
Abstract
There is unequivocal evidence that high-density lipoprotein (HDL) cholesterol levels in plasma are inversely associated with the risk of cardiovascular disease (CVD). Studies of families with inherited HDL disorders and genetic association studies in general (and patient) population samples have identified a large number of factors that control HDL cholesterol levels. However, they have not resolved why HDL cholesterol and CVD are inversely related. A growing body of evidence from nongenetic studies shows that HDL in patients at increased risk of CVD has lost its protective properties and that increasing the cholesterol content of HDL does not result in the desired effects. Hopefully, these insights can help improve strategies to successfully intervene in HDL metabolism. It is clear that there is a need to revisit the HDL hypothesis in an unbiased manner. True insights into the molecular mechanisms that regulate plasma HDL cholesterol and triglycerides or control HDL function could provide the handholds that are needed to develop treatment for, e.g., type 2 diabetes and the metabolic syndrome. Especially genome-wide association studies have provided many candidate genes for such studies. In this review we have tried to cover the main molecular studies that have been produced over the past few years. It is clear that we are only at the very start of understanding how the newly identified factors may control HDL metabolism. In addition, the most recent findings underscore the intricate relations between HDL, triglyceride, and glucose metabolism indicating that these parameters need to be studied simultaneously.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 General
Ever since plasma HDL cholesterol concentration was found to be inversely correlated with the risk of cardiovascular disease (CVD), there has been a strong interest in the biological mechanisms that can explain this correlation. From a large number of studies in humans, animals, and tissue culture, it has become clear that HDL exerts many beneficial functions with prominent roles in cellular cholesterol efflux and protection against inflammation. For recent overviews on this topic, see Luscher et al. (2014) and Rye and Barter (2014).
Paradoxically and not always recognized, however, is that rare inborn errors of HDL metabolism have illustrated that an almost complete loss of HDL or very high HDL cholesterol levels do not automatically translate in accelerated or protection from atherosclerosis, respectively. These observations may be related to the small numbers of patients available for studies or the absence/presence of concomitant established risk factors for CVD (such as increased LDL cholesterol, smoking, etc.) in these individuals. However, these findings by themselves indicate that the relation of HDL cholesterol with atherosclerosis is not as straightforward as for LDL cholesterol since in this case increases and decreases are always associated with increased and decreased risk, respectively.
Genetic approaches are frequently used to study whether changes in plasma HDL cholesterol concentration affect atherosclerosis. Such studies are conducted in families, larger patient population samples sharing large-impact mutations in HDL genes, as well as general population samples. Using candidate gene approaches, these studies mostly generated contrasting or confusing results [a short summary of these findings can be found in Chapman et al. (2011)]. Illustrative in this regard were investigations into variation at the locus encoding for the ATP-binding cassette transporter A1 (ABCA1). While a complete loss of ABCA1 function causes near HDL deficiency and often accelerated atherosclerosis in Tangier patients who are referred to the clinic, studies in general population samples indicated that ABCA1 gene variation is not necessarily related with plasma HDL cholesterol concentration and risk of CVD (for review, see Frikke-Schmidt (2011)).
More recently, it has become possible to study the impact of whole-genome variation on complex diseases which has shed light on our understanding whether or not genes and their products are related to plasma lipid traits and the risk of CVD. In this regard, particularly Mendelian randomization studies showed that genetic variation associated with increased HDL cholesterol does not protect from atherosclerosis (Voight et al. 2012). In this case, it concerned a study of common variants in HDL candidate genes; however, more recently, it was also shown that low-frequency coding variants (frequencies between 0.1 and 2 %) with relatively large effects on HDL cholesterol and/or triglycerides were also not associated with risk for coronary heart disease (Peloso et al. 2014).
These and other studies have placed HDL cholesterol as a pharmaceutical target under heavy fire especially in the context of several large clinical trials that tested drugs which increased HDL cholesterol but did reduce atherosclerosis (for reviews on CETP inhibitors and the use of niacin: Ginsberg and Reyes-Soffer 2013; Rader and Degoma 2014). While there is still hope for HDL-related interventions as outlined in a recent review (van Capelleveen et al. 2014), it is clear that there is a need to revisit the mechanisms that have been put forward to explain the unequivocal relation between HDL cholesterol and risk of CVD in epidemiological studies.
To date, it is repeatedly been pointed out that a focus on the cholesterol content of HDL should maybe be replaced by a focus on the functions that are associated with this lipoprotein (Feig et al. 2014; Peloso et al. 2014; Luscher et al. 2014; Riwanto and Landmesser 2013), but unfortunately, HDL functionality studies have thus far not provided a solution to the problems encountered. So far it is not clear which of HDL properties should and could be targeted. There is evidence for a focus on HDL as an acceptor of cholesterol (Khera et al. 2011) but later studies were not able to confirm this (Li et al. 2013a, b). In other words, the association with CVD risk has only been firmly established for the concentration of cholesterol in HDL while at the same time there is little evidence of a causal relation between HDL and CVD (Vergeer et al. 2010). Thus, it is possible that HDL cholesterol is a proxy for an unknown correlating factor. A renewed focus on the clinical application of HDL-based strategies for certain indications on the basis of functional properties of HDL but also on significant preclinical and clinical data, as was recently suggested, may hopefully bring relief (Gordts et al. 2013).
