
Abstract
High-density lipoproteins (HDLs) exert many beneficial effects which may help to protect against the development or progression of atherosclerosis or even facilitate lesion regression. These activities include promoting cellular cholesterol efflux, protecting low-density lipoproteins (LDLs) from modification, preserving endothelial function, as well as anti-inflammatory and antithrombotic effects. However, questions remain about the relative importance of these activities for atheroprotection. Furthermore, the many molecules (both lipids and proteins) associated with HDLs exert both distinct and overlapping activities, which may be compromised by inflammatory conditions, resulting in either loss of function or even gain of dysfunction. This complexity of HDL functionality has so far precluded elucidation of distinct structure–function relationships for HDL or its components. A better understanding of HDL metabolism and structure–function relationships is therefore crucial to exploit HDLs and its associated components and cellular pathways as potential targets for anti-atherosclerotic therapies and diagnostic markers.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Atherosclerosis
- Cholesterol efflux
- Endothelial function
- Foam cells
- High-density lipoproteins
- Inflammation
- Macrophages
- Oxidation
- Thrombosis
- Smooth muscle cells
1 Introduction
Atherothrombotic diseases, such as coronary heart disease, stroke, and peripheral arterial disease, are the world’s leading cause of death. The first-line therapy for prevention and treatment of coronary heart disease is low-density lipoprotein (LDL) lowering by statin therapy. Although statin drugs are very effective in reducing plasma LDL cholesterol concentrations, the risk of major cardiovascular events is only decreased by 15–40 % (Baigent et al. 2005; Ridker et al. 2010). Additional therapeutic approaches are needed to minimize this considerable remaining cardiovascular risk in statin-treated patients. It has been postulated that part of this residual risk may be explained by low circulating levels of high-density lipoproteins (HDLs). Clinical and large population-based epidemiological studies have consistently shown that low HDL cholesterol is independently associated with an increased risk of atherothrombotic diseases (Di Angelantonio et al. 2009; Barter et al. 2007a). However, this long-standing inverse relationship between HDL and atherosclerotic cardiovascular disease has been challenged by recent findings. Indeed, high plasma concentrations of HDL cholesterol might not be beneficial for certain subgroups of patients. Thus, in high-risk postinfarction patients characterized by high total cholesterol and C-reactive protein, elevated levels of HDL cholesterol were paradoxically found to be related to poor clinical outcome (Corsetti et al. 2006). Similarly, the inverse relationship between HDL cholesterol levels and major cardiovascular events was found lost in patients with established coronary artery disease undergoing diagnostic angiography or coronary artery bypass grafting (Angeloni et al. 2013; Silbernagel et al. 2013). Moreover, several large randomized clinical trials assessing the effects of new therapies targeting HDL metabolism, such as cholesteryl ester transfer protein (CETP) inhibitors and niacin, reported disappointing results. The Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial revealed that the combination of atorvastatin with the CETP inhibitor torcetrapib increased HDL cholesterol levels by 72.1 % relative to atorvastatin alone (Barter et al. 2007b). This raise in HDL cholesterol upon torcetrapib administration did, however, not lower the risk of the major cardiovascular outcome (Barter et al. 2007b). On the contrary, patients in the atorvastatin/torcetrapib group unexpectedly experienced significantly more adverse events (Barter et al. 2007b). More recently, another phase 3 clinical trial involving patients with a recent acute coronary syndrome, named dal-OUTCOMES, was designed to evaluate the effects of the CETP inhibitor dalcetrapib (Schwartz et al. 2012). Addition of dalcetrapib to statin therapy raised HDL cholesterol levels by 31–40 % but failed to reduce the risk of recurrent cardiovascular events (Schwartz et al. 2012). Niacin is another effective agent for raising HDL cholesterol and also lowering LDL cholesterol, triglycerides, and lipoprotein(a). Data from the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) trial, which included patients with established cardiovascular disease, showed that adding extended-release niacin to statin therapy was not effective in reducing the risk of cardiovascular events during a follow-up of 36 months despite a 25 % increase in HDL cholesterol (Boden et al. 2011).
Genetic studies also questioned a role for HDLs in atherothrombotic diseases. Lifelong reductions of plasma HDL cholesterol levels due to heterozygosity for a loss-of-function mutation in ABCA1 did not affect the risk for ischemic heart disease (Frikke-Schmidt et al. 2008). Likewise, genetic variation in the apolipoprotein A-I (apoA-I) gene, APOA1, affecting apoA-I and HDL cholesterol levels did not predict ischemic heart disease or myocardial infarction in the general population (Haase et al. 2010). A more recent Mendelian randomization analysis demonstrated that there is no significant association between risk of myocardial infarction and a loss-of-function SNP in the gene coding for endothelial lipase (LIPG Asn396Ser), which results in elevated levels of HDL cholesterol (Voight et al. 2012). Hence, there is ongoing discussion whether HDLs are causally involved in the development of atherothrombotic diseases or are merely an innocent bystander. On the other hand, there is considerable evidence from in vitro and in vivo studies that HDL particles isolated from human plasma as well as artificially reconstituted HDLs (rHDLs) exert multiple atheroprotective effects (Annema and von Eckardstein 2013).
The above-outlined controversies have shifted the interest of the cardiovascular research field to HDL functionality, meaning that the functional quality of the particles might be more important than the absolute amount of HDL cholesterol. Currently, the available HDL-targeted therapies for atherothrombotic diseases raise plasma HDL cholesterol levels per se. Nonetheless, plasma concentrations of HDL cholesterol do not necessarily reflect the functional capacity of HDL particles. In addition, there is evidence that the anti-atherogenic functions of HDLs are impaired in patients with cardiovascular disease (Annema and von Eckardstein 2013), and these dysfunctional HDLs might even promote atherothrombotic diseases. Therefore, a better understanding of the molecular mechanisms behind the protective functions of HDLs in the context of atherothrombotic diseases is fundamental for successful exploitation of HDLs for the treatment of coronary heart disease.
2 Interactions of HDLs with the Endothelium
2.1 Preservation of Endothelial Integrity
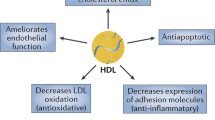
Perturbations in the function of endothelial cells lining the lumen of blood vessels are an important hallmark of the earliest stages of the atherosclerotic process. HDLs may improve or help reverse endothelial dysfunction by several endothelial-vasoprotective means. Several in vitro studies indicate that HDLs are able to counteract endothelial cell death. Treatment of endothelial cells with HDLs caused a marked attenuation of apoptosis in response to pro-atherogenic signals such as oxidized LDL (Suc et al. 1997; de Souza et al. 2010; Kimura et al. 2001), tumor necrosis factor α (TNFα) (Sugano et al. 2000; Riwanto et al. 2013), and growth factor deprivation (Kimura et al. 2001; Nofer et al. 2001b; Riwanto et al. 2013). Although the underlying mechanism has not been fully elucidated, it has been suggested that HDLs regulate endothelial cell survival by interfering with critical steps in apoptotic pathways. Several studies reported that HDLs block the activation of the mitochondrial pathway of apoptosis by preventing depolarization of the mitochondrial transmembrane and subsequent mitochondrial release of cytochrome c and apoptosis-inducing factor into the cytoplasm (Nofer et al. 2001b; de Souza et al. 2010). As a consequence, HDLs inhibit the cytochrome c-initiated caspase activation cascade (Sugano et al. 2000; Nofer et al. 2001b; de Souza et al. 2010). Moreover, levels of the apoptosis-inducing protein truncated Bid are reduced in endothelial cells cultured in the presence of HDLs, whereas endothelial expression of the antiapoptotic protein Bcl-xL is enhanced (Riwanto et al. 2013). The capacity of HDLs to attenuate apoptosis in endothelial cells requires induction of phosphatidylinositol 3-kinase (PI3K)/Akt/endothelial nitric oxide synthase (eNOS) signaling (Nofer et al. 2001b; Riwanto et al. 2013) and was attributed to apoA-I (Radojkovic et al. 2009) as well as HDL-associated lysophospholipids (Kimura et al. 2001; Nofer et al. 2001b). More recently, a lower clusterin (or apoJ) and increased apoC-III content in the HDLs of patients with stable coronary artery disease or acute coronary syndrome was found to be associated with reduced antiapoptotic activities in human endothelial cells (Riwanto et al. 2013).
Disruption of vascular endothelium integrity is another central event promoting atherosclerotic plaque formation. Sphingosine-1-phosphate (S1P), a bioactive sphingolipid metabolite, has been identified as an important endothelial barrier function-enhancing agonist (Garcia et al. 2001). In plasma S1P preferentially associates with HDL particles via binding to apoM (Christoffersen et al. 2011). In vitro studies provided evidence that delivery of S1P carried by apoM-containing HDLs to S1P1 receptors on endothelial cells leads to activation of Akt and eNOS, which in turn promotes endothelial barrier integrity (Argraves et al. 2008; Wilkerson et al. 2012). Interestingly, the lung vascular barrier function, as determined by Evan’s blue extravasation, is decreased in mice that lack S1P in their HDL fraction due to deficiency of apoM (Christoffersen et al. 2011). These data underline a significant role of HDL-associated S1P in endothelial barrier protection in vivo. In this context it is important to highlight the submicromolar concentration of S1P in HDLs which amounts to only 5–10 % of HDL particle concentration but is in the range of the affinity constants of the G-protein-coupled S1P receptors (Annema and von Eckardstein 2013; Karuna et al. 2011). Moreover, erythrocytes release S1P in response to HDLs (Lucke and Levkau 2010) so that in vivo the local concentration of S1P may be considerably higher than in isolated HDLs.
Acceleration of reendothelialization after vascular injury is another mechanism proposed to contribute to the positive influence of HDLs on endothelial function. In this respect, HDLs might be specifically important in driving endothelial cell migration during repair. Incubation of cultured endothelial cells with HDLs stimulated cell migration in response to wounding (Seetharam et al. 2006). Moreover, hepatic expression of human apoA-I rescued the impaired carotid artery reendothelialization following perivascular electric injury in apoA-I knockout mice (Seetharam et al. 2006). The promotion of endothelial cell migration by HDLs has been demonstrated to involve scavenger receptor BI (SR-BI)-mediated activation of Rac-directed formation of actin-based lamellipodia (Seetharam et al. 2006). An SR-BI-dependent mechanism for the promigratory actions of HDL was confirmed by experiments showing that the capacity of HDLs to stimulate endothelial cell migration was reduced by transfection of endothelial cells with siRNA specific to PDZ domain-containing protein 1 (PDZK1), an SR-BI adaptor protein that controls steady-state SR-BI levels, and that the reendothelialization capacity of injured vessels is lost in mice in which the pdzk1 gene has been disrupted (Zhu et al. 2008). Other studies suggested that HDL-induced endothelial cell migration is mediated by stimulation of Akt/eNOS phosphorylation through HDL-bound S1P in an S1P1-dependent manner and that this process is facilitated by endothelial lipase (Tatematsu et al. 2013).
Besides endothelial cell migration, also recruitment and incorporation of bone marrow-derived endothelial progenitor cells (EPCs) to sites of vessel injury contributes to the repair of the damaged vessel wall. HDLs activate the PI3K/Akt pathway in EPCs resulting in enhanced EPC proliferation, migration, and survival (Sumi et al. 2007; Petoumenos et al. 2009; Zhang et al. 2010). Moreover, preincubation of EPCs with HDL augmented their adhesive capacity to endothelial cells and potentiated their transendothelial migration (Petoumenos et al. 2009). Mice that were injected with rHDLs had an increased number of circulating EPCs and showed improved reendothelialization after carotid artery injury (Petoumenos et al. 2009). Additional mouse studies indicated that infusion of rHDLs enhanced bone marrow progenitor cell mobilization, neovascularization, and functional recovery after hind limb ischemia in wild-type mice (Sumi et al. 2007). In contrast, no beneficial effects of rHDL treatment on ischemia-induced angiogenesis were observed in eNOS knockout mice (Sumi et al. 2007), suggesting an essential role for eNOS in mediating HDL-stimulated EPC recruitment and the subsequent functional effects on the ischemic limb. There is some evidence that HDLs also affect circulating EPCs in humans. In patients with coronary artery disease, higher HDL cholesterol levels were significantly correlated with higher circulating EPC numbers (Petoumenos et al. 2009). These findings are corroborated by data from a study in patients with type 2 diabetes mellitus which found that rHDL infusion at a dose of 80 mg/kg body weight markedly increased circulating EPC levels compared to baseline (van Oostrom et al. 2007).
