
Abstract
Genome-wide comparison of phylogenetic trees is becoming an increasingly common approach in evolutionary genomics, and a variety of approaches for such comparison have been developed. In this article we present several methods for comparative analysis of large numbers of phylogenetic trees. To compare phylogenetic trees taking into account the bootstrap support for each internal branch, the boot-split distance (BSD) method is introduced as an extension of the previously developed split distance (SD) method for tree comparison. The BSD method implements the straightforward idea that comparison of phylogenetic trees can be made more robust by treating tree splits differentially depending on the bootstrap support. Approaches are also introduced for detecting treelike and netlike evolutionary trends in the phylogenetic Forest of Life (FOL), i.e., the entirety of the phylogenetic trees for conserved genes of prokaryotes. The principal method employed for this purpose includes mapping quartets of species onto trees to calculate the support of each quartet topology and so to quantify the tree and net contributions to the distances between species. We describe the applications methods used to analyze the FOL and the results obtained with these methods. These results support the concept of the Tree of Life (TOL) as a central evolutionary trend in the FOL as opposed to the traditional view of the TOL as a “species tree.”
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
With the advances of genomics, phylogenetics entered a new era that is noted by the availability of extensive collections of phylogenetic trees for thousands of individual genes. Examples of such tree collections are the phylomes that encompass trees for all sufficiently widespread genes in a given genome [1,2,3,4] or the “Forest of Life” (FOL) that consists of all trees for widespread genes in a representative set of organisms [5]. It has been known since the early days of phylogenetics that trees built on the same set of species often have different topologies, especially when the set includes distant species, most notably, in prokaryotes [6, 7]. The availability of “forests” consisting of numerous phylogenetic trees exacerbated the problem as an enormous diversity of tree topologies has been revealed. The inconsistency between trees has several major sources: (1) problems with ortholog identification caused primarily by cryptic paralogy; (2) various artifacts of phylogenetic analysis, such as long branch attraction (LBA); (3) horizontal gene transfer (HGT); and (4) other evolutionary processes distorting the vertical, treelike pattern such as incomplete lineage sorting and hybridization [1, 8,9,10]. In order to obtain robust results in genome-level phylogenetic analysis, for instance, to classify phylogenetic trees into clusters with (partially) congruent topologies or to identify common trends among multiple trees, reliable methods for comparing trees are indispensable.
The number and diversity of tree comparison methods and software have substantially increased in the last few years. The tree comparison methods variously use tree bipartitions, such as partition or symmetric difference metrics [11] and split distance [12]; distance between nodes such as the path length metrics [13], nodal distance [12, 14], and nodal distance for rooted trees [15]; comparison of evolutionary units such as triplets and quartets [16]; subtransfer operations such as subtree transfer distance [17], nearest-neighbor interchanging [18], subtree prune and regraft (SPR) using a rooted reference tree [19], SPR for unrooted trees [20] and tree bisection and reconnection (TBR) [17], and matching pair (MP) distance [21]; (dis)agreement methods such as agreement subtrees [22], disagree [12], corresponding mapping [23], and congruence index [24]; tree reconciliation [25]; and topological and branch lengths methods such as K-tree score [26]. Several algorithms have been proposed to analyze with multi-family trees. For example, the From Multiple to Single (FMTS) algorithm systematically prunes each gene copy from a multi-family tree to obtain all possible single-gene trees [12] and an algorithm implemented in TreeKO prunes nodes from the input rooted trees in which duplication and speciation events are labeled [27]. Another algorithm employs a variant of the classical Robinson-Foulds method to compare phylogenetic networks [28]. However, to the best of our knowledge, none of the available metrics for tree comparison takes into account the robustness of the branches, a feature that appears important to minimize the impact of artifacts (unreliable parts of a tree) on the outcome of comparative tree analysis. Here, we present the boot-split distance (BSD) method that calculates distances between phylogenetic trees with weighting based on bootstrap values. This method is implemented in the program TOPD/FMTS [12]. In our recent research, we used the BSD method combined with classical multidimensional scaling (CMDS) analysis to explore the main trends in the phylogenetic FOL and to explore the “Tree of Life” (TOL) concept in light of comparative genomics [5, 29].
Since the time (ca 1838) when Darwin drew the famous sketch of an evolutionary tree in his notebook on transmutation of species, with the legend “I think…,” the thinking on the “Tree of Life” (TOL) has evolved substantially. The first phylogenetic revolution, brought about by the pioneering work of Zuckerkandl and Pauling [30] and later Woese and coworkers [31], was the establishment of molecular sequences as the principal material for phylogenetic tree construction. The second revolution has been triggered by the advent of comparative genomics when it has been realized that HGT, at least among prokaryotes, was much more common than previously suspected. The first revolution was a triumph of the tree thinking, when a well-resolved TOL started to appear within reach. The second revolution undermines the very foundation of the TOL concept and threatens to destroy it altogether [32,33,34].
The current views of evolutionary biologists on the TOL span the entire range from acceptance to complete rejection, with a host of moderate positions. The following rough classification may be used to summarize these positions (a) acceptance of the TOL as the dominant trend in evolution: HGT is considered to be rare and overhyped, and most of the observed “transfers” are deemed to be artifacts [35,36,37,38]; (b) the TOL is the common history of the (nearly) nontransferable core of genes, surrounded by “vines” of HGT [39,40,41,42,43,44,45,46,47,48,49,50]; (c) each gene has its own evolutionary history blending HGT and vertical inheritance; a statistical trend might exist in the maze of gene histories, and it could even be treelike [5, 29, 51, 52]; and (d) ubiquity of HGT renders the TOL concept totally obsolete (prokaryotic species and higher taxa do not exist, and microbial “taxonomy” is created by a pattern of biased HGT) [32, 34, 53,54,55,56,57,58].
We found that, although different trends and patterns have to be invoked to describe the FOL in its entirety, the main, most robust trend is the “statistical TOL,” i.e., the signal of coherent topology that is discernible in a large fraction of the trees in the FOL, in particular, among the nearly universal trees (NUTs) [59, 60].
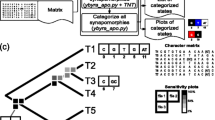
We further explored the FOL by analysis of species quartets [61]. A quartet is a group of four species which is the minimum evolutionary unit in unrooted phylogenetic trees; each quartet can assume three unrooted tree topologies [16]. We described a quantitative measure of the tree and net signals in evolution that is derived from an analysis of all quartets of species in all trees of the FOL. The results of this analysis indicate that, although diverse routes of netlike evolution jointly dominate the FOL, the pattern of treelike evolution that recapitulates the consensus topology of the NUTs is the single most prominent, coherent trend. Here, we report an extended version of these methodologies introduced to analyze the FOL and its trends, as well as new concepts of prokaryotic evolution under the FOL perspective (Fig. 1).