In this light, the current chapter focuses on recent studies that have shed new light on how genes and/or their products affect HDL metabolism. The first section shortly describes established regulators of HDL metabolism as a general framework to help understand the new insights that are described in the second section.
2 Determinants of Plasma HDL Cholesterol Levels
Twin studies have indicated that genetic and environmental parameters equally contribute to the levels of cholesterol in HDL in the blood (Goode et al. 2007). This paragraph only shortly describes the major primary and secondary regulators of HDL cholesterol. It is important to underscore, however, that changes in HDL cholesterol without changes in other plasma lipid traits are very rare. They are mostly seen in the context of changes in plasma triglycerides. At the population level, genome-wide association (GWA) studies have recently confirmed that in many cases, genetic variation is associated with changes in more than one lipid trait (Teslovich et al. 2010; Willer et al. 2013).
2.1 Established Primary Regulators of Plasma HDL Cholesterol
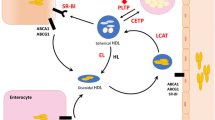
For the de novo production of HDL, the small intestine and liver need to produce apolipoprotein (apo) A-I, ATP-binding cassette protein A1, and lecithin–cholesterol acyltransferase encoded by the APOAI, ABCA1, and LCAT genes, respectively. When the production of any of these proteins is attenuated (through functional large-impact mutations), it immediately translates into a reduction of HDL cholesterol in the circulation. Other established modulators of HDL are cholesteryl ester transfer protein (encoded by CETP) and scavenger receptor class B member 1 (SRB1, encoded by SCARB1). While CETP mediates the transfer of cholesterol ester from HDL to triglyceride-rich lipoproteins in exchange for triglycerides (thereby controlling the levels of cholesteryl ester in HDL), SRB1 mediates the selective cellular uptake of cholesteryl ester in the liver and steroidogenic organs. Figure 1 gives a schematic representation of how the above genes relate to HDL biology.

A schematic presentation of factors that control HDL synthesis/binding and/or HDL-mediated cellular cholesterol homeostasis. The genes in the white boxes encode for key HDL proteins and enzymes, while the genes in the blue boxes encode for less established (new) factors that affect HDL metabolism. The roles of the genes in the gray boxes are addressed but solid evidence has not yet been provided. Abbreviations (proteins encoded by the gene names): ABCA1 ATP-binding cassette A1, ABCG1 ATP-binding cassette G1, APOAI apolipoprotein A-I, APOF apolipoprotein F, APOM apolipoprotein M, BMP1 bone morphogenetic protein-1, GLCE glucuronic acid epimerase, LCAT lecithin–cholesterol acyltransferase, PCPE2 procollagen C-proteinase enhancer 2, PCYT1 CTP:phosphocholine cytidylyltransferase alpha, SCARB1 encoding for scavenger receptor class B member 1
2.2 Established Secondary Regulators of Plasma HDL Cholesterol
There are a large number of other factors that affect HDL cholesterol concentration through modulating the lipolysis of plasma triglycerides [reviewed in Oldoni et al. (2014)]. Figure 2 only illustrates the major players. Most of these regulate the activity of lipoprotein lipase (LPL), the sole enzyme capable of breaking down triglycerides (packaged in chylomicrons and very-low-density lipoproteins (VLDL)) in the circulation. These include genes that encode for LPL’s cofactor apoC-II (APOCII) as well as apoA-V (APOAV) and inhibitors of the LPL reaction (APOCIII, ANGPTL3, ANGPTL4). More recently, also GPI-anchored HDL-binding protein 1 (GPIHBP1) was shown to affect HDL concentration through ultimately its effect on LPL activity. In addition, hepatic lipase (encoded by LIPC) and endothelial lipase (LIPG) also exert marked effects on HDL cholesterol concentration mainly through modulating HDL phospholipids and HDL triglycerides, respectively.