2.2 Preservation of Endothelial Function
Other reports on HDL functionality in endothelial cells established that HDLs have direct effects on vascular endothelium-relaxing properties. HDLs induce endothelium-derived vasorelaxation in aortic segments ex vivo in a dose-dependent manner (Nofer et al. 2004). Mechanistically, binding of HDL to SR-BI and the lysophospholipid receptor S1P3 on endothelial cells leads to PI3K-stimulated activation of Akt and mitogen-activated protein (MAP) kinase pathways, and Akt in turn phosphorylates eNOS at Ser-1177 to increase endothelial nitric oxide (NO) production and ultimately endothelium-dependent vasorelaxation (Nofer et al. 2004; Mineo et al. 2003; Yuhanna et al. 2001). The stimulatory effect of HDLs on NO-dependent vasorelaxation could be mimicked by three HDL-associated lysophospholipids, i.e., sphingosylphosphorylcholine, S1P, and lysosulfatide (Nofer et al. 2004). In addition to eNOS activation, modulation of Akt and MAP signaling by HDLs has been reported to delay the degradation of eNOS mRNA in endothelial cells, thereby upregulating eNOS protein levels (Ramet et al. 2003). Dietary oxysterols, in particular 7-oxysterols, impair endothelial vasorelaxant function by disrupting the active dimeric form of eNOS and by inducing production of reactive oxygen species (ROS) (Terasaka et al. 2010). HDLs can counteract the negative effects of 7-oxysterols on endothelial eNOS function by promoting efflux of 7-oxysterols from endothelial cells via ATP-binding cassette transporter G1 (ABCG1) (Terasaka et al. 2010). The same researchers revealed that HDL-mediated removal of cholesterol and oxysterols from endothelial cells through ABCG1 could reverse the cholesterol loading-induced inactivation of eNOS resulting from its increased interaction with caveolin (Terasaka et al. 2010). Similar to NO, there is evidence to suggest that the release of the endothelium-derived vasodilator prostacyclin can be influenced by HDLs. In human umbilical vein endothelial cells, HDLs elevated cyclooxygenase-2 protein expression and thereby induced prostacyclin synthesis via a pathway that involved PKC, ERK1/2, and sphingosine kinase SphK-2 (Xiong et al. 2014). The human relevance of these findings is underscored by studies demonstrating that infusion of rHDLs in hypercholesterolemic patients led to normalization of endothelium-dependent vasodilation in response to acetylcholine (Spieker et al. 2002). Moreover, heterozygosity for a loss-of-function mutation in ABCA1 has been reported to be associated with low levels of HDL cholesterol as well as an impaired endothelium-dependent forearm vasodilatory response, a phenotype which could be restored by intravenous administration of apoA-I/phosphatidylcholine discs (Bisoendial et al. 2003).
3 Anti-inflammatory Effects
3.1 Suppression of Myelopoiesis
Vascular inflammation, one of the main forces driving the formation of atherosclerotic plaques, is accompanied by the rolling, firm adhesion, and subsequent migration of monocytes across the vascular endothelium. A first critical determinant for monocyte transmigration is the number of monocytes present in the circulation. HDL-mediated cholesterol efflux pathways appear to play a prime role in the proliferation of myeloid cells in the bone marrow, cell mobilization from the bone marrow, and subsequent myeloid differentiation, at least in mice. Mice that lack both ABCA1 and ABCG1 have increased numbers of myeloid progenitor cells in bone marrow, blood, spleen, and liver, suggestive of aberrant myeloproliferation, myeloid progenitor cell mobilization, and extramedullary hematopoiesis (Yvan-Charvet et al. 2010; Westerterp et al. 2012). As a consequence monocyte counts in blood are elevated in ABCA1 x ABCG1 double knockout mice (Yvan-Charvet et al. 2010). In addition, the myeloproliferative phenotype and the enhanced mobilization of hematopoietic progenitor cells associated with bone marrow deficiency of ABCA1 and ABCG1 were reversed in mice carrying a human apoA-I transgene, and in high cholesterol diet-fed LDL receptor knockout mice, this was associated with less severe atherosclerosis (Yvan-Charvet et al. 2010; Westerterp et al. 2012). Correspondingly, raising HDL cholesterol levels in other murine models of myeloproliferative disorders also efficiently reduced the appearance of hematopoietic progenitor cells in the circulation (Westerterp et al. 2012). Direct proof for a link between HDL and myeloproliferation was provided by experiments showing that incubation of myeloid progenitor cells with HDLs ex vivo suppressed cell expansion (Yvan-Charvet et al. 2010). Taken together, these findings indicate that HDLs may beneficially impact the number of circulating monocytes. In humans there is some evidence for a correlation between HDL cholesterol levels and monocyte numbers in patients with familial hypercholesterolemia (Tolani et al. 2013). However, only few and inconsistent data on the association between monocyte counts and risk of atherosclerotic vascular disease in humans has been reported. In addition, it will be important to consider the heterogeneity of monocytes for any association with coronary heart disease (Zawada et al. 2012).
3.2 Suppression of Monocyte Extravasation
The arrest of circulating monocytes on inflamed endothelial monolayers is triggered by chemokines and their corresponding receptors, which both have been demonstrated to be subject to regulation by HDLs. Infusions of lipid-free apoA-I in apoE-deficient mice that were fed a high-fat diet reduced the relative abundance of cells stained positive for the chemokine receptors CCR2 and CX3CR1 in atherosclerotic plaques in the aortic sinus and decreased the circulating levels of the chemokines CCL2 and CCL5 (Bursill et al. 2010). Moreover, in vitro rHDLs were found to downregulate cell surface expression of chemokine receptors on monocytes and to render human monocytes and endothelial cells partly resistant to phytohemagglutinin- or cytokine-induced expression of chemokines (Bursill et al. 2010; Spirig et al. 2013). Although it was shown that HDLs counterbalance the upregulation of chemokines in response to pro-inflammatory cytokines by reducing nuclear factor-κβ (NF-κβ) pathway activation, how HDLs modulate monocyte expression of chemokine receptors on the molecular level remains to be established (Bursill et al. 2010). In addition to these effects on monocytes and endothelial cells, HDLs were also able to limit the production of CCL2 in vascular smooth muscle cells (VSMCs) exposed to thrombin (Tolle et al. 2008). The study identified HDL as a negative regulator of thrombin-induced activation of Rac1-dependent NADPH oxidases and subsequent ROS generation. This process is mediated by binding of HDL to SR-BI on VSMCs, and moreover lysosphingolipids present in HDL particles can act on local S1P3 receptor to inhibit CCL2 expression (Tolle et al. 2008).
Once monocytes are attracted by chemokines to inflamed endothelium, monocyte–endothelial cell interactions are mediated by several cell adhesion molecules. Experiments in vitro provided compelling evidence that cytokine-induced adhesion molecule expression on the endothelial cell surface is diminished in the presence of either native HDLs (Cockerill et al. 1995; Ashby et al. 1998) or rHDLs (Baker et al. 1999; Clay et al. 2001). Notably, the effects of HDLs on endothelial expression of adhesion molecules not only occurred in cultured cells but also in the in vivo setting under inflammatory conditions. For example, a single injection of discoidal rHDL particles in pigs prevented upregulation of E-selectin expression in response to intradermal interleukin-1α (Cockerill et al. 2001). Moreover, daily treatment of normocholesterolemic rabbits with rHDLs or lipid-free apoA-I markedly attenuated periarterial collar-induced endothelial expression of vascular cell adhesion molecule 1 (VCAM-1) and intercellular adhesion molecule 1 as well as concomitant neutrophil infiltration into the vessel wall and generation of ROS (Nicholls et al. 2005). The positive effects of lipid-free apoA-I on pro-inflammatory vascular changes were still observed when administered as a single low dose 3 h after insertion of the non-occlusive periarterial collar (Puranik et al. 2008), indicating that infusion of apoA-I may also be able to rescue preestablished vascular inflammation. In patients suffering from claudication, treatment with rHDLs led to a significant reduction in the percentage of VCAM-1 positive cells in femoral artery lesions (Shaw et al. 2008). Diminution of adhesion molecule expression on endothelial cells by HDLs may involve the activation of multiple receptors and signaling cascades. There are data supporting that HDLs alter adhesion molecule expression as a consequence of perturbation of cellular sphingolipid metabolism and the NF-κβ signaling pathway. HDL3 was found to impede TNFα-induced upregulation of VCAM-1 and E-selectin expression in human umbilical vein endothelial cells by antagonizing activation of sphingosine kinase and subsequent intracellular formation of S1P (Xia et al. 1999). Furthermore, it has been documented that in cultured endothelial cells, incubation with HDLs prevented NF-κβ activation, movement of p65 to the nucleus, and expression of NF-κβ target genes in response to TNFα (Xia et al. 1999; Park et al. 2003; Kimura et al. 2006; McGrath et al. 2009). The basal molecular mechanisms of the beneficial effects of HDL on endothelial inflammation are dependent on an increase in the expression of 3β-hydroxysteroid-Δ24 reductase that results in activation of PI3K and downstream activation of eNOS and heme oxygenase-1 (HO-1) (McGrath et al. 2009; Wu et al. 2013). Specific knockdown of SR-BI or S1P1 receptor by treatment of human endothelial cells with small interfering RNAs against SR-BI and PDZK1 or S1P1, respectively, markedly reduced the ability of HDLs to inhibit VCAM-1 expression and NF-κβ activation (Kimura et al. 2006; Wu et al. 2013), suggesting that both receptors may be critical for the anti-inflammatory endothelial actions of HDLs. Corresponding with the possible involvement of the S1P1 receptor, the inhibitory activity of HDLs on the expression of endothelial cell adhesion molecules has been proposed to be a function of biologically active lysosphingolipids carried by HDL particles (Nofer et al. 2003; Kimura et al. 2006). Besides the lipid component, the anti-inflammatory properties of HDLs could also be related to the microRNA content of HDL particles. Recently, it has been reported that transfer of microRNA-223 by HDLs to human aortic endothelial cells significantly repressed expression of intracellular adhesion molecule 1 (Tabet et al. 2014).
Monocytes bind to adhesion molecules on activated endothelial cells through monocytic cell surface receptors, such as CD11b. Ex vivo incubation of stimulated human peripheral blood monocytes with apoA-I in the lipid-free or HDL-associated state caused a reduction in cell surface expression of CD11b (Murphy et al. 2008). These results might be directly translatable to the human in vivo situation, as evidenced by the observation that infusion of rHDLs in patients with atherosclerosis disease in the femoral artery led to a significantly lower expression of CD11b on circulating monocytes (Shaw et al. 2008). The fact that cellular depletion of cholesterol by cyclodextrin mimicked the protective effects of apoA-I on the expression of monocyte CD11b and that inhibition of CD11b expression is lost in the presence of an antibody against ABCA1 or in monocytes derived from Tangier disease patients is indicative of a role for ABCA1-mediated cholesterol efflux in the mechanism of monocyte deactivation by apoA-I (Murphy et al. 2008).
3.3 Interference with Macrophage Differentiation and Activation
Monocytes that have entered the arterial intima subsequently differentiate into resident macrophages of at least two major subtypes that differentially produce a variety of pro- and anti-inflammatory mediators. In the subendothelial space, the macrophages become activated and comprise key targets for the anti-inflammatory action of HDLs. Incubation with HDLs primed isolated murine bone marrow macrophages into alternative M2 macrophages with anti-inflammatory features (Sanson et al. 2013). Moreover, HDLs were potent inhibitors of interferon-gamma-mediated expression of pro-inflammatory M1 markers in isolated mouse macrophages, and, interestingly, experiments performed with macrophages from STAT6-deficient mice demonstrated that STAT6 plays an essential role in the HDL-mediated shift of the macrophage phenotype to M2 (Sanson et al. 2013). However, other investigators could not replicate these observations, when using primary human monocyte-derived macrophages (Colin et al. 2014). A recent study provided novel mechanistic insights into the anti-inflammatory effects of HDLs in macrophages. Thus, administration of native HDL, rHDL, or apoA-I to mice challenged with a toll-like receptor (TLR) agonist protected from liver damage and reduced serum levels of pro-inflammatory cytokines, which was supposed to be due the ability of HDLs to reduce cytokine production in activated macrophages (De Nardo et al. 2014). The anti-inflammatory action of HDL particles on macrophages was explained by their ability to induce the expression of activating transcription factor 3 (ATF3), which functions as a repressor of pro-inflammatory target genes (De Nardo et al. 2014).
During the past decades, a considerable amount of data has been published revealing that natural regulatory T cells, which dampen inflammatory responses, may help decrease the atherosclerotic disease burden. The findings of a recent murine study imply that apoA-I could promote the expansion of regulatory T cells. Subcutaneous injection of human lipid-free apoA-I in LDL receptor/apoA-I double knockout mice fed an atherogenic diet restored the regulatory T-cell population in the lymph nodes (Wilhelm et al. 2010). In these animals the raise in the number of regulatory T cells in the lymph nodes was accompanied by a reversal of the inflammatory and autoimmune phenotype.
