault V, Leon A et al (2019) Cbs overdosage is necessary and sufficient to induce cognitive phenotypes in mouse models of Down syndrome and interacts genetically with Dyrk1a. Hum Mol Genet 28:1561–1577. https://doi.org/10.1093/hmg/ddy447
Szabó C (2007) Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov 6:917–935. https://doi.org/10.1038/nrd2425
Szabo C (2017) Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: mechanisms and implications. Am J Physiol, Cell Physiol 312:C3–C15. https://doi.org/10.1152/ajpcell.00282.2016
Kimura Y, Goto Y-I, Kimura H (2010) Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal 12:1–13. https://doi.org/10.1089/ars.2008.2282
Nagy P, Winterbourn CC (2010) Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem Res Toxicol 23:1541–1543. https://doi.org/10.1021/tx100266a
Corsello T, Komaravelli N, Casola A (2018) Role of hydrogen sulfide in NRF2- and sirtuin-dependent maintenance of cellular redox balance. Antioxidants 7:129. https://doi.org/10.3390/antiox7100129
Eleftheriadis T, Pissas G, Nikolaou E et al (2020) Mistimed H2S upregulation, Nrf2 activation and antioxidant proteins levels in renal tubular epithelial cells subjected to anoxia and reoxygenation. Biom Rep. https://doi.org/10.3892/br.2020.1309
Yang G, Zhao K, Ju Y et al (2013) Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal 18:1906–1919. https://doi.org/10.1089/ars.2012.4645
Hourihan JM, Kenna JG, Hayes JD (2013) The gasotransmitter hydrogen sulfide induces Nrf2-target genes by inactivating the Keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between Cys-226 and Cys-613. Antioxid Redox Signal 19:465–481. https://doi.org/10.1089/ars.2012.4944
Tebay LE, Robertson H, Durant ST et al (2015) Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med 88:108–146. https://doi.org/10.1016/j.freeradbiomed.2015.06.021
Dulak J, Deshane J, Jozkowicz A, Agarwal A (2008) Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation 117:231–241. https://doi.org/10.1161/CIRCULATIONAHA.107.698316
Loboda A, Jazwa A, Grochot-Przeczek A et al (2008) Heme oxygenase-1 and the vascular bed: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 10:1767–1812. https://doi.org/10.1089/ars.2008.2043
Loboda A, Damulewicz M, Pyza E et al (2016) Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci 73:3221–3247. https://doi.org/10.1007/s00018-016-2223-0
Kozakowska M, Ciesla M, Stefanska A et al (2012) Heme oxygenase-1 inhibits myoblast differentiation by targeting myomirs. Antioxid Redox Signal 16:113–127. https://doi.org/10.1089/ars.2011.3964
Kozakowska M, Pietraszek-Gremplewicz K, Ciesla M et al (2018) Lack of heme oxygenase-1 induces inflammatory reaction and proliferation of muscle satellite cells after cardiotoxin-induced skeletal muscle injury. Am J Pathol 188:491–506. https://doi.org/10.1016/j.ajpath.2017.10.017
Alves de Souza RW, Gallo D, Lee GR et al (2021) Skeletal muscle heme oxygenase-1 activity regulates aerobic capacity. Cell Rep 35:109018. https://doi.org/10.1016/j.celrep.2021.109018
Pietraszek-Gremplewicz K, Kozakowska M, Bronisz-Budzynska I et al (2018) Heme oxygenase-1 influences satellite cells and progression of duchenne muscular dystrophy in mice. Antioxid Redox Signal 29:128–148. https://doi.org/10.1089/ars.2017.7435
Xiao Q, Ying J, Xiang L, Zhang C (2018) The biologic effect of hydrogen sulfide and its function in various diseases. Medicine 97:e13065. https://doi.org/10.1097/MD.0000000000013065