A schematic presentation of factors that affect HDL through roles in the conversion and remodeling of HDL or through effects on triglyceride/glucose metabolism. The genes in the white boxes encode for established factors of HDL conversion/remodeling or plasma TG hydrolysis. The genes in the blue boxes encode for less established (new) factors. Abbreviations (proteins encoded by the gene names): ANGPTL angiopoietin-like protein, TRIB1 tribbles homolog 1, TTC39B tetratricopeptide repeat domain 39B, PPP1R3B glycogen-targeting PP1 subunit G(L), GALNT2 ppGalNAc-T2, GCKR glucokinase (hexokinase 4) regulator
2.3 Missing Heritability
So far, studies of over 40 genes have provided solid evidence that their products affect plasma HDL cholesterol concentration. With so many established genes, one may expect these accounting for the estimated 50 % heritability of this trait. This appears however not to be the case when GWA data are analyzed with the current statistical methods and datasets: The most recent of meta-analysis indicates that common genetic variation can only explain 12 % variation of HDL cholesterol levels while in this study, both variations in established loci as well as newly identified loci (n = 46) were taken into account (Willer et al. 2013). However, in these calculations, gene–gene and gene–environment interactions are not for. In addition, the estimated impact of gene variation on the phenotype is based on the presence and frequency of such variations and these are not constant factors over the genome.
From a different angle, candidate gene resequencing studies in individuals with very high or low HDL cholesterol (selected from the general population) have shown that apparent functional mutations are only found in a few percent of the cases (Cohen et al. 2004; Haase et al. 2012). Also in individuals that were referred to the clinic because of extreme levels of HDL cholesterol, resequencing studies of candidate genes only provided satisfying clues in a minority of the subjects studied (Candini et al. 2010; Holleboom et al. 2011a, b; Kiss et al. 2007). It may be noted, however, that most studies conducted thus far focused only on APOAI, LCAT, and ABCA1 leaving ample room for large-effect variants in other genes. Another study focused on the origin of high HDL cholesterol levels through sequencing CETP, LIPG, and GALNT, showed an enrichment of rare coding and splicing mutations in 171 probands (Tietjen et al. 2012). A second study conducted a search for mutations in 197 lipid-related genes in 80 individuals with extreme HDL cholesterol phenotypes. The outcome was that multiple mutations in different genes combined could be responsible for extreme low or high HDL cholesterol levels (Motazacker et al. 2013). Although a polygenic origin of a complex trait like HDL cholesterol level appears logical, especially in view of similar studies for plasma triglycerides (Johansen et al. 2010) or LDL cholesterol (Talmud et al. 2013), this needs to be confirmed. Larger comprehensive resequencing efforts are warranted to study to what extent large-impact mutations in established and candidate genes can explain HDL cholesterol concentration in plasma and how such mutations relate to CVD risk.
3 Novel Insight into HDL Biology
As indicated above, GWA studies have identified many genomic regions that affect plasma lipid traits. One of the most famous papers in this regard published in 2010 (Teslovich et al. 2010) already listed 38 loci with HDL cholesterol as lead trait. Importantly, these and many other GWA studies rediscovered many of the established “HDL genes” which underscored the potential importance of the newly identified loci. The most comprehensive meta-analysis in this field of research to date lists an additional 46 loci that are associated with HDL cholesterol (Willer et al. 2013). This section tries to capture the novel molecular insights that have been published over the last few years. Many studies focused on genes that on the basis of their proximity to genetic markers in GWA studies were “annotated as HDL genes.” In addition, other studies (not initiated through genetic insight) which produced interesting novel insight in HDL biology are shortly discussed.
3.1 De Novo Synthesis of HDL and HDL Binding
3.1.1 Bone Morphogenetic Protein-1 and Procollagen C-Proteinase Enhancer-2
In 2007, bone morphogenetic protein-1 encoded by BMP-1 (aliases OI13, PCOLC, PCP, PCP2, TLD) was identified as the metalloproteinase that stimulates the conversion of newly secreted proapoA-I to its phospholipid-binding form (Chau et al. 2007). While these results were obtained through in vitro studies, early genetic association studies showed the possible involvement of procollagen C-proteinase enhancer-2 (encoded by PCPE2 or PCOLCE2) in modulating HDL cholesterol levels (Hinds et al. 2004). It was subsequently shown that PCPE2 together with BMP1 and proapoAI forms a ternary complex and that it is involved in the regulation of apoA-I posttranslational processing (Zhu et al. 2009). In a later study, it was shown that Pcpe2 KO mice have strongly increased plasma apoA-I and HDL cholesterol levels compared with wild-type littermates, regardless of gender or diet (Francone et al. 2011). Changes in HDL particle size and electrophoretic mobility observed in Pcpe2 KO mice suggest that the presence of proapoA-I impairs the maturation of HDL. Although initially picked up by GWA studies, PCPE2 is not listed in the subsequent respective meta-analyses (Teslovich et al. 2010; Willer et al. 2013), while BMP1 is also not listed in the latter studies. So far, the role of BMP1 or PCPE2 in posttranslational processing in humans has not been reported which in the light of the larger GWA studies performed is now dependent on the identification of large-impact mutations in individuals with HDL disorders.