4 Effects of HDLs on Vascular Lipid and Lipoprotein Homeostasis
4.1 From LDL Retention to Atheroma Formation
4.1.1 Initial Lipid Accumulation in Atherosclerosis-Prone Arterial Intima: A Sign of LDL Retention
The intima is disproportionally thick at the outer curvatures of the proximal branches of coronary and carotid arteries, where turbulent flow conditions prevail and the pulse wave amplitude is relatively high (Wentzel et al. 2012). The thickening is a physiological response to these conditions and ensues when the vascular smooth muscle cells (VSMCs) divide and secrete various components of the extracellular matrix at the stressed sites. One major factor explaining the relationship between the thickness of the intima and its susceptibility to atherosclerotic lesion development is the lack of a capillary delivery system and lack of a lymphatic drainage system in this tissue (Hulten and Levin 2009; Eliska et al. 2006). As a consequence, the inflowing apoB100-containing lipoprotein particles (notably LDL and also VLDL remnants) are only slowly conveyed by the intimal fluid from the subendothelial superficial layer into the deeper intimal layers and ultimately to the medial layer of the arterial wall where lymphatic capillaries are present. Indeed, in cholesterol-fed rabbits, intimal LDL concentration initially increases in atherosclerosis-susceptible sites, where their intimal residence time of the LDL particles is long, the transit time being calculated to be hours (Schwenke and Carew 1989a, b).
Since progressive accumulation of LDL cholesterol is a key feature of atherogenesis, we can deduce that some intimal LDL particles never leave the site. The constant inflow without compensatory outflow of LDL particles results in a dramatic increase in their concentration in the intimal fluid to reach values equaling or even exceeding (by twofold) those in the corresponding human blood plasma (Smith 1990). Thus, the concentration of LDL in the arterial intimal fluid is 10–20 times higher than in the extracellular fluids of other extrahepatic tissues, in which its concentration is about 1/10th of that in the corresponding plasma, so rendering the intima a truly “hypercholesterolemic” site.
During their lengthy passage across the thick avascular tissue, the LDL particles tend to bind to the subendothelial extracellular proteoglycan matrix, a phenomenon known as retention of LDL (Williams and Tabas 1995). The proteoglycans form a tight network that is negatively charged. To this network the LDL particles bind via ionic interactions, when positively charged lysine and arginine residues located in the proteoglycan binding site of apoB100 interact with the negatively charged sulfate groups of the glycosaminoglycan components of the proteoglycans (Camejo et al. 1998). The prolonged residence time of LDL particles in the intima, whether the particles are in solution or trapped in the matrix, enhances their exposure to a variety of local LDL-modifying factors. Thus, during their long extracellular residence time, the LDL particles are modified, for example, by oxygen radicals secreted by the resident cells of the intima, notably by endothelial cells, and also by SMCs. It is of particular relevance that the endothelial cells become activated to secrete pro-oxidative compounds at sites with turbulent flow, i.e., at the atherogenesis-prone sites of the arterial tree (Nigro et al. 2011). Moreover, the subendothelially located LDL particles are not protected by the powerful antioxidant systems present in the circulating blood plasma, and they also gradually become depleted of their own antioxidant molecules capable of protecting them from free radical attack and oxidation (Esterbauer et al. 1991). In addition to the oxidative processes, various hydrolytic (both proteolytic and lipolytic) enzymes may also modify the LDL particles in the intima (Tabas 1999). Importantly, the oxidative and hydrolytic changes of LDL particles render them unstable, trigger their aggregation and fusion into lipid droplets, and markedly strengthen their binding to arterial proteoglycans (Oorni et al. 2000). These in vitro observations have their close counterparts in various animal models, and also in the human carotid arteries, in which tiny proteoglycan-, collagen-, and elastin-associated lipid deposits, detectable only by aid of special electron microscopic techniques, are located in the subendothelial extracellular space even before any microscopic signs of atherosclerosis are visible (Nievelstein-Post et al. 1994; Tamminen et al. 1999; Pasquinelli et al. 1989).
4.1.2 Formation of a Fatty Streak
As stated above, atherosclerosis typically develops at certain predilection sites as a response to the hemodynamic stress and other types of injury affecting the endothelial cells. Importantly, in such stressed areas with disturbed flow conditions, endothelial inflammatory signaling pathways become activated and a local pro-inflammatory environment is created (Nigro et al. 2011). We can consider this local scenario as a singular starting condition to the development of atherosclerosis. Thus, the flow-mediated preconditioning of the intima renders it a suitable soil for the retention, modification, and accumulation of LDL particles. Because the subendothelially located modified LDL particles and the products released from them are pro-inflammatory, they are likely to have profound local cell-activating effects and to worsen the inflammation by inducing local generation of various LDL-modifying enzymes and agents and so creating a positive feedback loop (Pentikainen et al. 2000; Tabas et al. 2007). Moreover, synthesis and secretion of pro-inflammatory cytokines and chemokines, such as the monocyte chemotactic protein-1 (MCP-1; CCL2), are induced (Libby 2002; Zernecke and Weber 2010). Indeed, monocytes preferentially adhere to the endothelium covering inflamed tissue sites, i.e., regarding atherogenesis, they tend to enter the intimal areas containing oxidized or otherwise modified LDL particles (Schmitt et al. 2014a, b).
The extracellularly located modified LDL particles, whether free floating in the intimal fluid or whether matrix bound, are ingested by intimal macrophages and, to some extent, also by intimal SMCs (Witztum 2005; Wang et al. 1996). On their surfaces, the macrophages express scavenger receptors, which recognize oxidized and otherwise modified LDL particles (Krieger and Herz 1994). The macrophages may also phagocytose modified LDL particles, provided the particles are in aggregate form or have been converted into droplets via fusion. Since uptake of LDL cholesterol by macrophages is not negatively feedback regulated by the inflowing cholesterol, uptake of LDL cholesterol, be it scavenger receptor dependent or mediated by other mechanisms, may continue until the cells are filled with cholesterol (Brown and Goldstein 1983). Of great interest, also unmodified native LDLs can be ingested by macrophages at a rate which is linear to their concentration in the extracellular fluid (Kruth 2011). Since the concentration of LDL in the intimal fluid is exceptionally high (see above), such micropinocytotic uptake of LDLs by macrophages may actually be of great significance in the atherosclerosis-prone arterial intima.
In the macrophages, the excess of intracellular cholesterol is esterified with fatty acids and stored in their cytoplasm as cholesteryl ester droplets (Brown and Goldstein 1983). Macrophages full of such droplets look foamy under the microscope and are called foam cells. When numerous foam cells form clusters, they have yellow appearance visible to the naked eye and the site appears as a fatty streak (Stary et al. 1994). Importantly, when fatty streaks develop in human carotid arteries, the subendothelial perifibrous lipid droplets disappear (Pasquinelli et al. 1989). This switch conceivably reflects uptake of the perifibrous droplets by the subendothelial macrophages. Thus, in a fatty streak the LDL-derived cholesterol is located mostly intracellularly. Taken together, an important role of subendothelial macrophages in the arterial intima is to clear unmodified or modified LDL particles from their surrounding and to store their cholesterol as cholesteryl esters. Indeed, foam cells in the arterial intima are the most defining feature of atherosclerosis.
HDLs directly bind unesterified cholesterol. As the consequence, HDL can mobilize unesterified cholesterol both from cells and extracellular locations (Freeman et al. 2014). Regarding cholesteryl esters, the extracellularly stored esters, present either in the core of the matrix-bound LDL particles or in the core of the perifibrous lipid droplets derived from modified LDL particles, are likely to fully resist transfer to HDLs. In sharp contrast, intracellular cholesterol stored as cholesteryl esters in the cytoplasmic lipid droplets of macrophage foam cells is sensitive to transfer to HDLs (Rosenson et al. 2012). Thus, regarding the return of LDL cholesterol from the arterial intima back to the circulating blood, it appears to be of fundamental importance that the LDL particles are first taken up and metabolized by intimal macrophages. Accordingly, this macrophage-dependent preparative step appears to be mandatory for the initiation of reverse cholesterol transport in the arterial intima. However, since the macrophages accumulate LDL-derived cholesterol, an imbalance between cholesterol influx and cholesterol efflux exists in the atherosclerosis-prone arterial intima. One reason for the relative inability of HDLs to effectively remove cholesterol from the intimal macrophages is their functional modification including proteolysis, oxidation, and lipolysis to various extents (Lee-Rueckert and Kovanen 2011; DiDonato et al. 2013).
4.1.3 Development of an Atheroma
Conversion of a fatty streak into an atheroma is the next phase in atherogenesis (Stary et al. 1995). It is characterized by extracellular accumulation of cholesterol in the deep layers of the intima, i.e., below the superficial foam cell layers. However, in sharp contrast to the superficial layers of the intima, where atherogenesis initiates and where macrophages swiftly arrive and efficiently remove the lipids so preventing their extracellular accumulation, in the deep layers, the macrophages arrive late (Nakashima et al. 2008). Accordingly, once formed, the deep extracellular lipids are not rapidly scavenged by macrophages. Rather, the continuous formation of lipid droplets without their concomitant removal leads to their progressive deposition, which ultimately leads to the appearance of “lipid lakes”. The lakes consist of huge numbers of such droplets, and together the lakes form a lipid core (Guyton and Klemp 1996). The presence of a lipid core is the hallmark of an atheroma.
During the process of lipid core formation, macrophages start to migrate from the superficial layer into the deep intimal area where they ingest the extracellular lipid droplets in the periphery of the lipid core and themselves become converted into foam cells. Since the deep intimal region is distant from the circulating blood, it easily becomes hypoxic (Hulten and Levin 2009). Although survival mechanisms are activated in macrophages trapped in such hypoxic areas (Ramkhelawon et al. 2013), promotion of lipid accumulation, ATP depletion, and immobilization of the macrophage foam cells is likely to ultimately lead to their death. Of note, death of a foam cell is bound to liberation of its cytoplasmic cholesteryl ester droplets into the extracellular space, where the droplets mix with the preexisting extracellular lipid droplets. Moreover, the nonviable macrophages (foam cells) fail to effectively remove the cellular debris derived from the dying or dead foam cells (Thorp et al. 2011), so leaving the formed debris deposited among the lipids of the growing core. A lipid core also containing remnants derived from dead cells is called a “necrotic lipid core”.
In summary, regarding cholesterol metabolism in the arterial intima, the superficial and the deep layers appear to be fundamentally different. Mechanistically, in the superficial compartment of the arterial intima, viable macrophages ingest and store LDL cholesterol as cholesteryl esters, and transfer it to HDLs for ultimate return to the circulation, continual hydrolysis of the cytoplasmic cholesteryl esters being the critical factor for the transfer of cholesterol from LDL to HDL. In contrast, in the deep intimal compartment, macrophages only inefficiently remove LDL cholesterol, and, provided a macrophage is converted into a foam cell, it may die and release its cholesteryl ester cargo into the extracellular space, where the cholesteryl esters are not available for removal by HDLs. Thus, in the absence of an efficient transfer of cholesterol from LDL to HDL, the necrotic lipid core grows progressively and may convert an atheroma into a clinically significant atherosclerotic lesion.
4.2 Inhibition of LDL Modification by HDLs
Resident macrophages in the subendothelial space take up oxidized LDLs via scavenger receptors and become macrophage foam cells, the hallmark cell type of atherosclerotic lesions. HDLs have been proven to help to reduce the formation of lipid-laden macrophage foam cells by rendering LDL particles resistant to oxidation. Preexposure of LDL particles to apoA-I or mature HDL particles completely prevented the oxidative modification of LDLs by cultured human aortic endothelial cells and SMCs (Navab et al. 2000). Moreover, the human artery wall cells were no longer able to oxidize LDLs isolated from mice and humans that received a single treatment with human apoA-I. Likewise, HDLs have been shown to possess potent antioxidative activity in chemical in vitro models for oxidative modifications of LDLs, using, for example, copper ions (Parthasarathy et al. 1990) or the more mild oxidizing agent 2,2′-azobis(2-amidinopropane) dihydrochloride (AAPH) (Kontush et al. 2003). At least part of the ability of HDLs to inhibit LDL oxidation was attributed to the capacity of apoA-I to remove seeding molecules present in freshly isolated LDLs (Navab et al. 2000). Notably, the reduction of lipid hydroperoxides to their corresponding hydroxides by HDLs was associated with the selective oxidation of Met residues in apoA-I to their Met sulfoxide form (Garner et al. 1998), suggesting that Met residues in apoA-I may act as endogenous antioxidants within the HDL particle.