Waz S, Heeba GH, Hassanin SO, Abdel-Latif RG (2021) Nephroprotective effect of exogenous hydrogen sulfide donor against cyclophosphamide-induced toxicity is mediated by Nrf2/HO-1/NF-κB signaling pathway. Life Sci 264:118630. https://doi.org/10.1016/j.lfs.2020.118630
Zhang C-Y, Li X-H, Zhang T et al (2013) Hydrogen sulfide upregulates heme oxygenase-1 expression in rats with volume overload-induced heart failure. Biomed Rep 1:454–458. https://doi.org/10.3892/br.2013.87
Hua W, Chen Q, Gong F et al (2013) Cardioprotection of H2S by downregulating iNOS and upregulating HO-1 expression in mice with CVB3-induced myocarditis. Life Sci 93:949–954. https://doi.org/10.1016/j.lfs.2013.10.007
Calvert JW, Jha S, Gundewar S et al (2009) Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res 105:365–374. https://doi.org/10.1161/CIRCRESAHA.109.199919
Qingyou Z, Junbao D, Weijin Z et al (2004) Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem Biophys Res Commun 317:30–37. https://doi.org/10.1016/j.bbrc.2004.02.176
Wang G, Li W, Chen Q et al (2015) Hydrogen sulfide accelerates wound healing in diabetic rats. Int J Clin Exp Pathol 8:5097–5104
Jain SK, Huning L, Micinski D (2014) Hydrogen sulfide upregulates glutamate-cysteine ligase catalytic subunit, glutamate-cysteine ligase modifier subunit, and glutathione and inhibits interleukin-1β secretion in monocytes exposed to high glucose levels. Metab Syndr Relat Disord 12:299–302. https://doi.org/10.1089/met.2014.0022
Chen X, Zhao X, Cai H et al (2017) The role of sodium hydrosulfide in attenuating the aging process via PI3K/AKT and CaMKKβ/AMPK pathways. Redox Biol 12:987–1003. https://doi.org/10.1016/j.redox.2017.04.031
Yang M, Huang Y, Chen J et al (2014) Activation of AMPK participates hydrogen sulfide-induced cyto-protective effect against dexamethasone in osteoblastic MC3T3-E1 cells. Biochem Biophys Res Commun 454:42–47. https://doi.org/10.1016/j.bbrc.2014.10.033
Han CY, Ki SH, Kim YW et al (2011) Ajoene, a stable garlic by-product, inhibits high fat diet-induced hepatic steatosis and oxidative injury through LKB1-dependent AMPK activation. Antioxid Redox Signal 14:187–202. https://doi.org/10.1089/ars.2010.3190
Gertz M, Steegborn C (2010) The lifespan-regulator p66Shc in mitochondria: redox enzyme or redox sensor? Antioxid Redox Signal 13:1417–1428. https://doi.org/10.1089/ars.2010.3147
Magenta A, Greco S, Capogrossi MC et al (2014) Nitric oxide, oxidative stress, and p 66 S h c interplay in diabetic endothelial dysfunction. Biomed Res Int 2014:1–16. https://doi.org/10.1155/2014/193095
D’Agostino M, Torcinaro A, Madaro L et al (2018) Role of miR-200c in myogenic differentiation impairment via p66Shc: implication in skeletal muscle regeneration of dystrophic mdx mice. Oxid Med Cell Longev 2018:1–10. https://doi.org/10.1155/2018/4814696
Xie Z-Z, Shi M-M, Xie L et al (2014) Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxid Redox Signal 21:2531–2542. https://doi.org/10.1089/ars.2013.5604
Chen H-Z, Wan Y-Z, Liu D-P (2013) Cross-talk between SIRT1 and p66Shc in vascular diseases. Trends Cardiovasc Med 23:237–241. https://doi.org/10.1016/j.tcm.2013.01.001
Buler M, Andersson U, Hakkola J (2016) Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB j 30:3942–3960. https://doi.org/10.1096/fj.201600410RR
Kuno A, Horio Y (2016) SIRT1: a novel target for the treatment of muscular dystrophies. Oxid Med Cell Longev 2016:6714686. https://doi.org/10.1155/2016/6714686