3.1.2 Apolipoprotein M
Originally identified as a protein associated with HDL in 1999 (Xu and Dahlback 1999), apoM was later implicated to play a role in the generation of prebeta-HDL and cholesterol efflux mediated by HDL (Wolfrum et al. 2005). These results have recently been supported by evidence in plasma of humans (Plomgaard et al. 2009) while others have found that human APOM gene variation affects HDL cholesterol (Park et al. 2013; Aung et al. 2013). Through carrying sphingosine-1-phosphate with effects on angiogenesis, endothelial cell migration, and inflammation, apoM (Christoffersen and Nielsen 2013) may be a link to many of the functions that are attributed to HDL. While there is thus substantial evidence for a role of apoM in human HDL metabolism, the gene has thus far not been picked up by GWA studies, while large-impact mutations in this gene have so far not been reported either.
3.1.3 CTP:Phosphocholine Cytidylyltransferase Alpha (CT Alpha)
Through an interest in the synthesis of phosphatidylcholine (the primary phospholipid in cellular membranes but also of HDL), the group of Dennis Vance studied a conditional PCTY1 knockout mouse and showed in 2004 that hepatic CTP:phosphocholine cytidylyltransferase alpha reduces HDL cholesterol and apoA-I while it also affected VLDL (Jacobs et al. 2004). In 2008, the same investigators subsequently showed that in hepatocytes isolated from these mice, ABCA1 expression was reduced. Other findings included normalization of plasma HDL and VLDL in these mice after adenoviral delivery of CT alpha which clearly implicated its importance in HDL genesis (Jacobs et al. 2008).
3.1.4 Apolipoprotein F
In proteomic studies, apoF has been identified to be associated with HDL. This protein is also known as lipid transfer inhibitor protein which is able to inhibit CETP. Plasma apoF levels were found to be positively associated with HDL cholesterol in males but not in females (Morton et al. 2008). The Rader group subsequently showed that overexpression of apoF in mice reduced HDL cholesterol levels by accelerating clearance from the circulation (Lagor et al. 2009). A later study showed that a murine apoF knockout model had no substantial effect on plasma lipid concentrations, HDL size, lipid, or protein (Lagor et al. 2012) although a reduced ability to promote cholesterol efflux was observed. Until now, no other studies on apoF have been published.
3.1.5 Glucuronic Acid Epimerase
Through studies in Turkish families, a linkage peak with low HDL cholesterol was identified on chromosome 15 which was >20 cM wide including the gene encoding hepatic lipase (LIPC), which has important functions in HDL metabolism. However, the same investigators suspected that variations in LIPC might not fully explain this linkage peak and sought additional gene(s) that might contribute to the peak. This is how they identified GLCE encoding glucuronic acid epimerase, a heparan sulfate proteoglycan biosynthetic enzyme (Hodoglugil et al. 2011). So far, no other studies on the role of this enzyme in HDL metabolism have been conducted while the gene was not found associated with HDL cholesterol levels in the latest meta-analyses GWA study (Willer et al. 2013).
3.1.6 Beta-Chain of ATP Synthase
In 2003, beta-chain of ATP synthase (encoded by ATPB5), a principal protein complex of the mitochondrial inner membrane, was surprisingly identified as a high-affinity HDL receptor for apoA-I receptor with a role in hepatic HDL endocytosis (Martinez et al. 2003). A subsequent study demonstrated a major role of cytoskeleton reorganization in F(1)-ATPase/P2Y(13)-dependent HDL endocytosis under the control of the small GTPase RhoA and its effector ROCK I (Malaval et al. 2009). Others showed that binding of HDL to this receptor triggers the generation of ADP, which via the activation of the purinergic receptor P2Y13 stimulates the uptake and transport of HDL and initially lipid-free apoA-I by endothelial cells (Cavelier et al. 2012). So far, there is no genetic support for the involvement of beta-chain of ATP synthase and/or P2Y(13), but even mild mutations could have in the case of ATPB5 lethal consequences. Most recently, it was reported that there may be a role for beta-chain of ATP synthase in regulating HDL cholesterol levels as mitochondrial inhibitor factor 1, which inhibits ATPB5 and can be measured in serum, is associated with HDL cholesterol levels (Genoux et al. 2013).
3.2 HDL Conversion and Remodeling
3.2.1 Angptl Family of Proteins
In the year 2000, ANGPTL4 was identified as a peroxisome proliferator-activated receptor α response gene (Kersten et al. 2000) while ANGPTL3 is since 2002 known for its effects on glucose and lipid metabolism through a hypolipidemic mouse strain (Koishi et al. 2002). Both Angptl3 and Angptl4 are known to inhibit LPL, thereby having an indirect effect on HDL cholesterol concentration in plasma, but Angptl3 may also exert direct effects on HDL remodeling through inhibition of endothelial lipase (Shimamura et al. 2007). In GWA studies published in 2008 and 2010, ANGPTL3 was shown to be associated primarily with triglycerides (Kathiresan et al. 2008; Teslovich et al. 2010) while more recently it was shown that human ANGPTL3 deficiency also causes marked reduction in HDL cholesterol (Musunuru et al. 2010). In 2010, ANGPTL4 was reported to be associated with HDL cholesterol as primary trait (Teslovich et al. 2010) while later on ANGPTL4 variants were found associated with lower triglycerides and elevated HDL cholesterol (Romeo et al. 2007). Another study showed that mutant alleles of ANGPTL3 and ANGPTL4 that were associated with low plasma triglyceride levels interfered either with the synthesis or secretion of the protein or with the ability of the ANGPTL protein to inhibit LPL (Romeo et al. 2009). More recently, ANGPTL8 (also known as betatrophin) was shown to also affect HDL cholesterol through affecting the activity of Angpl3 (Quagliarini et al. 2012). In addition, low-frequency variants in ANGPTL8 were shown to affect HDL cholesterol levels (Peloso et al. 2014). For a recent review on the current eight Angptl proteins, please see Santulli (2014).