A schematic of the methods and concepts involved in the FOL analysis
2 Materials
2.1 The Forest of Life (FOL) and Nearly Universal Trees (NUTs)
We analyzed the set of 6901 phylogenetic trees from [5] that were obtained as follows. Clusters of orthologous genes were obtained from the COG [62] and EggNOG [63] databases from 100 prokaryotic species (59 bacteria and 41 archaea). The species were selected to represent the taxonomic diversity of Archaea and Bacteria (for the complete list of species, see Additional File 1). The BeTs algorithm [62] was used to identify the orthologs with the highest mean similarity to other members of the same cluster (“index orthologs”), so the final clusters contained 100 or fewer genes, with no more than one representative of each species. The sequences in each cluster were aligned using the Muscle program [64] with default parameters and refined using Gblocks [65]. The program Multiphyl [66], which selects the best of 88 amino acid substitution models, was used to reconstruct the maximum likelihood tree of each cluster. The nearly universal trees (NUTs) are defined as trees from COGs that are represented in more than 90% of the species included in the study.
3 Methods
3.1 Boot-Split Distance: A Method to Compare Phylogenetic Trees Taking into Account Bootstrap Support
3.1.1 Boot-Split Distance (BSD)
The BSD method compares trees based on the original split distance (SD) [12] method. Both methods work by collecting all possible binary splits of the two compared trees and calculating the fraction of equal splits, i.e., those splits that are present in both trees (different splits refer to splits that are present in only one of the two trees). Instead of considering all branches as being equal as is the case in SD, the BSD method takes into account the bootstrap values to increase or decrease the SD value proportionally to the robustness of individual internal branches. The BSD value is the average of the BSD in the equal splits (eBSD) and the BSD in the different splits (Eq. 1). Equations 2 and 3 give the formulas to calculate the eBSD and dBSD values, respectively.
Here e is the sum of bootstrap values of equal splits, d is the sum of bootstrap value of different splits, a is the sum of all bootstrap values, M e is the mean bootstrap value of equal splits, and M d is the mean bootstrap value of different splits.
The BSD algorithm proceeds in four basic steps to compare pairs of trees (Fig. 2). The first step is to obtain all possible splits from both trees. This procedure implies a binary split of the tree at each internal branch, so that the tree is partitioned into two parts each of which contains at least two species. Then, the common set of leaves between the two trees is obtained, that is, the set of shared species. Only trees with a common leaf set of at least four species can be compared. The third step consists in pruning all splits to the common leaf set of species; at this step, species that are present in only one of the two compared trees are removed from the split list. After this procedure, in partially overlapping trees, the algorithm checks whether each of the splits remains a valid partition, that is, a partition that separates at least two species from the rest of the tree. If a split is not a valid partition, it is removed. Finally, the algorithm calculates the BSD using Eqs. 1–3.