Du C, Lin X, Xu W et al (2019) Sulfhydrated sirtuin-1 increasing its deacetylation activity is an essential epigenetics mechanism of anti-atherogenesis by hydrogen sulfide. Antioxid Redox Signal 30:184–197. https://doi.org/10.1089/ars.2017.7195
Li X, Zhang K-Y, Zhang P et al (2014) Hydrogen sulfide inhibits formaldehyde-induced endoplasmic reticulum stress in PC12 cells by upregulation of SIRT-1. PLoS ONE 9:e89856. https://doi.org/10.1371/journal.pone.0089856
Suo R, Zhao Z-Z, Tang Z-H et al (2013) Hydrogen sulfide prevents H2O2-induced senescence in human umbilical vein endothelial cells through SIRT1 activation. Mol Med Rep 7:1865–1870. https://doi.org/10.3892/mmr.2013.1417
Geng B, Yang J, Qi Y et al (2004) H2S generated by heart in rat and its effects on cardiac function. Biochem Biophys Res Commun 313:362–368. https://doi.org/10.1016/j.bbrc.2003.11.130
Bian J-S, Yong QC, Pan T-T et al (2006) Role of hydrogen sulfide in the cardioprotection caused by ischemic preconditioning in the rat heart and cardiac myocytes. J Pharmacol Exp Ther 316:670–678. https://doi.org/10.1124/jpet.105.092023
Minamishima S, Bougaki M, Sips PY et al (2009) Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3 dependent mechanism in mice. Circulation 120:888–896. https://doi.org/10.1161/CIRCULATIONAHA.108.833491
Xie H, Xu Q, Jia J et al (2015) Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem Biophys Res Commun 458:632–638. https://doi.org/10.1016/j.bbrc.2015.02.017
Zhou X, An G, Chen J (2014) Hydrogen sulfide improves left ventricular function in smoking rats via regulation of apoptosis and autophagy. Apoptosis 19:998–1005. https://doi.org/10.1007/s10495-014-0978-z
Barr LA, Shimizu Y, Lambert JP et al (2015) Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide 46:145–156. https://doi.org/10.1016/j.niox.2014.12.013
Rapposelli S (2011) Novel adenosine 5′-triphosphate-sensitive potassium channel ligands: a patent overview (2005–2010). Expert Opin Ther Pat 21:355–379. https://doi.org/10.1517/13543776.2011.553601
Roy Chowdhury U, Dosa PI, Fautsch MP (2017) ATP sensitive potassium channel openers: a new class of ocular hypotensive agents. Exp Eye Res 158:85–93. https://doi.org/10.1016/j.exer.2016.04.020
Liu Z, Cai H, Dang Y et al (2016) Adenosine triphosphate-sensitive potassium channels and cardiomyopathies (Review). Mol Med Rep 13:1447–1454. https://doi.org/10.3892/mmr.2015.4714
Voitychuk OI, Strutynskyi RB, Yagupolskii LM et al (2011) Sarcolemmal cardiac KATP channels as a target for the cardioprotective effects of the fluorine-containing pinacidil analogue, flocalin: cardioprotective effects of flocalin. Br J Pharmacol 162:701–711. https://doi.org/10.1111/j.1476-5381.2010.01072.x
McNair A, Andreasen F, Nielsen PE (1983) Antihypertensive effect of diazoxide given intravenously in small repeated doses. Eur J Clin Pharmacol 24:151–156. https://doi.org/10.1007/BF00613809
Jahangir A, Terzic A (2005) K channel therapeutics at the bedside. J Mol Cell Cardiol 39:99–112. https://doi.org/10.1016/j.yjmcc.2005.04.006
Bienengraeber M, Olson TM, Selivanov VA et al (2004) ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet 36:382–387. https://doi.org/10.1038/ng1329
Graciotti L, Becker J, Granata AL et al (2011) Dystrophin is required for the normal function of the cardio-protective KATP channel in cardiomyocytes. PLoS ONE 6:e27034. https://doi.org/10.1371/journal.pone.0027034
Hodgson DM (2003) Cellular remodeling in heart failure disrupts KATP channel-dependent stress tolerance. EMBO J 22:1732–1742. https://doi.org/10.1093/emboj/cdg192