In addition to apoA-I, antioxidant properties of HDLs are conferred by paraoxonase 1 (PON1), an antioxidant enzyme transported in the circulation by HDL particles. The inhibitory effect of HDLs on LDL oxidation by human artery wall cells or copper could be mimicked by incubation of LDLs with purified PON1 (Navab et al. 2000; Liu et al. 2008). After treatment of oxidized phospholipids with PON1, oxygenated polyunsaturated fatty acids were no longer detectable (Watson et al. 1995a), indicating that PON1 was able to destroy these multi-oxygenated biologically active molecules which are abundantly formed during the atherogenic oxidation of LDL particles. Studies in transgenic and knockout mice on the role of PON1 in the pathogenesis of atherosclerosis have supported the protective effects of PON1 on LDL oxidation. The lack of PON1 in the HDL fraction of PON1 knockout mice resulted in an inability of these HDL particles to prevent the formation of oxidized LDLs, and accordingly pon1-null mice were more susceptible to atherosclerosis than their wild-type littermates (Shih et al. 1998). Expression of human PON1 in transgenic mice led to the opposite phenotype. On the atherosclerosis-prone apoE −/− background, mice transgenic for human PON1 developed significantly less atherosclerotic plaques in the aorta than the control mice. Moreover, in vitro PON1-enriched HDLs from PON1 transgenic/apoE knockout were more potent inhibitors of copper-catalyzed oxidation of LDL particles (Tward et al. 2002).
Another antioxidant enzyme that has been related to the cardioprotective antioxidative function of HDL particles is lipoprotein-associated phospholipase A2 (Lp-PLA2), also known as platelet-activating factor acetylhydrolase (PAF-AH). In human plasma a small proportion of the circulating Lp-PLA2 enzyme activity is found in HDL particles (Tellis and Tselepis 2009). Inhibition of the enzymatic activity of Lp-PLA2 rendered HDLs unable to prevent monocyte binding induced by oxidized LDLs in endothelial cells, and supplementation of the dysfunctional HDL particles with purified Lp-PLA2 restored the normal protective function (Watson et al. 1995b). In mice, adenovirus-mediated expression of human Lp-PLA2 leads to a reduction in the autoantibody titers against oxidized LDLs and malondialdehyde (MDA)-modified LDLs (Quarck et al. 2001; Noto et al. 2003). Despite the fact that the antioxidative potential of HDL-bound Lp-PLA2 has been well documented, several lines of evidence suggest that the presence of Lp-PLA2 in LDL particles may actually stimulate atherogenesis. The pro-atherogenic actions of Lp-PLA2 in LDLs are thought to arise from oxidized free fatty acids and lysophospholipids, two inflammatory mediators which are released after the hydrolysis of oxidized phospholipids by Lp-PLA2 (Tellis and Tselepis 2009). LDLs are the preferred lipoprotein carriers for Lp-PLA2 in the circulation, which would explain why previous prospective cohort studies paradoxically linked high plasma Lp-PLA2 levels with adverse cardiovascular outcomes (Brilakis et al. 2005; Koenig et al. 2004; Sabatine et al. 2007). In support of the hypothesis that Lp-PLA2 is atheroprotective only when incorporated in HDL particles, Rallidis et al. found that elevated total plasma Lp-PLA2 mass and activity increased the risk for future cardiac death in patients with stable coronary artery disease, while Lp-PLA2 mass and activity in apoB-depleted plasma showed a significant association with future cardiac death in the opposite direction (Rallidis et al. 2012).
The protective effect of HDLs on LDL oxidation has been tentatively ascribed to the antioxidant activities of lecithin-cholesterol acyltransferase (LCAT). Similar to HDLs, purified LCAT has been shown to diminish the formation of both lipid hydroperoxides and conjugated dienes in copper-oxidized LDLs (Vohl et al. 1999). Moreover, adenovirus-mediated transfer of the human LCAT gene into mice with combined leptin and LDL receptor deficiency decreased circulating levels of autoantibodies binding to MDA-LDL by 40 % and accordingly limited the accumulation of oxidized LDLs in the arterial wall (Mertens et al. 2003).
4.3 Macrophage Cholesterol Efflux and Reverse Cholesterol Transport
The uptake of modified LDLs in arterial macrophages leads to the cellular accumulation of cholesterol and oxidized lipids. This process causes the macrophages to transform into lipid-laden macrophage foam cells which can no longer escape the arterial wall (Potteaux et al. 2011). The excessive cellular cholesterol in macrophage foam cells can be efficiently removed by the potent cholesterol acceptors lipid-free apoA-I and HDLs to be transported back to the liver by a process termed reverse cholesterol transport.
4.3.1 Transendothelial HDL Transport
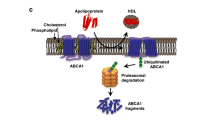
In order to reach these resident macrophage foam cells in the innermost layer of the arterial wall, apoA-I and HDLs first need to passage across the endothelial cell layer. There is cell culture evidence that lipid-free apoA-I binds to specific saturable high-affinity binding sites on aortic endothelial cells, which is followed by internalization, transcytosis, and delivery of lipidated apoA-I to the basolateral side (Rohrer et al. 2006). A similar phenomenon was observed when cultivated aortic endothelial cells were treated with mature HDLs (Rohrer et al. 2009). However, the mechanisms involved in regulating this endothelial transport of apoA-I and HDLs are not fully understood. The ABCA1 transporter is thought to play major role in the apical-to-basolateral transport of apoA-I in aortic endothelial cells (Cavelier et al. 2006). Induction of ABCA1 expression by a combination of oxysterol and 9-cis-retinoic acid increased the binding and internalization of apoA-I in endothelial cells, whereas knockdown of endothelial ABCA1 by RNA interference reduced the cellular uptake and transcytosis of apoA-I (Cavelier et al. 2006). Conversely, siRNA-mediated gene silencing experiments revealed that the cell surface binding and transport of HDLs through aortic endothelial cells are highly dependent on SR-BI and ABCG1, but not on ABCA1 (Rohrer et al. 2009). Subsequent research has demonstrated expression of the ectopic β-chain of F0F1 ATPase (β-ATPase) on the endothelial cell surface (Cavelier et al. 2012). The results of this study further suggest that the binding of lipid-free apoA-I to endothelial cell surface β-ATPase facilitates the uptake and transport of lipidated apoA-I and mature HDLs by enhancing β-ATPase-mediated hydrolysis of extracellular ATP into ADP and inducing consecutive activation of the purinergic P2Y12 receptor (Cavelier et al. 2012). More recently, transendothelial transport of HDL particles was found to be modulated by endothelial lipase both through its enzymatic lipolytic activity and its ability to bridge the binding of lipoproteins to the endothelial surface (Robert et al. 2013). Initiation of an atherosclerosis-related inflammatory phenotype after stimulation of vascular endothelial cells with interleukin-6 significantly increased the binding, cell association, and transport of HDLs, which was linked mechanistically to the increase in endothelial lipase expression in response to interleukin-6 (Robert et al. 2013).
4.3.2 Cholesterol Efflux from Macrophages
Once apoA-I and HDLs have reached macrophage foam cells in the atheromatous vessel wall, macrophage cholesterol efflux can be elicited via several different pathways. It is well documented that lipid-free apoA-I and pre-beta HDL particles are able to remove cholesterol and phospholipids from macrophage foam cells via the ABCA1 transporter (Wang et al. 2000; Wang and Tall 2003). It has been postulated that apoA-I-dependent cholesterol efflux from cells is mediated either by a direct protein–protein interaction between apoA-I and ABCA1 or indirectly by ABCA1-induced changes in the membrane cholesterol distribution (Wang and Tall 2003). The ability of ABCA1 to mobilize cellular lipids to apoA-I is essential for the initial lipidation of apoA-I. The ABCG1 transporter is responsible for a major part of the macrophage cholesterol efflux towards mature HDL particles (Wang et al. 2004). Since cellular removal of cholesterol via ABCG1 was not accompanied by specific binding of HDLs to the plasma membrane (Wang et al. 2004), the molecular basis of efflux of cholesterol mediated by ABCG1, like ABCA1, is not fully understood and warrants further investigation. Another cell surface receptor involved in macrophage cholesterol efflux elicited by mature HDLs is SR-BI. Binding of HDL to macrophage SR-BI facilitates a bidirectional exchange of unesterified cholesterol and other lipids between the cell membrane and HDL acceptor particles according to the cholesterol concentration gradient (de La Llera-Moya et al. 2001). This cholesterol concentration gradient between cells and HDL not only allows cholesterol transfer via SR-BI but also passive diffusion of cholesterol molecules to nearby HDL particles by receptor-independent processes (von Eckardstein et al. 2001; Yancey et al. 2003). The HDL-associated enzyme LCAT catalyzes the esterification of cholesterol in HDL particles (Calabresi and Franceschini 2010). The generated cholesteryl esters leave the particle surface and form the core of the maturing HDL particle; hence, a local gradient of unesterified cholesterol is created that maintains a continuous flow of unesterified cholesterol from macrophages towards HDLs (Calabresi and Franceschini 2010).
4.3.3 Exit from the Arterial Wall
There is little reported data on the route used by HDLs to leave the arterial intima. Recent lines of evidence support a role for the lymphatic vasculature in the transport of HDLs from the interstitial space back to the bloodstream. Following injection of fluorescently labeled HDLs in the mouse footpad, HDLs were subsequently found in the draining lymph node and its afferent and efferent lymphatic vessels (Lim et al. 2013). Moreover, the movement of cholesterol from macrophages in the footpad to the lymph and plasma was substantially reduced in mice with surgical interruption of the afferent lymphatic vessels or in Chy mutant mice that lack dermal lymphatic capillaries (Lim et al. 2013; Martel et al. 2013). The importance of the lymphatic system in the reentry of cholesterol originating from macrophages via HDLs in the circulation was further confirmed by additional experiments showing that the appearance of macrophage-derived cholesterol in the plasma was impaired after implantation of [3H]-cholesterol-labeled macrophages in the tail skin of apoA-I transgenic mice with microsurgical ablation of the major lymphatic conduits in the tail (Martel et al. 2013). To provide more direct proof for the involvement of the lymphatic vasculature in the transport of cholesterol from the atherosclerotic plaque, aortic segments with advanced atherosclerotic lesions were loaded with D6-cholesterol and transplanted in apoE knockout mice (Martel et al. 2013). Inhibition of regrowth of a functional lymphatic vasculature in the aortic donor wall by an antibody blocking the function of vascular endothelial growth factor receptor 3 markedly suppressed the apoE-induced removal of D6-cholesterol from the transplanted aorta (Martel et al. 2013), hence emphasizing the pivotal role of the lymphatic system for the egress of cholesterol from the aortic wall. The entry of HDL particles into lymphatic vessels appears to be primarily dependent on SR-BI. Experiments in cultured cells demonstrate that the internalization and transcytosis of HDL particles by lymphatic endothelial cells are diminished by an SR-BI blocking antibody or a selective SR-BI inhibitor (Lim et al. 2013). In agreement, lymphatic transport of HDLs is compromised in SR-BI-null mice and in mice treated with an antibody against SR-BI (Lim et al. 2013). Although not formally tested, it is also possible that, in order to reach the circulation again, HDL particles undergo transcytosis through the luminal endothelial cell barrier or transfer cholesterol to endothelial cells for delivery to circulating HDLs. The importance of a well-regulated homeostasis between entry and exit of HDL particles into and from the arterial wall, respectively, is indicated by the enrichment of modified and dysfunctional HDLs and apoA-I in atherosclerotic lesions (DiDonato et al. 2013, 2014; Huang et al. 2014).
4.3.4 Delivery of Cholesterol to the Liver and Intestine
After having left the arterial wall and entered the circulation, the HDL particles transport the cholesterol either directly or indirectly via apoB-containing lipoproteins to the liver for either secretion into bile or for de novo assembly of lipoproteins. The first direct way by which HDL can deliver cholesterol to the liver is via SR-BI, a cell surface receptor that binds HDLs and mediates the selective uptake of HDL-associated cholesteryl esters and the subsequent resecretion of cholesteryl ester-poor HDLs (Acton et al. 1996). Secondly, HDL particles can be taken up from the circulation by the liver via HDL holoparticle endocytosis (i.e., uptake of the whole HDL particle). The binding of apoA-I to the ectopic β-chain of F0F1 ATPase on hepatocytes triggers extracellular production of ADP, which in turn acts on the P2Y13 receptor to stimulate HDL holoparticle endocytosis (Martinez et al. 2003; Jacquet et al. 2005). In humans, the major fraction of the cholesteryl esters in HDL particles are shuttled via the action of CETP to apoB-containing lipoproteins (Charles and Kane 2012). These cholesteryl esters originating from HDLs are internalized as part of apoB-containing lipoproteins by hepatic LDL receptors. Following hepatic uptake, HDL-derived cholesterol is, at least in part, targeted for fecal excretion via the biliary route (Lewis and Rader 2005; Nijstad et al. 2011). Hepatic cholesterol can either be secreted directly in the free form into the bile by the ABCG5/G8 heterodimer or alternatively be converted by cholesterol 7α-hydroxylase (CYP7A1) and other enzymes into bile acids for hepatic excretion via the bile salt export pump (BSEP) (Dikkers and Tietge 2010). Secretion into bile enables cholesterol to move from the hepatocyte to the intestinal lumen for final excretion via the stool. This major HDL-driven pathway to eliminate atherogenic cholesterol from the body is also known as reverse cholesterol transport (Annema and Tietge 2012). Although reverse cholesterol transport from plaque macrophages to feces only represents a very small proportion of the total reverse cholesterol transport flux from peripheral tissues, it is key component of the reverse transport potentially capable of initiating and maintaining the regression of atherosclerosis (Lee-Rueckert et al. 2013).