3.2.2 Tribbles Homolog 1
Variation at the TRIB1 gene locus has been reported to be associated with triglycerides (as lead trait), HDL cholesterol, total cholesterol, and LDL cholesterol (Kathiresan et al. 2008; Teslovich et al. 2010). It is one of the few genes subsequently studied in depth in mouse models, in vitro experiments, and further human genetic association studies. Viral-mediated overexpression in the liver of mice simultaneously reduced plasma triglycerides and cholesterol of all major lipoproteins (including HDL). On the other hand, TRIB1 knockout mice showed elevated levels of triglycerides (Burkhardt et al. 2010) without statistically significant effects on HDL cholesterol. These effects were related to an on VLDL production but it is not known what mechanisms are responsible. Further epidemiological studies confirmed that TRIB1 is associated with HDL cholesterol but interestingly without affecting apoA-I concentration (Varbo et al. 2011). More recently it was shown that Trib1 is important to adipose tissue maintenance and suppression of metabolic disorders by controlling the differentiation of tissue-resident M2-like macrophages (Satoh et al. 2013). In addition, a TRIB1 single-nucleotide polymorphism was found associated with nonalcoholic fatty liver disease in humans while the same investigators showed that TRIB1 expression affects hepatic lipogenesis and glycogenesis through multiple molecular interactions (including a molecular interaction with MLXIPL or CHREBP, a hepatic lipogenic master regulator) (Ishizuka et al. 2014). In addition to a role in lipid metabolism, there is a substantial literature on the role of tribbles in cancer (Liang et al. 2013).
3.2.3 Tetratricopeptide Repeat Domain/Glycogen-Targeting PP1 Subunit G(L)
TTC39B was discovered through GWA studies in 2009 (Kathiresan et al. 2009). When knocked down in mice (using a viral-mediated strategy), it was shown to increase HDL cholesterol levels (Teslovich et al. 2010). However, no studies have since revealed insight into how tetratricopeptide repeat domain 39B may affect lipid metabolism while another GWA study could not replicate the effect of TTC39B variation on plasma lipid levels (Dumitrescu et al. 2011).
PPP1R3B was also found to be associated with HDL cholesterol as primary trait (Teslovich et al. 2010), and in the same paper, viral-mediated overexpression was shown to reduce HDL cholesterol levels in mice. The gene encoding for glycogen-targeting PP1 (protein phosphatase 1) subunit G(L) has also been associated with type 2 diabetes and maturity-onset diabetes of the young but sequence variants at the PPP1R3B locus were not found to be related to diabetes in mostly Caucasian families (Dunn et al. 2006). Other studies proposed a role for this gene in inflammation (Dehghan et al. 2011), Alzheimer’s disease (Kamboh et al. 2012), and hepatic steatosis (Palmer et al. 2013). As for TTC39B, there are no mechanistic studies that have revealed insight into how PPP1R3B gene products may affect lipid metabolism.
3.2.4 ppGalNAc-T2
Variation at the GALNT2 locus was also shown to be associated with HDL cholesterol (Teslovich et al. 2010). Viral-mediated overexpression and knockdown were shown to decrease and increase HDL cholesterol levels in mice, respectively. In a later study a rare functional GALNT2 variant, identified in two index cases with very high HDL cholesterol, was reported to affect HDL metabolism indirectly via an effect on the lipolysis of triglycerides (Holleboom et al. 2011a, b). This was suggested to be in part due to attenuated glycosylation of apoC-III which impaired its capacity to inhibit LPL in the catabolism of triglycerides. Other investigators reported that GALNT2 regulates plasma lipid levels through the glycosylation of ANGTPL3, another inhibitor of the LPL reaction (Schjoldager et al. 2010). Recently, ppGalNAc-T2 was identified as a novel regulator of insulin signaling (Marucci et al. 2013) while there appears to be a role for GALNT2 in hypertension too (Pendergrass et al. 2013; McDonough et al. 2013).