Jiang B, Tang G, Cao K et al (2010) Molecular mechanism for H2S-induced activation of KATP channels. Antioxid Redox Signal 12:1167–1178. https://doi.org/10.1089/ars.2009.2894
Gade AR, Kang M, Akbarali HI (2013) Hydrogen sulfide as an allosteric modulator of ATP-sensitive potassium channels in colonic inflammation. Mol Pharmacol 83:294–306. https://doi.org/10.1124/mol.112.081596
Mys L, Goshovska Y, Strutynska N et al (2022) Pyridoxal-5-phosphate induced cardioprotection in aging associated with up-expression of cystathionine-γ-lyase, 3-mercaptopyruvate sulfurtransferase, and ATP-sensitive potassium channels. Eur J Clin Investig. https://doi.org/10.1111/eci.13683
Liang W, Chen J, Mo L et al (2016) ATP-sensitive K+ channels contribute to the protective effects of exogenous hydrogen sulfide against high glucose-induced injury in H9c2 cardiac cells. Int J Mol Med 37:763–772. https://doi.org/10.3892/ijmm.2016.2467
Testai L, Marino A, Piano I et al (2016) The novel H2S-donor 4-carboxyphenyl isothiocyanate promotes cardioprotective effects against ischemia/reperfusion injury through activation of mitoK ATP channels and reduction of oxidative stress. Pharmacol Res 113:290–299. https://doi.org/10.1016/j.phrs.2016.09.006
Fioranelli M, Bottaccioli AG, Bottaccioli F et al (2018) Stress and inflammation in coronary artery disease: a review psychoneuroendocrineimmunology-based. Front Immunol 9:2031. https://doi.org/10.3389/fimmu.2018.02031
Benedetti F, Curreli S, Krishnan S et al (2017) Anti-inflammatory effects of H2S during acute bacterial infection: a review. J Transl Med 15:100. https://doi.org/10.1186/s12967-017-1206-8
Bourque C, Zhang Y, Fu M et al (2018) H2S protects lipopolysaccharide-induced inflammation by blocking NFκB transactivation in endothelial cells. Toxicol Appl Pharmacol 338:20–29. https://doi.org/10.1016/j.taap.2017.11.004
Peterson JM, Wang DJ, Shettigar V et al (2018) NF-κB inhibition rescues cardiac function by remodeling calcium genes in a Duchenne muscular dystrophy model. Nat Commun 9:3431. https://doi.org/10.1038/s41467-018-05910-1
Zhang Y, Li H, Zhao G et al (2015) Hydrogen sulfide attenuates the recruitment of CD11b+Gr-1+ myeloid cells and regulates Bax/Bcl-2 signaling in myocardial ischemia injury. Sci Rep 4:4774. https://doi.org/10.1038/srep04774
Tokuda K, Kida K, Marutani E et al (2012) Inhaled hydrogen sulfide prevents endotoxin-induced systemic inflammation and improves survival by altering sulfide metabolism in mice. Antioxid Redox Signal 17:11–21. https://doi.org/10.1089/ars.2011.4363
Zhou X, Cao Y, Ao G et al (2014) CaMKKβ-dependent activation of AMP-activated protein kinase is critical to suppressive effects of hydrogen sulfide on neuroinflammation. Antioxid Redox Signal 21:1741–1758. https://doi.org/10.1089/ars.2013.5587
Han SJ, Noh MR, Jung J-M et al (2017) Hydrogen sulfide-producing cystathionine γ-lyase is critical in the progression of kidney fibrosis. Free Radic Biol Med 112:423–432. https://doi.org/10.1016/j.freeradbiomed.2017.08.017
Wang Y, Xing Q-Q, Tu J-K et al (2019) Involvement of hydrogen sulfide in the progression of renal fibrosis. Chin Med J (Engl) 132:2872–2880. https://doi.org/10.1097/CM9.0000000000000537
Tran BH, Yu Y, Chang L et al (2019) A novel liposomal S-propargyl-cysteine: a sustained release of hydrogen sulfide reducing myocardial fibrosis via TGF-β1/Smad pathway. Int J Nanomed 14:10061–10077. https://doi.org/10.2147/IJN.S216667