5 Atherothrombosis
The most important mechanism of acute coronary syndromes, i.e., unstable angina, acute myocardial infarction, and sudden cardiac death, is rupture or erosion of a coronary atheroma (Libby 2013b). The risk of atheromatous rupture or erosion appears to critically depend on both the cellular and extracellular compositions of the atheroma. The superficial layer of an atheroma separating a necrotic lipid core from the circulating blood is called the “fibrous cap”. Although a cap usually contains foam cells, its critical components are VSMCs, which synthesize and secrete collagen and other components of the extracellular matrix. It is current understanding that the cap is thinning as the necrotic lipid core is growing. Whether the thinning is a response to the enlargement of the necrotic core, or whether thinning is an independent process and just allows the core to expand, has not been established.
As expected, a lesion with a thick fibrous cap and a small lipid core is stable, whereas a lesion with a thin fibrous cap and a large lipid core is unstable and prone to rupture. Indeed, it is now well recognized that acute coronary syndromes most commonly result from rupture of a thin-cap fibroatheroma (TCFA), which is characterized by a large necrotic core with an overlying thin fibrous cap measuring less than 65 μm (Sakakura et al. 2013; Fujii et al. 2013).
Provided a stable lesion with a thick fibrous cap causes significant stenosis of the arterial lumen, and turbulent flow conditions are created, some endothelial cells may detach, that is, the plaque surface becomes eroded (Libby 1995; Farb et al. 1996). Apoptosis of endothelial cells also leads to endothelial denudation, i.e., to plaque erosion when, for example, subendothelial mast cells degrade the basement membrane of endothelial cells (Durand et al. 2004; Mayranpaa et al. 2006). The exposed subendothelial structures are prothrombotic and a platelet-rich thrombus is formed locally. Since the activated platelets secrete platelet-derived growth factor which stimulates SMCs to divide and generate extracellular matrix, a neointima is formed at the site of erosion. This is one mechanism by which the fibrotic cap of a stenosing plaque becomes thicker and the plaque grows into the lumen and so aggravates the local stenosis.
When an unstable TCFA ruptures, the flowing blood becomes exposed to the necrotic lipid core. The various components of the core, notably tissue factor released by macrophages, apoptotic bodies, remnants of dead cells, and extracellularly located lipids, all are contributing to the strong thrombogenic response usually associated with a plaque rupture (Fuster et al. 2005; Corti et al. 2003). Thus, the platelet-rich arterial thrombus growing at the site of plaque rupture easily becomes occlusive, even if the plaque only had caused a mild stenosis which was hemodynamically nonsignificant.
In view of the importance of the thickness of collagen-rich matrix of the fibrous cap in determining plaque stability and instability, it is essential to understand the factors that may regulate its thickness. Regarding such regulation, two related observations made in advanced human atherosclerotic plaques are of prime importance. First, irrespective of the structure of an atherosclerotic plaque, the actual site of atheromatous ulceration with ensuing local formation of a platelet-rich thrombus is characterized by an inflammatory process (van der Wal et al. 1994). Thus, atheromatous coronary and carotid ulcerations have invariably shown the presence of inflammatory cell infiltrates, composed of macrophages, T cells, and mast cells (Kovanen et al. 1995; Bui et al. 2009). When compared with the evolving atherosclerotic plaques, which contain inflammatory cell infiltrates, composed of various subsets of macrophages and T cells and also of mast cells and neutrophils (Woollard 2013), the vulnerable plaques appear to be equally, if not even more immunologically, active. The second critical aspect of a vulnerable atherosclerotic plaque is the fact that SMCs are dying in their fibrous caps (Bennett 1999).
Fundamentally, the various activities of SMCs tend to thicken the cap, while those of the inflammatory cells tend to oppose them. The net production of matrix components depends on the number of the matrix-producing SMCs and the ability of the individual cells to produce such components. The number of SMCs depends on the balance of cell replication and cell death. In human atherosclerotic plaques, the infiltrating lymphocytes are of the Th1 subpopulation of CD4+ cells and the pro-inflammatory Th1-derived cytokines, notably interferon-gamma, dominate (Hansson 2005). This cytokine is able to inhibit the capacity of SMCs to produce collagen (Libby 1995) and together with TNFα and interleukin-1β (IL-1β) contribute to their apoptotic death. Thus, activation of Th1 cells, together with IL-1β-producing macrophages and TNFα-producing mast cells within the cap of an atheroma, could lower the local rate of collagen production by lowering the number of SMCs and by inhibiting the rate of synthesis in those SMCs that survive (Kaartinen et al. 1996). Importantly, the fibrous caps often contain cholesterol crystals, which have the potential to activate the NLRP3 inflammasome in macrophages and so trigger release of IL-1β from the macrophages (Rajamaki et al. 2010; Duewell et al. 2010). Since it is unlikely that HDLs are capable of removing crystalline cholesterol from the intima, the crystals, once formed, may permanently stimulate cap macrophages to secrete IL-1β and so contribute to the ongoing cap thinning.
The various components of the extracellular matrix are degraded by members of the superfamily of matrix metalloproteinases (MMPs) primarily produced by the macrophages of the caps (Newby 2005). For the MMPs to become active, they need to be proteolytically activated. Among the proteolytic enzymes capable of activating plaque MMPs are plasmin, chymase, and tryptase, the two latter ones being neutral proteases secreted by activated mast cells in atherosclerotic lesions (Johnson et al. 1998). Once activated, interstitial collagenase, MMP-1, can degrade the otherwise protease-resistant fibrillar collagen fibers into fragments, and another member of the family, MMP-9, can break collagen fragments further. Importantly, stromelysin, MMP-3, has a broad spectrum and can degrade other components of the extracellular matrix as well, including proteoglycans and elastin (Libby 2013a). Cathepsins are another group of enzymes capable of effectively degrading the various components of the fibrous cap (Fonovic and Turk 2014). Thus, the presence of inflammatory cells in the caps of advanced plaques tends to render the plaque unstable and prone to rupture by both decreasing the production and increasing the degradation of the extracellular matrix of the fibrous cap.
5.1 Effects of HDLs on Smooth Muscle Cells
VSCMs can indirectly influence the development of atherosclerosis by the secretion of growth factors and vasoactive agents, and the protective effects of HDLs on atherosclerotic lesion formation might, at least partly, be related to their impact on the secretory function of VSMCs. Exposure of aortic SMCs to mildly oxidized LDLs enhanced the production and release of platelet-derived growth factor and basic fibroblast growth factor, while addition of HDLs abolished the stimulatory effect of oxidized LDLs on growth factor secretion by these VSMCs (Cucina et al. 1998, 2006). Conversely, treatment of rabbit SMCs in culture with HDLs potentiated the synthesis of the vasodilator prostacyclin via a mechanism involving MAP kinase kinase signaling and increased cyclooxygenase-2 expression (Vinals et al. 1997, 1999). VSMCs are also considered to protect the atherosclerotic plaque against destabilization and rupture. In this respect, HDLs regulate both the proliferation and migration of VSMCs. Nofer and colleagues showed that HDLs accelerate proliferation of VSMCs by driving the cell cycle forward in the G1 phase via cyclin D1-induced phosphorylation of pRb (Nofer et al. 2001a). Elevated cyclin D1 expression in VSMCs in response to treatment with HDLs in turn relies on activation of the Raf/MEK/ERK signal transduction pathway (Nofer et al. 2001a). On the other hand, the platelet-derived growth factor-mediated migration of VCMCs is limited by HDLs through the interaction of HDL-bound S1P with the S1P2 receptor (Tamama et al. 2005). Finally, HDLs were found to suppress inflammatory responses in VSMCs. Oxidized LDLs, via formation of ROS, induced NF-κβ activation in cultured VSMCs, while their pretreatment with HDLs inhibited the intracellular rise in ROS and the subsequent upregulation of NF-κβ elicited by the oxidized LDLs (Robbesyn et al. 2003).
5.2 Antithrombotic Effects of HDLs
5.2.1 HDLs and Platelets
The majority of acute coronary syndromes are caused by the rupture of an atherosclerotic plaque in a coronary artery and subsequent formation of an occlusive coronary thrombus. There appears to be a relationship between plasma HDLs and the thrombogenic potential of blood, as evidenced by the fact that the total thrombus area in an ex vivo flow chamber model for coronary thrombosis was lower in human subjects with higher HDL cholesterol levels (Naqvi et al. 1999). HDLs have several favorable effects on platelets that might explain its interference with thrombus formation in the coronary vasculature. First, it has been reported that HDLs can block the activation and aggregation of platelets. In a small clinical trail, 13 patients with type 2 diabetes mellitus were randomized to a single infusion of rHDLs (CSL-111) or placebo (Calkin et al. 2009). Study results established that a 40 % increase in HDL cholesterol levels in patients with type 2 diabetes mellitus was associated with a 50–75 % reduction in platelet aggregation (Calkin et al. 2009). A similar decrease in the aggregation of platelets was observed when isolated human platelets were incubated with rHDLs or native HDLs in vitro (Calkin et al. 2009; Nofer et al. 1998; Badrnya et al. 2013). Native HDLs and rHDLs not only modulate platelet aggregation but also prevent platelet degranulation and spreading as well as adhesion of platelets to fibrinogen (Calkin et al. 2009; Nofer et al. 1998; Badrnya et al. 2013). In addition, rHDLs limited the incorporation of platelets in growing thrombi under flow conditions in vitro (Calkin et al. 2009). The ability of HDLs to block platelet aggregation was proposed to be due to a reduction in the availability of the second messengers 1,2-diacylglycerol and inositol 1,4,5-trisphosphate, a decreased mobilization of intracellular calcium, activation of protein kinase C, and elevated Na+/H+ exchange (Nofer et al. 1996; 1998). The role of SR-BI for the anti-aggregatory effect of HDLs is controversial. One study reported that inhibition of platelet reactivity by HDLs results from cholesterol depletion in lipid rafts but that this is independent of ABCG1 and SR-BI (Calkin et al. 2009). In contrast, Brodde et al. found that SR-BI is the receptor responsible for binding of HDL to platelets and that HDLs are no longer able to attenuate thrombin-induced adhesion of platelets to fibrinogen when using platelets isolated from SR-BI knockout mice (Brodde et al. 2011). The inhibitory activity of HDLs on platelets is mimicked by soybean phosphocholine (Calkin et al. 2009) and phosphatidylserine-containing liposomes (Brodde et al. 2011), underlining the importance of the phospholipid component in this antithrombotic function of HDLs. As a final point, HDLs may regulate the platelet coagulant activity by reducing the synthesis of platelet-activating factor in endothelial cells (Sugatani et al. 1996).
Another step of the arterial atherothrombotic process in which HDLs may interfere is the production of platelets from bone marrow megakaryocyte progenitor cells. In vitro experiments have revealed that rHDLs promote the removal of cholesterol from megakaryocyte progenitor cells in an ABCG4-dependent fashion, thereby decreasing cellular proliferation (Murphy et al. 2013). Consistent with these observations, intravenous administration of rHDLs resulted in a decrease in the amount of bone marrow megakaryocyte progenitor cells and concomitantly reduced circulating platelets in Ldlr −/− mice transplanted with wild-type bone marrow, while rHDLs lost their effects on bone marrow megakaryocyte progenitor cells and platelet numbers when injected into Ldlr −/− mice that were transplanted with ABCG4-deficient bone marrow (Murphy et al. 2013). A small placebo-controlled clinical study demonstrated that a single injection of rHDLs significantly reduced platelet counts in patients with peripheral vascular disease (Murphy et al. 2013). The importance of deregulation of cholesterol homeostasis in megakaryocyte progenitor cells for atherothrombosis was supported by evidence that selective disruption of ABCG4 in bone marrow cells of LDL receptor knockout mice accelerated the progression of atherosclerotic lesion formation and was associated with marked thrombocytosis and an increased tendency to develop arterial thrombosis (Murphy et al. 2013).
5.2.2 HDLs and Coagulation
Various publications have described an impact of HDLs on the extrinsic coagulation pathway that might result in a decreased tendency for thrombosis. Inhibition of the catalytic activity of the tissue factor–factor VIIA complex by HDLs has been shown to impede the activation of the procoagulant factor X (Carson 1981). Besides direct inactivation of tissue factor and factor Va, HDLs are able to modulate the tissue factor-activated coagulation cascade by downregulating tissue factor expression in thrombin-stimulated endothelial cells through suppression of RhoA and induction of PI3K (Viswambharan et al. 2004). In addition, incorporation of anionic phospholipids into apoA-I-containing rHDL particles resulted in the loss of their procoagulant properties, revealing that, by scavenging anionic phospholipids, apoA-I may control blood coagulation (Oslakovic et al. 2009). HDLs have also been shown to promote the ability of the anticoagulant activated protein C and its cofactor protein S to cause inactivation of factor Va through Arg306 cleavage, and in normal healthy subjects, plasma levels of apoA-I were positively correlated with the anticoagulant potency of activated protein C and protein S in a prothrombin-time clotting assay (Griffin et al. 1999). Of note, these findings were later questioned by another study revealing that the previously observed capacity of the HDL fraction to boost the activated protein C system was not an actual property of HDLs, but instead was due to co-isolation of anionic phospholipid membranes with the HDL fraction during ultracentrifugation (Oslakovic et al. 2010).