3.2.5 Glucokinase (Hexokinase 4) Regulator
GCKR (also known as GKRP) encodes for a regulatory protein that inhibits glucokinase in the liver and pancreatic islet cells by binding non-covalently to form an inactive complex with the enzyme. This gene is considered a susceptibility gene candidate for a form of maturity-onset diabetes of the young. GCKR was picked up in both GWA studies with a focus on plasma lipids but also with type 2 diabetes (van de Bunt and Gloyn 2010). Varbo et al. showed that a GCKR SNP was associated with increased triglycerides, decreased HDL cholesterol, and remarkably increased apoA-I (Varbo et al. 2011). Several genetic association studies have shown that GCKR variants are associated with triglycerides, insulin resistance (Shen et al. 2013), type 2 diabetes fasting plasma glucose (Li et al. 2013a, b), hyperglycemia (Stancakova et al. 2012), and nonalcoholic fatty liver disease (Lin et al. 2014). In 2013, the crystal structure was resolved (Pautsch et al. 2013) while two potent small-molecule GK–GKRP disruptors were recently reported to normalize blood glucose levels in several rodent models of diabetes (Lloyd et al. 2013).
Conclusions and Perspectives
The harvest of screening the most recent published literature for novel HDL influencing genes is extensive. However, it also shows that there are only very few mechanistic clues on how “the new kids on the block” (primarily identified through GWA studies) affect HDL cholesterol or HDL function.
It is interesting that none of the genes encoding for the proteins and enzymes that have been discussed in the section on the de novo synthesis of HDL are listed in the current largest GWA studies. This may be surprising in light of the evidence for the roles of especially bone morphogenetic protein-1, procollagen C-proteinase enhancer 2, apoM, and CT alpha in HDL metabolism. It shows that GWA strategies, like any approach, come with limitations. Thus, prioritizing research interest on the basis of the top ranking GWA hits is not necessarily the most promising route to go.
In the section on (new) insights in HDL conversion and remodeling, it is clear that the factors discussed are only indirectly associated with HDL cholesterol levels through effects on triglyceride and/or glucose metabolism. It may be noted in this regard that HDL cholesterol was proposed to be the lead trait for ANGPTL4, TTC39B, PPP1R3B, and GALNT2, in GWA studies which highlights a need for more comprehensive studies with focus on the mechanistic relations between HDL cholesterol, triglycerides, and glucose metabolism. Such studies may need the use of genome-scale metabolic maps.
Finally, this review underlines an urgent need of mechanistic studies. But where to start? Answering this question is today’s challenge as evidence-based prioritization tools and high-throughput functional studies are yet to come.
Abbreviations
- CVD:
-
Cardiovascular disease
- GWA:
-
Genome-wide association
- HDL:
-
High-density lipoprotein
- LDL:
-
Low-density lipoprotein
- VLDL:
-
Very-low-density lipoprotein
References
Aung LH et al (2013) Association of the apolipoprotein M gene polymorphisms and serum lipid levels. Mol Biol Rep 40(2):1843–1853
Burkhardt R et al (2010) Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice. J Clin Invest 120(12):4410–4414
Candini C et al (2010) Identification and characterization of novel loss of function mutations in ATP-binding cassette transporter A1 in patients with low plasma high-density lipoprotein cholesterol. Atherosclerosis 213(2):492–498
Cavelier C et al (2012) The beta-chain of cell surface F(0)F(1) ATPase modulates apoA-I and HDL transcytosis through aortic endothelial cells. Arterioscler Thromb Vasc Biol 32(1):131–139
Chapman MJ et al (2011) Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J 32(11):1345–1361
Chau P et al (2007) Bone morphogenetic protein-1 (BMP-1) cleaves human proapolipoprotein A1 and regulates its activation for lipid binding. Biochemistry 46(28):8445–8450
Christoffersen C, Nielsen LB (2013) Apolipoprotein M: bridging HDL and endothelial function. Curr Opin Lipidol 24(4):295–300
Cohen JC et al (2004) Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 305(5685):869–872
Dehghan A et al (2011) Meta-analysis of genome-wide association studies in >80,000 subjects identifies multiple loci for C-reactive protein levels. Circulation 123(7):731–738
Dumitrescu L et al (2011) Genetic determinants of lipid traits in diverse populations from the population architecture using genomics and epidemiology (PAGE) study. PLoS Genet 7(6):e1002138
Dunn JS et al (2006) Examination of PPP1R3B as a candidate gene for the type 2 diabetes and MODY loci on chromosome 8p23. Ann Hum Genet 70(Pt 5):587–593
Feig JE et al (2014) High-density lipoprotein and atherosclerosis regression: evidence from preclinical and clinical studies. Circ Res 114(1):205–213