Meng G, Zhu J, Xiao Y et al (2015) Hydrogen sulfide donor GYY4137 protects against myocardial fibrosis. Oxid Med Cell Longev 2015:691070. https://doi.org/10.1155/2015/691070
Cai W-J, Wang M-J, Moore PK et al (2007) The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res 76:29–40. https://doi.org/10.1016/j.cardiores.2007.05.026
Papapetropoulos A, Pyriochou A, Altaany Z et al (2009) Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA 106:21972–21977. https://doi.org/10.1073/pnas.0908047106
Zhou Y, Li X, Xue W-L et al (2022) YB-1 recruits Drosha to promote splicing of pri-miR-192 to mediate the proangiogenic effects of H2S. Antioxid Redox Signal. https://doi.org/10.1089/ars.2021.0105
Xue W-L, Chen R-Q, Zhang Q-Q et al (2020) Hydrogen sulfide rescues high glucose-induced migration dysfunction in HUVECs by upregulating miR-126-3p. Am J Physiol Cell Physiol 318:C857–C869. https://doi.org/10.1152/ajpcell.00406.2019
Chen NC, Yang F, Capecci LM et al (2010) Regulation of homocysteine metabolism and methylation in human and mouse tissues. FASEB J 24:2804–2817. https://doi.org/10.1096/fj.09-143651
Lu F, Lu B, Zhang L et al (2020) Hydrogen sulphide ameliorating skeletal muscle atrophy in db/db mice via Muscle RING finger 1 S-sulfhydration. J Cell Mol Med 24:9362–9377. https://doi.org/10.1111/jcmm.15587
Ellwood RA, Hewitt JE, Torregrossa R et al (2021) Mitochondrial hydrogen sulfide supplementation improves health in the C. elegans Duchenne muscular dystrophy model. Proc Natl Acad Sci USA 118:e2018342118. https://doi.org/10.1073/pnas.2018342118
Veeranki S, Tyagi SC (2015) Role of hydrogen sulfide in skeletal muscle biology and metabolism. Nitric Oxide 46:66–71. https://doi.org/10.1016/j.niox.2014.11.012
Zhang Y, Masters L, Wang Y et al (2021) Cystathionine gamma-lyase/H2S signaling facilitates myogenesis under aging and injury condition. FASEB J 35:e21511. https://doi.org/10.1096/fj.202002675R
Du J, Li W, Yang J et al (2013) Hydrogen sulfide is endogenously generated in rat skeletal muscle and exerts a protective effect against oxidative stress. Chin Med J (Engl) 126:930–936
Islam KN, Polhemus DJ, Donnarumma E et al (2015) Hydrogen sulfide levels and nuclear factor-erythroid 2-related factor 2 (NRF2) activity are attenuated in the setting of critical limb ischemia (CLI). J Am Heart Assoc 4:e001986. https://doi.org/10.1161/JAHA.115.001986
Bitar MS, Nader J, Al-Ali W et al (2018) Hydrogen sulfide donor NaHS improves metabolism and reduces muscle atrophy in type 2 diabetes: implication for understanding sarcopenic pathophysiology. Oxid Med Cell Longev 2018:6825452. https://doi.org/10.1155/2018/6825452
Kanwar YS, Manaligod JR, Wong PW (1976) Morphologic studies in a patient with homocystinuria due to 5, 10-methylenetetrahydrofolate reductase deficiency. Pediatr Res 10:598–609. https://doi.org/10.1203/00006450-197606000-00008
Ishii I, Akahoshi N, Yamada H et al (2010) Cystathionine gamma-Lyase-deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem 285:26358–26368. https://doi.org/10.1074/jbc.M110.147439
Parsanathan R, Jain SK (2018) Hydrogen sulfide increases glutathione biosynthesis, and glucose uptake and utilisation in C2C12 mouse myotubes. Free Radic Res 52:288–303. https://doi.org/10.1080/10715762.2018.1431626
Zhao L, Liu X, Zhang J et al (2020) Hydrogen sulfide alleviates skeletal muscle fibrosis via attenuating inflammation and oxidative stress. Front Physiol 11:533690. https://doi.org/10.3389/fphys.2020.533690