6 Myocardial Injury
The main pathophysiological manifestation of coronary heart disease is myocardial damage due to ischemia–reperfusion. It has been postulated that HDLs can provide protection against myocardial injury following ischemia–reperfusion. In an in vivo murine ischemia–reperfusion model, it was found that the infarcted area was decreased when mice were treated 30 min before ischemia–reperfusion with either human HDLs or S1P (Theilmeier et al. 2006). In this regard, HDL or S1P injection mitigated the inflammatory response to ischemia–reperfusion as manifested by a lower accumulation of leukocytes in the infarcted area (Theilmeier et al. 2006). Furthermore, cardiomyocytes were better able to resist ischemia–reperfusion-induced apoptosis in the presence of HDLs or S1P (Theilmeier et al. 2006), potentially due to a higher tolerance of HDL-treated cardiomyocytes to hypoxia-induced opening of the mitochondrial permeability transition pore (Frias et al. 2013). S1P3 was identified as the receptor responsible for the beneficial effect of HDLs and S1P on reperfusion injury (Theilmeier et al. 2006). The finding that HDLs ameliorate myocardial ischemia–reperfusion damage has subsequently been confirmed and extended by others. Ex vivo exposure of isolated hearts from wild-type mice to HDLs provided dose-dependent cardioprotection from reperfusion injury (Frias et al. 2013). On the other hand, HDL-mediated protection of isolated hearts or cardiomyocytes against hypoxia was lost in mice deficient in tumor necrosis factor and in mice with cardiomyocyte-restricted knockout of signal transducer and activator of transcription 3 (STAT3) (Frias et al. 2013), suggesting a role for the survivor activating factor enhancement pathway which is initiated by tumor necrosis factor and involves activation of STAT3. In another study, HDL cholesterol levels were raised in LDL receptor-deficient mice using adenovirus-mediated gene transfer of human apoA-I (Gordts et al. 2013). After induction of myocardial infarction by permanent ligation of the left anterior descending coronary artery, mice that were injected with an adenovirus expressing human apoA-I exhibited a higher survival rate, reduced infarct expansion, attenuated dilatation of the left ventricle, as well as improved systolic and diastolic cardiac function when compared with controls (Gordts et al. 2013).
Conclusions
HDL particles exert many effects which may help either to protect arteries from the development, progression, and complication of atherosclerosis or even facilitate repair and regression of lesions. Nevertheless, HDL has not yet been successfully exploited for preventive or curative therapy of cardiovascular diseases. One potential reason for this shortfall is the structural and functional complexity of HDL particles which is further increased in several inflammatory conditions including CAD itself and which is not discerned by the biomarker HDL cholesterol. Moreover, the relative importance of the many physiological and pathological activities of normal and dysfunctional HDL, respectively, for the pathogenesis of atherosclerosis is unknown. The elucidation of structure–function relationships of HDL-associated molecules is essential to exploit HDL for the development of anti-atherogenic drugs as well as of diagnostic biomarkers for the identification, personalized treatment stratification, and monitoring of patients at increased cardiovascular risk. In addition to the metabolism of HDL or its anti-atherogenic molecules, also the cellular pathways positively regulated by HDL are interesting targets for anti-atherosclerotic therapies.
Abbreviations
- AAPH:
-
2,2′-Azobis(2-amidinopropane) dihydrochloride
- ApoA-I:
-
Apolipoprotein A-I
- ABCA1:
-
ATP-binding cassette transporter A1
- ABCG1:
-
ATP-binding cassette transporter G1
- ATF3:
-
Activating transcription factor 3
- β-ATPase:
-
β-Chain of F0F1 ATPase
- BSEP:
-
Bile salt export pump
- CETP:
-
Cholesteryl ester transfer protein
- CYP7A1:
-
Cholesterol 7α-hydroxylase
- eNOS:
-
Endothelial nitric oxide synthase
- EPC:
-
Endothelial progenitor cell
- HDL:
-
High-density lipoprotein
- HO-1:
-
Heme oxygenase-1
- IL-1β:
-
Interleukin-1β
- LCAT:
-
Lecithin-cholesterol acyltransferase
- LDL:
-
Low-density lipoprotein
- Lp-PLA2 :
-
Lipoprotein-associated phospholipase A2
- MAP:
-
Mitogen-activated protein
- MCP-1:
-
Monocyte chemotactic protein-1
- MDA:
-
Malondialdehyde
- MMP:
-
Matrix metalloproteinase
- NF-κβ:
-
Nuclear factor-κβ
- NO:
-
Nitric oxide
- PAF-AH:
-
Platelet-activating factor acetylhydrolase
- PDZK1:
-
PDZ domain-containing protein 1
- PI3K:
-
Phosphatidylinositol 3-kinase
- PON1:
-
Paraoxonase 1
- rHDL:
-
Reconstituted high-density lipoprotein
- ROS:
-
Reactive oxygen species
- S1P:
-
Sphingosine-1-phosphate
- SMC:
-
Smooth muscle cell
- SR-BI:
-
Scavenger receptor BI
- STAT3:
-
Signal transducer and activator of transcription 3
- TCFA:
-
Thin-cap fibroatheroma
- TLR:
-
Toll-like receptor
- TNFα:
-
Tumor necrosis factor α
- VCAM-1:
-
Vascular cell adhesion molecule 1
- VSMC:
-
Vascular smooth muscle cell
References
Acton S, Rigotti A, Landschulz KT et al (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271(5248):518–520
Angeloni E, Paneni F, Landmesser U et al (2013) Lack of protective role of HDL-C in patients with coronary artery disease undergoing elective coronary artery bypass grafting. Eur Heart J 34(46):3557–3562
Annema W, Tietge UJ (2012) Regulation of reverse cholesterol transport - a comprehensive appraisal of available animal studies. Nutr Metab (Lond) 9(1):25
Annema W, von Eckardstein A (2013) High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ J 77(10):2432–2448
Argraves KM, Gazzolo PJ, Groh EM et al (2008) High density lipoprotein-associated sphingosine 1-phosphate promotes endothelial barrier function. J Biol Chem 283(36):25074–25081
Ashby DT, Rye KA, Clay MA et al (1998) Factors influencing the ability of HDL to inhibit expression of vascular cell adhesion molecule-1 in endothelial cells. Arterioscler Thromb Vasc Biol 18(9):1450–1455
Badrnya S, Assinger A, Volf I (2013) Native high density lipoproteins (HDL) interfere with platelet activation induced by oxidized low density lipoproteins (OxLDL). Int J Mol Sci 14(5):10107–10121
Baigent C, Keech A, Kearney PM et al (2005) Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 366(9493):1267–1278
Baker PW, Rye KA, Gamble JR et al (1999) Ability of reconstituted high density lipoproteins to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J Lipid Res 40(2):345–353
Barter P, Gotto AM, LaRosa JC et al (2007a) HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med 357(13):1301–1310
Barter PJ, Caulfield M, Eriksson M et al (2007b) Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 357(21):2109–2122
Bennett MR (1999) Apoptosis of vascular smooth muscle cells in vascular remodelling and atherosclerotic plaque rupture. Cardiovasc Res 41(2):361–368
Bisoendial RJ, Hovingh GK, Levels JH et al (2003) Restoration of endothelial function by increasing high-density lipoprotein in subjects with isolated low high-density lipoprotein. Circulation 107(23):2944–2948
Boden WE, Probstfield JL, Anderson T et al (2011) Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 365(24):2255–2267
Brilakis ES, McConnell JP, Lennon RJ et al (2005) Association of lipoprotein-associated phospholipase A2 levels with coronary artery disease risk factors, angiographic coronary artery disease, and major adverse events at follow-up. Eur Heart J 26(2):137–144
Brodde MF, Korporaal SJ, Herminghaus G et al (2011) Native high-density lipoproteins inhibit platelet activation via scavenger receptor BI: role of negatively charged phospholipids. Atherosclerosis 215(2):374–382
Brown MS, Goldstein JL (1983) Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem 52:223–261
Bui QT, Prempeh M, Wilensky RL (2009) Atherosclerotic plaque development. Int J Biochem Cell Biol 41(11):2109–2113
Bursill CA, Castro ML, Beattie DT et al (2010) High-density lipoproteins suppress chemokines and chemokine receptors in vitro and in vivo. Arterioscler Thromb Vasc Biol 30(9):1773–1778
Calabresi L, Franceschini G (2010) Lecithin:cholesterol acyltransferase, high-density lipoproteins, and atheroprotection in humans. Trends Cardiovasc Med 20(2):50–53
Calkin AC, Drew BG, Ono A et al (2009) Reconstituted high-density lipoprotein attenuates platelet function in individuals with type 2 diabetes mellitus by promoting cholesterol efflux. Circulation 120(21):2095–2104
Camejo G, Hurt-Camejo E, Wiklund O et al (1998) Association of apo B lipoproteins with arterial proteoglycans: pathological significance and molecular basis. Atherosclerosis 139(2):205–222
Carson SD (1981) Plasma high density lipoproteins inhibit the activation of coagulation factor X by factor VIIa and tissue factor. FEBS Lett 132(1):37–40
Cavelier C, Rohrer L, von Eckardstein A (2006) ATP-Binding cassette transporter A1 modulates apolipoprotein A-I transcytosis through aortic endothelial cells. Circ Res 99(10):1060–1066
Cavelier C, Ohnsorg PM, Rohrer L et al (2012) The beta-chain of cell surface F(0)F(1) ATPase modulates apoA-I and HDL transcytosis through aortic endothelial cells. Arterioscler Thromb Vasc Biol 32(1):131–139
Charles MA, Kane JP (2012) New molecular insights into CETP structure and function: a review. J Lipid Res 53(8):1451–1458
Christoffersen C, Obinata H, Kumaraswamy SB et al (2011) Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci USA 108(23):9613–9618
Clay MA, Pyle DH, Rye KA et al (2001) Time sequence of the inhibition of endothelial adhesion molecule expression by reconstituted high density lipoproteins. Atherosclerosis 157(1):23–29
Cockerill GW, Rye KA, Gamble JR et al (1995) High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol 15(11):1987–1994
Cockerill GW, Huehns TY, Weerasinghe A et al (2001) Elevation of plasma high-density lipoprotein concentration reduces interleukin-1-induced expression of E-selectin in an in vivo model of acute inflammation. Circulation 103(1):108–112
Colin S, Fanchon M, Belloy L et al (2014) HDL does not influence the polarization of human monocytes toward an alternative phenotype. Int J Cardiol 172(1):179–184
Corsetti JP, Zareba W, Moss AJ et al (2006) Elevated HDL is a risk factor for recurrent coronary events in a subgroup of non-diabetic postinfarction patients with hypercholesterolemia and inflammation. Atherosclerosis 187(1):191–197
Corti R, Fuster V, Badimon JJ (2003) Pathogenetic concepts of acute coronary syndromes. J Am Coll Cardiol 41(4 Suppl S):7S–14S
Cucina A, Pagliei S, Borrelli V et al (1998) Oxidised LDL (OxLDL) induces production of platelet derived growth factor AA (PDGF AA) from aortic smooth muscle cells. Eur J Vasc Endovasc Surg 16(3):197–202
Cucina A, Scavo MP, Muzzioli L et al (2006) High density lipoproteins downregulate basic fibroblast growth factor production and release in minimally oxidated-LDL treated smooth muscle cells. Atherosclerosis 189(2):303–309
de La Llera-Moya M, Connelly MA, Drazul D et al (2001) Scavenger receptor class B type I affects cholesterol homeostasis by magnifying cholesterol flux between cells and HDL. J Lipid Res 42(12):1969–1978
De Nardo D, Labzin LI, Kono H et al (2014) High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3. Nat Immunol 15(2):152–160
de Souza JA, Vindis C, Negre-Salvayre A et al (2010) Small, dense HDL 3 particles attenuate apoptosis in endothelial cells: pivotal role of apolipoprotein A-I. J Cell Mol Med 14(3):608–620
Di Angelantonio E, Sarwar N, Perry P et al (2009) Major lipids, apolipoproteins, and risk of vascular disease. JAMA 302(18):1993–2000
DiDonato JA, Huang Y, Aulak KS et al (2013) Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation 128(15):1644–1655
DiDonato JA, Aulak K, Huang Y et al (2014) Site-specific nitration of apolipoprotein A-I at tyrosine 166 is both abundant within human atherosclerotic plaque and dysfunctional. J Biol Chem 289(15):10276–10292
Dikkers A, Tietge UJ (2010) Biliary cholesterol secretion: more than a simple ABC. World J Gastroenterol 16(47):5936–5945
Duewell P, Kono H, Rayner KJ et al (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464(7293):1357–1361
Durand E, Scoazec A, Lafont A et al (2004) In vivo induction of endothelial apoptosis leads to vessel thrombosis and endothelial denudation: a clue to the understanding of the mechanisms of thrombotic plaque erosion. Circulation 109(21):2503–2506