Francone OL et al (2011) Disruption of the murine procollagen C-proteinase enhancer 2 gene causes accumulation of proapoA-I and increased HDL levels. J Lipid Res 52(11):1974–1983
Frikke-Schmidt R (2011) Genetic variation in ABCA1 and risk of cardiovascular disease. Atherosclerosis 218(2):281–282
Genoux A et al (2013) Serum IF1 concentration is independently associated to HDL levels and to coronary heart disease: the GENES study. J Lipid Res 54(9):2550–2558
Ginsberg HN, Reyes-Soffer G (2013) Niacin: a long history, but a questionable future. Curr Opin Lipidol 24(6):475–479
Goode EL et al (2007) Heritability of longitudinal measures of body mass index and lipid and lipoprotein levels in aging twins. Twin Res Hum Genet 10(5):703–711
Gordts SC et al (2013) Pleiotropic effects of HDL: towards new therapeutic areas for HDL-targeted interventions. Curr Mol Med 14(4):481–503
Haase CL et al (2012) Population-based resequencing of APOA1 in 10,330 individuals: spectrum of genetic variation, phenotype, and comparison with extreme phenotype approach. PLoS Genet 8(11):e1003063
Hinds DA et al (2004) Application of pooled genotyping to scan candidate regions for association with HDL cholesterol levels. Hum Genomics 1(6):421–434
Hodoglugil U et al (2011) Glucuronic acid epimerase is associated with plasma triglyceride and high-density lipoprotein cholesterol levels in Turks. Ann Hum Genet 75(3):398–417
Holleboom AG et al (2011a) Heterozygosity for a loss-of-function mutation in GALNT2 improves plasma triglyceride clearance in man. Cell Metab 14(6):811–818
Holleboom AG et al (2011b) High prevalence of mutations in LCAT in patients with low HDL cholesterol levels in The Netherlands: identification and characterization of eight novel mutations. Hum Mutat 32(11):1290–1298
Ishizuka Y et al (2014) TRIB1 down-regulates hepatic lipogenesis and glycogenesis via multiple molecular interactions. J Mol Endocrinol 52(2):145–158
Jacobs RL et al (2004) Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J Biol Chem 279(45):47402–47410
Jacobs RL et al (2008) Hepatic CTP:phosphocholine cytidylyltransferase-alpha is a critical predictor of plasma high density lipoprotein and very low density lipoprotein. J Biol Chem 283(4):2147–2155
Johansen CT et al (2010) Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet 42(8):684–687
Kamboh MI et al (2012) Genome-wide association study of Alzheimer’s disease. Transl Psychiatry 2:e117
Kathiresan S et al (2008) Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet 40(2):189–197
Kathiresan S et al (2009) Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet 41(1):56–65
Kersten S et al (2000) Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J Biol Chem 275(37):28488–28493
Khera AV et al (2011) Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364(2):127–135
Kiss RS et al (2007) Genetic etiology of isolated low HDL syndrome: incidence and heterogeneity of efflux defects. Arterioscler Thromb Vasc Biol 27(5):1139–1145
Koishi R et al (2002) Angptl3 regulates lipid metabolism in mice. Nat Genet 30(2):151–157
Lagor WR et al (2009) Overexpression of apolipoprotein F reduces HDL cholesterol levels in vivo. Arterioscler Thromb Vasc Biol 29(1):40–46
Lagor WR et al (2012) The effects of apolipoprotein F deficiency on high density lipoprotein cholesterol metabolism in mice. PLoS ONE 7(2):e31616
Li H et al (2013a) Association of glucokinase regulatory protein polymorphism with type 2 diabetes and fasting plasma glucose: a meta-analysis. Mol Biol Rep 40(6):3935–3942
Li XM et al (2013b) Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol 33(7):1696–1705
Liang KL et al (2013) Tribbles in acute leukemia. Blood 121(21):4265–4270
Lin YC et al (2014) Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am J Clin Nutr 99(4):869–874
Lloyd DJ et al (2013) Antidiabetic effects of glucokinase regulatory protein small-molecule disruptors. Nature 504(7480):437–440
Luscher TF et al (2014) High-density lipoprotein: vascular protective effects, dysfunction, and potential as therapeutic target. Circ Res 114(1):171–182
Malaval C et al (2009) RhoA/ROCK I signalling downstream of the P2Y13 ADP-receptor controls HDL endocytosis in human hepatocytes. Cell Signal 21(1):120–127
Martinez LO et al (2003) Ectopic beta-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature 421(6918):75–79
Marucci A et al (2013) Role of GALNT2 in the modulation of ENPP1 expression, and insulin signaling and action: GALNT2: a novel modulator of insulin signaling. Biochim Biophys Acta 1833(6):1388–1395
McDonough CW et al (2013) Atenolol induced HDL-C change in the pharmacogenomic evaluation of antihypertensive responses (PEAR) study. PLoS ONE 8(10):e76984
Morton RE et al (2008) Lipid transfer inhibitor protein (apolipoprotein F) concentration in normolipidemic and hyperlipidemic subjects. J Lipid Res 49(1):127–135
Motazacker MM et al (2013) Evidence of a polygenic origin of extreme high-density lipoprotein cholesterol levels. Arterioscler Thromb Vasc Biol 33(7):1521–1528