Yang R, Jia Q, Li Y, Mehmood S (2020) Protective effect of exogenous hydrogen sulfide on diaphragm muscle fibrosis in streptozotocin-induced diabetic rats. Exp Biol Med (Maywood) 245:1280–1289. https://doi.org/10.1177/1535370220931038
Ichinoseki-Sekine N, Smuder AJ, Morton AB et al (2021) Hydrogen sulfide donor protects against mechanical ventilation-induced atrophy and contractile dysfunction in the rat diaphragm. Clin Transl Sci 14:2139–2145. https://doi.org/10.1111/cts.13081
Longchamp A, Mirabella T, Arduini A et al (2018) Amino acid restriction triggers angiogenesis via GCN2/ATF4 regulation of VEGF and H2S production. Cell 173:117-129.e14. https://doi.org/10.1016/j.cell.2018.03.001
Majumder A, Singh M, George AK et al (2018) Hydrogen sulfide improves postischemic neoangiogenesis in the hind limb of cystathionine‐β‐synthase mutant mice via PPAR‐γ/VEGF axis. Physiol Rep 6:e13858. https://doi.org/10.14814/phy2.13858
Cain C, Devarakonda T, Thompson J et al (2019) Prevention and treatment of Duchenne cardiomyopathy with hydrogen sulfide-donor therapy. FASEB J. https://doi.org/10.1096/fasebj.2019.33.1_supplement.831.5
Panza E, Vellecco V, Iannotti FA et al (2021) Duchenne’s muscular dystrophy involves a defective transsulfuration pathway activity. Redox Biol 45:102040. https://doi.org/10.1016/j.redox.2021.102040
Saclier M, Ben Larbi S, My Ly H et al (2021) Interplay between myofibers and pro-inflammatory macrophages controls muscle damage in mdx mice. J Cell Sci 134:jcs258429. https://doi.org/10.1242/jcs.258429
Rodríguez-Cruz M, Sanchez R, Escobar RE et al (2015) Evidence of insulin resistance and other metabolic alterations in boys with duchenne or becker muscular dystrophy. Int J Endocrinol 2015:867273. https://doi.org/10.1155/2015/867273
Podkalicka P, Mucha O, Kaziród K et al (2022) miR-378 affects metabolic disturbances in the mdx model of Duchenne muscular dystrophy. Sci Rep 12:3945. https://doi.org/10.1038/s41598-022-07868-z
Tomasova L, Drapala A, Jurkowska H et al (2017) Na2S, a fast-releasing H2S donor, given as suppository lowers blood pressure in rats. Pharmacol Rep 69:971–977. https://doi.org/10.1016/j.pharep.2017.03.021
Rose P, Dymock BW, Moore PK (2015) GYY4137, a novel water-soluble, H2S-releasing molecule. Methods Enzymol 554:143–167. https://doi.org/10.1016/bs.mie.2014.11.014
Huang CW, Feng W, Peh MT et al (2016) A novel slow-releasing hydrogen sulfide donor, FW1256, exerts anti-inflammatory effects in mouse macrophages and in vivo. Pharmacol Res 113:533–546. https://doi.org/10.1016/j.phrs.2016.09.032
Xu S, Yang C-T, Meng F-H et al (2016) Ammonium tetrathiomolybdate as a water-soluble and slow-release hydrogen sulfide donor. Bioorg Med Chem Lett 26:1585–1588. https://doi.org/10.1016/j.bmcl.2016.02.005
Dyson A, Dal-Pizzol F, Sabbatini G et al (2017) Ammonium tetrathiomolybdate following ischemia/reperfusion injury: chemistry, pharmacology, and impact of a new class of sulfide donor in preclinical injury models. PLoS Med 14:e1002310. https://doi.org/10.1371/journal.pmed.1002310
Landis PS (1965) The chemistry of 1,2-dithiole-3-thiones. Chem Rev 65:237–245. https://doi.org/10.1021/cr60234a004
Urquhart MC, Ercole F, Whittaker MR et al (2018) Recent advances in the delivery of hydrogen sulfide via a macromolecular approach. Polym Chem 9:4431–4439. https://doi.org/10.1039/C8PY00938D