Eliska O, Eliskova M, Miller AJ (2006) The absence of lymphatics in normal and atherosclerotic coronary arteries in man: a morphologic study. Lymphology 39(2):76–83
Esterbauer H, Puhl H, Dieber-Rotheneder M et al (1991) Effect of antioxidants on oxidative modification of LDL. Ann Med 23(5):573–581
Farb A, Burke AP, Tang AL et al (1996) Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation 93(7):1354–1363
Fonovic M, Turk B (2014) Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta 1840(8):2560–2570
Freeman SR, Jin X, Anzinger JJ et al (2014) ABCG1-mediated generation of extracellular cholesterol microdomains. J Lipid Res 55(1):115–127
Frias MA, Pedretti S, Hacking D et al (2013) HDL protects against ischemia reperfusion injury by preserving mitochondrial integrity. Atherosclerosis 228(1):110–116
Frikke-Schmidt R, Nordestgaard BG, Stene MC et al (2008) Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 299(21):2524–2532
Fujii K, Hao H, Ohyanagi M et al (2013) Intracoronary imaging for detecting vulnerable plaque. Circ J 77(3):588–595
Fuster V, Moreno PR, Fayad ZA et al (2005) Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol 46(6):937–954
Garcia JG, Liu F, Verin AD et al (2001) Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108(5):689–701
Garner B, Waldeck AR, Witting PK et al (1998) Oxidation of high density lipoproteins. II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J Biol Chem 273(11):6088–6095
Gordts SC, Muthuramu I, Nefyodova E et al (2013) Beneficial effects of selective HDL-raising gene transfer on survival, cardiac remodelling and cardiac function after myocardial infarction in mice. Gene Ther 20(11):1053–1061
Griffin JH, Kojima K, Banka CL et al (1999) High-density lipoprotein enhancement of anticoagulant activities of plasma protein S and activated protein C. J Clin Invest 103(2):219–227
Guyton JR, Klemp KF (1996) Development of the lipid-rich core in human atherosclerosis. Arterioscler Thromb Vasc Biol 16(1):4–11
Haase CL, Tybjaerg-Hansen A, Grande P et al (2010) Genetically elevated apolipoprotein A-I, high-density lipoprotein cholesterol levels, and risk of ischemic heart disease. J Clin Endocrinol Metab 95(12):E500–E510
Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352(16):1685–1695
Huang Y, DiDonato JA, Levison BS et al (2014) An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat Med 20(2):193–203
Hulten LM, Levin M (2009) The role of hypoxia in atherosclerosis. Curr Opin Lipidol 20(5):409–414
Jacquet S, Malaval C, Martinez LO et al (2005) The nucleotide receptor P2Y13 is a key regulator of hepatic high-density lipoprotein (HDL) endocytosis. Cell Mol Life Sci 62(21):2508–2515
Johnson JL, Jackson CL, Angelini GD et al (1998) Activation of matrix-degrading metalloproteinases by mast cell proteases in atherosclerotic plaques. Arterioscler Thromb Vasc Biol 18(11):1707–1715
Kaartinen M, Penttila A, Kovanen PT (1996) Mast cells in rupture-prone areas of human coronary atheromas produce and store TNF-alpha. Circulation 94(11):2787–2792
Karuna R, Park R, Othman A et al (2011) Plasma levels of sphingosine-1-phosphate and apolipoprotein M in patients with monogenic disorders of HDL metabolism. Atherosclerosis 219(2):855–863
Kimura T, Sato K, Kuwabara A et al (2001) Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J Biol Chem 276(34):31780–31785
Kimura T, Tomura H, Mogi C et al (2006) Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. J Biol Chem 281(49):37457–37467
Koenig W, Khuseyinova N, Lowel H et al (2004) Lipoprotein-associated phospholipase A2 adds to risk prediction of incident coronary events by C-reactive protein in apparently healthy middle-aged men from the general population: results from the 14-year follow-up of a large cohort from southern Germany. Circulation 110(14):1903–1908
Kontush A, Chantepie S, Chapman MJ (2003) Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler Thromb Vasc Biol 23(10):1881–1888
Kovanen PT, Kaartinen M, Paavonen T (1995) Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation 92(5):1084–1088
Krieger M, Herz J (1994) Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP). Annu Rev Biochem 63:601–637
Kruth HS (2011) Receptor-independent fluid-phase pinocytosis mechanisms for induction of foam cell formation with native low-density lipoprotein particles. Curr Opin Lipidol 22(5):386–393
Lee-Rueckert M, Kovanen PT (2011) Extracellular modifications of HDL in vivo and the emerging concept of proteolytic inactivation of prebeta-HDL. Curr Opin Lipidol 22(5):394–402
Lee-Rueckert M, Blanco-Vaca F, Kovanen PT et al (2013) The role of the gut in reverse cholesterol transport–focus on the enterocyte. Prog Lipid Res 52(3):317–328
Lewis GF, Rader DJ (2005) New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res 96(12):1221–1232
Libby P (1995) Molecular bases of the acute coronary syndromes. Circulation 91(11):2844–2850
Libby P (2002) Inflammation in atherosclerosis. Nature 420(6917):868–874
Libby P (2013a) Collagenases and cracks in the plaque. J Clin Invest 123(8):3201–3203
Libby P (2013b) Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med 368(21):2004–2013
Lim HY, Thiam CH, Yeo KP et al (2013) Lymphatic vessels are essential for the removal of cholesterol from peripheral tissues by SR-BI-mediated transport of HDL. Cell Metab 17(5):671–684
Liu Y, Mackness B, Mackness M (2008) Comparison of the ability of paraoxonases 1 and 3 to attenuate the in vitro oxidation of low-density lipoprotein and reduce macrophage oxidative stress. Free Radic Biol Med 45(6):743–748
Lucke S, Levkau B (2010) Endothelial functions of sphingosine-1-phosphate. Cell Physiol Biochem 26(1):87–96
Martel C, Li W, Fulp B et al (2013) Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J Clin Invest 123(4):1571–1579
Martinez LO, Jacquet S, Esteve JP et al (2003) Ectopic beta-chain of ATP synthase is an apolipoprotein A-I receptor in hepatic HDL endocytosis. Nature 421(6918):75–79
Mayranpaa MI, Heikkila HM, Lindstedt KA et al (2006) Desquamation of human coronary artery endothelium by human mast cell proteases: implications for plaque erosion. Coron Artery Dis 17(7):611–621
McGrath KC, Li XH, Puranik R et al (2009) Role of 3beta-hydroxysteroid-delta 24 reductase in mediating antiinflammatory effects of high-density lipoproteins in endothelial cells. Arterioscler Thromb Vasc Biol 29(6):877–882
Mertens A, Verhamme P, Bielicki JK et al (2003) Increased low-density lipoprotein oxidation and impaired high-density lipoprotein antioxidant defense are associated with increased macrophage homing and atherosclerosis in dyslipidemic obese mice: LCAT gene transfer decreases atherosclerosis. Circulation 107(12):1640–1646
Mineo C, Yuhanna IS, Quon MJ et al (2003) High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J Biol Chem 278(11):9142–9149
Murphy AJ, Woollard KJ, Hoang A et al (2008) High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol 28(11):2071–2077
Murphy AJ, Bijl N, Yvan-Charvet L et al (2013) Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nat Med 19(5):586–594
Nakashima Y, Wight TN, Sueishi K (2008) Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovasc Res 79(1):14–23
Naqvi TZ, Shah PK, Ivey PA et al (1999) Evidence that high-density lipoprotein cholesterol is an independent predictor of acute platelet-dependent thrombus formation. Am J Cardiol 84(9):1011–1017
Navab M, Hama SY, Cooke CJ et al (2000) Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: step 1. J Lipid Res 41(9):1481–1494
Newby AC (2005) Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev 85(1):1–31
Nicholls SJ, Dusting GJ, Cutri B et al (2005) Reconstituted high-density lipoproteins inhibit the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation 111(12):1543–1550
Nievelstein-Post P, Mottino G, Fogelman A et al (1994) An ultrastructural study of lipoprotein accumulation in cardiac valves of the rabbit. Arterioscler Thromb 14(7):1151–1161
Nigro P, Abe J, Berk BC (2011) Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal 15(5):1405–1414
Nijstad N, Gautier T, Briand F et al (2011) Biliary sterol secretion is required for functional in vivo reverse cholesterol transport in mice. Gastroenterology 140(3):1043–1051
Nofer JR, Tepel M, Kehrel B et al (1996) High density lipoproteins enhance the Na+/H+ antiport in human platelets. Thromb Haemost 75(4):635–641
Nofer JR, Walter M, Kehrel B et al (1998) HDL3-mediated inhibition of thrombin-induced platelet aggregation and fibrinogen binding occurs via decreased production of phosphoinositide-derived second messengers 1,2-diacylglycerol and inositol 1,4,5-tris-phosphate. Arterioscler Thromb Vasc Biol 18(6):861–869
Nofer JR, Junker R, Pulawski E et al (2001a) High density lipoproteins induce cell cycle entry in vascular smooth muscle cells via mitogen activated protein kinase-dependent pathway. Thromb Haemost 85(4):730–735
Nofer JR, Levkau B, Wolinska I et al (2001b) Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J Biol Chem 276(37):34480–34485
Nofer JR, Geigenmuller S, Gopfert C et al (2003) High density lipoprotein-associated lysosphingolipids reduce E-selectin expression in human endothelial cells. Biochem Biophys Res Commun 310(1):98–103
Nofer JR, van der Giet M, Tolle M et al (2004) HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J Clin Invest 113(4):569–581
Noto H, Hara M, Karasawa K et al (2003) Human plasma platelet-activating factor acetylhydrolase binds to all the murine lipoproteins, conferring protection against oxidative stress. Arterioscler Thromb Vasc Biol 23(5):829–835
Oorni K, Pentikainen MO, Ala-Korpela M et al (2000) Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: molecular mechanisms and effects on matrix interactions. J Lipid Res 41(11):1703–1714
Oslakovic C, Krisinger MJ, Andersson A et al (2009) Anionic phospholipids lose their procoagulant properties when incorporated into high density lipoproteins. J Biol Chem 284(9):5896–5904
Oslakovic C, Norstrom E, Dahlback B (2010) Reevaluation of the role of HDL in the anticoagulant activated protein C system in humans. J Clin Invest 120(5):1396–1399
Park SH, Park JH, Kang JS et al (2003) Involvement of transcription factors in plasma HDL protection against TNF-alpha-induced vascular cell adhesion molecule-1 expression. Int J Biochem Cell Biol 35(2):168–182
Parthasarathy S, Barnett J, Fong LG (1990) High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein. Biochim Biophys Acta 1044(2):275–283
Pasquinelli G, Preda P, Vici M et al (1989) Electron microscopy of lipid deposits in human atherosclerosis. Scanning Microsc 3(4):1151–1159
Pentikainen MO, Oorni K, Ala-Korpela M et al (2000) Modified LDL - trigger of atherosclerosis and inflammation in the arterial intima. J Intern Med 247(3):359–370
Petoumenos V, Nickenig G, Werner N (2009) High-density lipoprotein exerts vasculoprotection via endothelial progenitor cells. J Cell Mol Med 13(11–12):4623–4635
Potteaux S, Gautier EL, Hutchison SB et al (2011) Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J Clin Invest 121(5):2025–2036
Puranik R, Bao S, Nobecourt E et al (2008) Low dose apolipoprotein A-I rescues carotid arteries from inflammation in vivo. Atherosclerosis 196(1):240–247
Quarck R, De Geest B, Stengel D et al (2001) Adenovirus-mediated gene transfer of human platelet-activating factor-acetylhydrolase prevents injury-induced neointima formation and reduces spontaneous atherosclerosis in apolipoprotein E-deficient mice. Circulation 103(20):2495–2500
Radojkovic C, Genoux A, Pons V et al (2009) Stimulation of cell surface F1-ATPase activity by apolipoprotein A-I inhibits endothelial cell apoptosis and promotes proliferation. Arterioscler Thromb Vasc Biol 29(7):1125–1130
Rajamaki K, Lappalainen J, Oorni K et al (2010) Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS ONE 5(7):e11765
Rallidis LS, Tellis CC, Lekakis J et al (2012) Lipoprotein-associated phospholipase A(2) bound on high-density lipoprotein is associated with lower risk for cardiac death in stable coronary artery disease patients: a 3-year follow-up. J Am Coll Cardiol 60(20):2053–2060
Ramet ME, Ramet M, Lu Q et al (2003) High-density lipoprotein increases the abundance of eNOS protein in human vascular endothelial cells by increasing its half-life. J Am Coll Cardiol 41(12):2288–2297
Ramkhelawon B, Yang Y, van Gils JM et al (2013) Hypoxia induces netrin-1 and Unc5b in atherosclerotic plaques: mechanism for macrophage retention and survival. Arterioscler Thromb Vasc Biol 33(6):1180–1188