Musunuru K et al (2010) Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med 363(23):2220–2227
Oldoni F et al (2014) Mendelian disorders of high-density lipoprotein metabolism. Circ Res 114(1):124–142
Palmer ND et al (2013) Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology 58(3):966–975
Park YJ et al (2013) The APOM polymorphism as a novel risk factor for dyslipidaemia in rheumatoid arthritis: a possible shared link between disease susceptibility and dyslipidaemia. Clin Exp Rheumatol 31(2):180–188
Pautsch A et al (2013) Crystal structure of glucokinase regulatory protein. Biochemistry 52(20):3523–3531
Peloso GM et al (2014) Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am J Hum Genet 94(2):223–232
Pendergrass SA et al (2013) Phenome-wide association study (PheWAS) for detection of pleiotropy within the population architecture using genomics and epidemiology (PAGE) network. PLoS Genet 9(1):e1003087
Plomgaard P et al (2009) Apolipoprotein M predicts pre-beta-HDL formation: studies in type 2 diabetic and nondiabetic subjects. J Intern Med 266(3):258–267
Quagliarini F et al (2012) Atypical angiopoietin-like protein that regulates ANGPTL3. Proc Natl Acad Sci USA 109(48):19751–19756
Rader DJ, Degoma EM (2014) Future of cholesteryl ester transfer protein inhibitors. Annu Rev Med 65:385–403
Riwanto M, Landmesser U (2013) High density lipoproteins and endothelial functions: mechanistic insights and alterations in cardiovascular disease. J Lipid Res 54(12):3227–3243
Romeo S et al (2007) Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet 39(4):513–516
Romeo S et al (2009) Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest 119(1):70–79
Rye KA, Barter PJ (2014) Cardioprotective functions of HDLs. J Lipid Res 55(2):168–179
Santulli G (2014) Angiopoietin-like proteins: a comprehensive look. Front Endocrinol (Lausanne) 5:4
Satoh T et al (2013) Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature 495(7442):524–528
Schjoldager KT et al (2010) O-glycosylation modulates proprotein convertase activation of angiopoietin-like protein 3: possible role of polypeptide GalNAc-transferase-2 in regulation of concentrations of plasma lipids. J Biol Chem 285(47):36293–36303
Shen Y et al (2013) GCKR variants increase triglycerides while protecting from insulin resistance in Chinese children. PLoS ONE 8(1):e55350
Shimamura M et al (2007) Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol 27(2):366–372
Stancakova A et al (2012) Hyperglycemia and a common variant of GCKR are associated with the levels of eight amino acids in 9,369 Finnish men. Diabetes 61(7):1895–1902
Talmud PJ et al (2013) Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet 381(9874):1293–1301
Teslovich TM et al (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466(7307):707–713
Tietjen I et al (2012) Segregation of LIPG, CETP, and GALNT2 mutations in Caucasian families with extremely high HDL cholesterol. PLoS ONE 7(8):e37437
van Capelleveen JC et al (2014) Novel therapies focused on the high-density lipoprotein particle. Circ Res 114(1):193–204
van de Bunt M, Gloyn AL (2010) From genetic association to molecular mechanism. Curr Diab Rep 10(6):452–466
Varbo A et al (2011) TRIB1 and GCKR polymorphisms, lipid levels, and risk of ischemic heart disease in the general population. Arterioscler Thromb Vasc Biol 31(2):451–457
Vergeer M et al (2010) The HDL hypothesis: does high-density lipoprotein protect from atherosclerosis? J Lipid Res 51(8):2058–2073
Voight BF et al (2012) Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 380(9841):572–580
Willer CJ et al (2013) Discovery and refinement of loci associated with lipid levels. Nat Genet 45(11):1274–1283
Wolfrum C et al (2005) Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat Med 11(4):418–422
Xu N, Dahlback B (1999) A novel human apolipoprotein (apoM). J Biol Chem 274(44):31286–31290
Zhu J et al (2009) Regulation of apoAI processing by procollagen C-proteinase enhancer-2 and bone morphogenetic protein-1. J Lipid Res 50(7):1330–1339
Acknowledgments
I wish to apologize to colleagues whose work may not be cited in this review: It remains an attempt to cover the existing literature. This work is supported by Fondation LeDucq (Transatlantic Network, 2009–2014), the Netherlands CardioVascular Research Initiative (CVON2012-17; Genius), and the European Union (COST BM0904; Resolve: FP7-305707; TransCard: FP7-603091–2).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this chapter
Cite this chapter
Kuivenhoven, J.A., Groen, A.K. (2015). Beyond the Genetics of HDL: Why Is HDL Cholesterol Inversely Related to Cardiovascular Disease?. In: von Eckardstein, A., Kardassis, D. (eds) High Density Lipoproteins. Handbook of Experimental Pharmacology, vol 224. Springer, Cham. https://doi.org/10.1007/978-3-319-09665-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-09665-0_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09664-3
Online ISBN: 978-3-319-09665-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)