Baskar R, Sparatore A, Del Soldato P, Moore PK (2008) Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative inhibit rat vascular smooth muscle cell proliferation. Eur J Pharmacol 594:1–8. https://doi.org/10.1016/j.ejphar.2008.07.029
Frantzias J, Logan J, Mollat P et al (2012) Hydrogen sulphide-releasing diclofenac derivatives inhibit breast cancer-induced osteoclastogenesis in vitro and prevent osteolysis ex vivo: S-NSAIDs inhibit osteoclast-tumour cell interaction. Br J Pharmacol 165:1914–1925. https://doi.org/10.1111/j.1476-5381.2011.01704.x
Liang D, Wu H, Wong MW, Huang D (2015) Diallyl trisulfide is a fast H2S donor, but diallyl disulfide is a slow one: the reaction pathways and intermediates of glutathione with polysulfides. Org Lett 17:4196–4199. https://doi.org/10.1021/acs.orglett.5b01962
Zhao Y, Yang C, Organ C et al (2015) Design, synthesis, and cardioprotective effects of n-mercapto-based hydrogen sulfide donors. J Med Chem 58:7501–7511. https://doi.org/10.1021/acs.jmedchem.5b01033
Foster JC, Powell CR, Radzinski SC, Matson JB (2014) S-Aroylthiooximes: a facile route to hydrogen sulfide releasing compounds with structure-dependent release kinetics. Org Lett 16:1558–1561. https://doi.org/10.1021/ol500385a
Martelli A, Testai L, Citi V et al (2013) Arylthioamides as H2S donors: l-cysteine-activated releasing properties and vascular effects in vitro and in vivo. ACS Med Chem Lett 4:904–908. https://doi.org/10.1021/ml400239a
Devarie-Baez NO, Bagdon PE, Peng B et al (2013) Light-induced hydrogen sulfide release from “Caged” gem-dithiols. Org Lett 15:2786–2789. https://doi.org/10.1021/ol401118k
Fukushima N, Ieda N, Sasakura K et al (2014) Synthesis of a photocontrollable hydrogen sulfide donor using ketoprofenate photocages. Chem Commun 50:587–589. https://doi.org/10.1039/C3CC47421F
Zheng Y, Yu B, Ji K et al (2016) Esterase-sensitive prodrugs with tunable release rates and direct generation of hydrogen sulfide. Angew Chem Int Ed 55:4514–4518. https://doi.org/10.1002/anie.201511244
Jimidar CC, Grunenberg J, Karge B et al (2022) Masked amino trimethyl lock (H2N-TML) systems: new molecular entities for the development of turn-on fluorophores and their application in hydrogen sulfide (H2S) imaging in human cells. Chem A Eur J. https://doi.org/10.1002/chem.202103525
Yang C-T, Chen L, Xu S et al (2017) Recent development of hydrogen sulfide releasing/stimulating reagents and their potential applications in cancer and glycometabolic disorders. Front Pharmacol 8:664. https://doi.org/10.3389/fphar.2017.00664
Acknowledgements
We would like to apologize for the inevitable omissions of many original works in the field of H2S that we were unable to cite due to space limitations. Figures were produced using BioRender.com.
Funding
This work was supported by the National Science Centre Grant #2019/35/B/NZ3/02817 (to AŁ).
Author information
Authors and Affiliations
Contributions
AŁ conceived the manuscript; KK, MM, and AŁ wrote the first manuscript draft; KK, MM, and AŁ generated the figures; JD commented on previous versions of the manuscript and provided critical editing; AŁ developed the manuscript to its final form. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaziród, K., Myszka, M., Dulak, J. et al. Hydrogen sulfide as a therapeutic option for the treatment of Duchenne muscular dystrophy and other muscle-related diseases. Cell. Mol. Life Sci. 79, 608 (2022). https://doi.org/10.1007/s00018-022-04636-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04636-0