Ridker PM, Genest J, Boekholdt SM et al (2010) HDL cholesterol and residual risk of first cardiovascular events after treatment with potent statin therapy: an analysis from the JUPITER trial. Lancet 376(9738):333–339
Riwanto M, Rohrer L, Roschitzki B et al (2013) Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation 127(8):891–904
Robbesyn F, Garcia V, Auge N et al (2003) HDL counterbalance the proinflammatory effect of oxidized LDL by inhibiting intracellular reactive oxygen species rise, proteasome activation, and subsequent NF-kappaB activation in smooth muscle cells. FASEB J 17(6):743–745
Robert J, Lehner M, Frank S et al (2013) Interleukin 6 stimulates endothelial binding and transport of high-density lipoprotein through induction of endothelial lipase. Arterioscler Thromb Vasc Biol 33(12):2699–2706
Rohrer L, Cavelier C, Fuchs S et al (2006) Binding, internalization and transport of apolipoprotein A-I by vascular endothelial cells. Biochim Biophys Acta 1761(2):186–194
Rohrer L, Ohnsorg PM, Lehner M et al (2009) High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter G1. Circ Res 104(10):1142–1150
Rosenson RS, Brewer HB Jr, Davidson WS et al (2012) Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125(15):1905–1919
Sabatine MS, Morrow DA, O’Donoghue M et al (2007) Prognostic utility of lipoprotein-associated phospholipase A2 for cardiovascular outcomes in patients with stable coronary artery disease. Arterioscler Thromb Vasc Biol 27(11):2463–2469
Sakakura K, Nakano M, Otsuka F et al (2013) Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ 22(6):399–411
Sanson M, Distel E, Fisher EA (2013) HDL induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PLoS ONE 8(8):e74676
Schmitt MM, Fraemohs L, Hackeng TM et al (2014a) Atherogenic mononuclear cell recruitment is facilitated by oxidized lipoprotein-induced endothelial junctional adhesion molecule-A redistribution. Atherosclerosis 234(2):254–264
Schmitt MM, Megens RT, Zernecke A et al (2014b) Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation 129(1):66–76
Schwartz GG, Olsson AG, Abt M et al (2012) Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med 367(22):2089–2099
Schwenke DC, Carew TE (1989a) Initiation of atherosclerotic lesions in cholesterol-fed rabbits. I. Focal increases in arterial LDL concentration precede development of fatty streak lesions. Arteriosclerosis 9(6):895–907
Schwenke DC, Carew TE (1989b) Initiation of atherosclerotic lesions in cholesterol-fed rabbits. II. Selective retention of LDL vs. selective increases in LDL permeability in susceptible sites of arteries. Arteriosclerosis 9(6):908–918
Seetharam D, Mineo C, Gormley AK et al (2006) High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res 98(1):63–72
Shaw JA, Bobik A, Murphy A et al (2008) Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res 103(10):1084–1091
Shih DM, Gu L, Xia YR et al (1998) Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature 394(6690):284–287
Silbernagel G, Schottker B, Appelbaum S et al (2013) High-density lipoprotein cholesterol, coronary artery disease, and cardiovascular mortality. Eur Heart J 34(46):3563–3571
Smith EB (1990) Transport, interactions and retention of plasma proteins in the intima: the barrier function of the internal elastic lamina. Eur Heart J 11(Suppl E):72–81
Spieker LE, Sudano I, Hurlimann D et al (2002) High-density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation 105(12):1399–1402
Spirig R, Schaub A, Kropf A et al (2013) Reconstituted high-density lipoprotein modulates activation of human leukocytes. PLoS ONE 8(8):e71235
Stary HC, Chandler AB, Glagov S et al (1994) A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, American heart association. Arterioscler Thromb 14(5):840–856
Stary HC, Chandler AB, Dinsmore RE et al (1995) A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, American heart association. Circulation 92(5):1355–1374
Suc I, Escargueil-Blanc I, Troly M et al (1997) HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler Thromb Vasc Biol 17(10):2158–2166
Sugano M, Tsuchida K, Makino N (2000) High-density lipoproteins protect endothelial cells from tumor necrosis factor-alpha-induced apoptosis. Biochem Biophys Res Commun 272(3):872–876
Sugatani J, Miwa M, Komiyama Y et al (1996) High-density lipoprotein inhibits the synthesis of platelet-activating factor in human vascular endothelial cells. J Lipid Mediat Cell Signal 13(1):73–88
Sumi M, Sata M, Miura S et al (2007) Reconstituted high-density lipoprotein stimulates differentiation of endothelial progenitor cells and enhances ischemia-induced angiogenesis. Arterioscler Thromb Vasc Biol 27(4):813–818
Tabas I (1999) Nonoxidative modifications of lipoproteins in atherogenesis. Annu Rev Nutr 19:123–139
Tabas I, Williams KJ, Boren J (2007) Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 116(16):1832–1844
Tabet F, Vickers KC, Cuesta Torres LF et al (2014) HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat Commun 5:3292
Tamama K, Tomura H, Sato K et al (2005) High-density lipoprotein inhibits migration of vascular smooth muscle cells through its sphingosine 1-phosphate component. Atherosclerosis 178(1):19–23
Tamminen M, Mottino G, Qiao JH et al (1999) Ultrastructure of early lipid accumulation in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 19(4):847–853
Tatematsu S, Francis SA, Natarajan P et al (2013) Endothelial lipase is a critical determinant of high-density lipoprotein-stimulated sphingosine 1-phosphate-dependent signaling in vascular endothelium. Arterioscler Thromb Vasc Biol 33(8):1788–1794
Tellis CC, Tselepis AD (2009) The role of lipoprotein-associated phospholipase A2 in atherosclerosis may depend on its lipoprotein carrier in plasma. Biochim Biophys Acta 1791(5):327–338
Terasaka N, Westerterp M, Koetsveld J et al (2010) ATP-binding cassette transporter G1 and high-density lipoprotein promote endothelial NO synthesis through a decrease in the interaction of caveolin-1 and endothelial NO synthase. Arterioscler Thromb Vasc Biol 30(11):2219–2225
Theilmeier G, Schmidt C, Herrmann J et al (2006) High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 114(13):1403–1409
Thorp E, Subramanian M, Tabas I (2011) The role of macrophages and dendritic cells in the clearance of apoptotic cells in advanced atherosclerosis. Eur J Immunol 41(9):2515–2518
Tolani S, Pagler TA, Murphy AJ et al (2013) Hypercholesterolemia and reduced HDL-C promote hematopoietic stem cell proliferation and monocytosis: studies in mice and FH children. Atherosclerosis 229(1):79–85
Tolle M, Pawlak A, Schuchardt M et al (2008) HDL-associated lysosphingolipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production. Arterioscler Thromb Vasc Biol 28(8):1542–1548
Tward A, Xia YR, Wang XP et al (2002) Decreased atherosclerotic lesion formation in human serum paraoxonase transgenic mice. Circulation 106(4):484–490
van der Wal AC, Becker AE, van der Loos CM et al (1994) Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 89(1):36–44
van Oostrom O, Nieuwdorp M, Westerweel PE et al (2007) Reconstituted HDL increases circulating endothelial progenitor cells in patients with type 2 diabetes. Arterioscler Thromb Vasc Biol 27(8):1864–1865
Vinals M, Martinez-Gonzalez J, Badimon JJ et al (1997) HDL-induced prostacyclin release in smooth muscle cells is dependent on cyclooxygenase-2 (Cox-2). Arterioscler Thromb Vasc Biol 17(12):3481–3488
Vinals M, Martinez-Gonzalez J, Badimon L (1999) Regulatory effects of HDL on smooth muscle cell prostacyclin release. Arterioscler Thromb Vasc Biol 19(10):2405–2411
Viswambharan H, Ming XF, Zhu S et al (2004) Reconstituted high-density lipoprotein inhibits thrombin-induced endothelial tissue factor expression through inhibition of RhoA and stimulation of phosphatidylinositol 3-kinase but not Akt/endothelial nitric oxide synthase. Circ Res 94(7):918–925
Vohl MC, Neville TA, Kumarathasan R et al (1999) A novel lecithin-cholesterol acyltransferase antioxidant activity prevents the formation of oxidized lipids during lipoprotein oxidation. Biochemistry 38(19):5976–5981
Voight BF, Peloso GM, Orho-Melander M et al (2012) Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 380(9841):572–580
von Eckardstein A, Nofer JR, Assmann G (2001) High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arterioscler Thromb Vasc Biol 21(1):13–27
Wang N, Tall AR (2003) Regulation and mechanisms of ATP-binding cassette transporter A1-mediated cellular cholesterol efflux. Arterioscler Thromb Vasc Biol 23(7):1178–1184
Wang Y, Lindstedt KA, Kovanen PT (1996) Phagocytosis of mast cell granule remnant-bound LDL by smooth muscle cells of synthetic phenotype: a scavenger receptor-mediated process that effectively stimulates cytoplasmic cholesteryl ester synthesis. J Lipid Res 37(10):2155–2166
Wang N, Silver DL, Costet P et al (2000) Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J Biol Chem 275(42):33053–33058
Wang N, Lan D, Chen W et al (2004) ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc Natl Acad Sci USA 101(26):9774–9779
Watson AD, Berliner JA, Hama SY et al (1995a) Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest 96(6):2882–2891
Watson AD, Navab M, Hama SY et al (1995b) Effect of platelet activating factor-acetylhydrolase on the formation and action of minimally oxidized low density lipoprotein. J Clin Invest 95(2):774–782
Wentzel JJ, Chatzizisis YS, Gijsen FJ et al (2012) Endothelial shear stress in the evolution of coronary atherosclerotic plaque and vascular remodelling: current understanding and remaining questions. Cardiovasc Res 96(2):234–243
Westerterp M, Gourion-Arsiquaud S, Murphy AJ et al (2012) Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell 11(2):195–206
Wilhelm AJ, Zabalawi M, Owen JS et al (2010) Apolipoprotein A-I modulates regulatory T cells in autoimmune LDLr-/-, ApoA-I-/- mice. J Biol Chem 285(46):36158–36169
Wilkerson BA, Grass GD, Wing SB et al (2012) Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: high density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J Biol Chem 287(53):44645–44653
Williams KJ, Tabas I (1995) The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol 15(5):551–561
Witztum JL (2005) You are right too! J Clin Invest 115(8):2072–2075
Woollard KJ (2013) Immunological aspects of atherosclerosis. Clin Sci (Lond) 125(5):221–235
Wu BJ, Chen K, Shrestha S et al (2013) High-density lipoproteins inhibit vascular endothelial inflammation by increasing 3beta-hydroxysteroid-Delta24 reductase expression and inducing heme oxygenase-1. Circ Res 112(2):278–288
Xia P, Vadas MA, Rye KA et al (1999) High density lipoproteins (HDL) interrupt the sphingosine kinase signaling pathway. A possible mechanism for protection against atherosclerosis by HDL. J Biol Chem 274(46):33143–33147
Xiong SL, Liu X, Yi GH (2014) High-density lipoprotein induces cyclooxygenase-2 expression and prostaglandin I-2 release in endothelial cells through sphingosine kinase-2. Mol Cell Biochem 389(1–2):197–207
Yancey PG, Bortnick AE, Kellner-Weibel G et al (2003) Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol 23(5):712–719
Yuhanna IS, Zhu Y, Cox BE et al (2001) High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med 7(7):853–857
Yvan-Charvet L, Pagler T, Gautier EL et al (2010) ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328(5986):1689–1693
Zawada AM, Rogacev KS, Schirmer SH et al (2012) Monocyte heterogeneity in human cardiovascular disease. Immunobiology 217(12):1273–1284
Zernecke A, Weber C (2010) Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc Res 86(2):192–201
Zhang Q, Yin H, Liu P et al (2010) Essential role of HDL on endothelial progenitor cell proliferation with PI3K/Akt/cyclin D1 as the signal pathway. Exp Biol Med (Maywood) 235(9):1082–1092
Zhu W, Saddar S, Seetharam D et al (2008) The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ Res 102(4):480–487
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution Noncommercial License, which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Copyright information
© 2015 The Author(s)
About this chapter
Cite this chapter
Annema, W., von Eckardstein, A., Kovanen, P.T. (2015). HDL and Atherothrombotic Vascular Disease. In: von Eckardstein, A., Kardassis, D. (eds) High Density Lipoproteins. Handbook of Experimental Pharmacology, vol 224. Springer, Cham. https://doi.org/10.1007/978-3-319-09665-0_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-09665-0_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-09664-3
Online ISBN: 978-3-319-09665-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)