The main algorithm of the BSD method. The algorithm to calculate the BSD between two trees includes four basic steps: (1) split both trees in all possible partitions, (2) read the common set of species of both trees, (3) prune the splits according with the common leaf set, and (4) calculate the BSD
3.1.2 The BSD Algorithm
There are three possible types of comparisons for trees that do not include paralogs, that is, include one and only one sequence from each of the constituent species (Fig. 3). In the first case, the two trees completely overlap, that is, consist of the same set of species (Fig. 3a). In this case, step 2, the pruning procedure, is not necessary, and the comparison involves only obtaining all possible splits and the calculation of the BSD. In the second case, one of the compared trees is a subset of the other tree (Fig. 3b). In this case, the splits are only pruned and occasionally removed from the bigger tree. In the third case, when the two trees partially overlap or when a tree is a subset of another tree, a pruning procedure is required. In the example shown in Fig. 4, after the pruning procedure (step 3), there is only one remaining split (split: AB|CD) that is repeated several times in both trees. The remaining AB|CD split in Tree 1 is separated by four nodes that have different bootstrap values. In this case, the bootstrap of the remaining split is calculated using Eq. 4, where n is the total number of nodes between the two sides of the split and BSi is the bootstrap value (adjusted to the 0–1 range) of the node i.

Examples of the BSD algorithm in single family trees. (a) Two trees of the same size. (b) Tree 1 is a subtree of the Tree 2. Two trees that partially overlap. SD split distance, BSD boot-split distance, eBSD BSD of equal splits, dBSD BSD of different splits, p number of equal splits, q number of different splits, m total number of splits, a sum of bootstraps in all splits, e sum of bootstraps in equal splits, d sum of bootstraps in different splits, M a mean bootstrap value, M e mean bootstrap value in equal splits, M d mean bootstrap value in different splits

Calculation of BSD for trees with an unequal numbers of species. The larger tree (1) is pruned prior to the calculation of BSD. The bootstrap value for the only shared internal branch is calculated according to Eq. 4
The bootstrap value associated with a particular branch of a binary tree is taken as a measure of the probability that the four subtrees on the opposite ends of this branch are partitioned correctly. To estimate the probability of the correct partitioning of an arbitrary set of four subtrees, the internal branch of the quartet tree is mapped onto each of the internal branches of the original tree. The quartet is considered to be resolved correctly if it is resolved correctly relative to any of these branches. Under the assumption that bootstrap probabilities on individual branches are independent, Eq. 4 is obtained as the estimate of the bootstrap probability for the internal branch of the quartet tree.
3.1.3 Using a Bootstrap Threshold: Pros and Cons
The key question regarding the BSD method is as follows: what is the best approach to phylogenetic tree comparison—using all branches, reliable or not, with the appropriate weighting, or using only branches supported by high bootstrap values? The first option is illustrated in Fig. 3, whereas Fig. 5 shows an example of a tree comparison that employs a bootstrap threshold of 70, i.e., only branches supported by a higher bootstrap are taken into account in the comparison. The second procedure appears reasonable and can be recommended in some cases. However, it is not advisable as a general approach because, when two large trees with varying bootstrap values are compared, using a strict threshold restricts the comparison to a small subset of robust branches, resulting in an artificially low BSD value. In other words, this procedure artificially inflates the similarity between the two trees by depreciating a large fraction of the branches. In addition, before considering the use of only most supported branches, one should take into account that the BSD method already uses bootstrap values to adjust the distance between trees, so if two trees are topologically similar (low SD) but supported by low bootstrap, the distance value increases (higher BSD), which is one of the advantages of the BSD method (see Eqs. 2 and 3).

Example of the BSD algorithm using a bootstrap cutoff. The figure shows the comparison of two phylogenetic trees that takes into account only those branches with bootstrap support greater than 70. SD split distance, BSD boot-split distance, eBSD BSD of equal splits, dBSD BSD of different splits, p number of equal splits, q number of different splits, m total number of splits, a sum of bootstraps in all splits, e sum of bootstraps in equal splits, d sum of bootstraps in different splits, M a mean bootstrap value, M e mean bootstrap value in equal splits, M d mean bootstrap value in different splits
3.1.4 Testing the BSD Method
The performance of the BSD method was compared with that of the original SD method implemented in the TOPD/FMTS program [12]. Figure 6 shows the correlation of SD and BSD for trees with a number of species from 4 to 15 (a) and from 16 to 100 (b) from a recent large-scale analysis of the FOL [5]. The three-way comparison of SD, BSD, and tree size (number of species) shows a positive correlation between SD and BSD for all tree sizes (R 2 = 0.8613 for trees with 4–16 species and R 2 = 0.7055 for trees with 16–100 species) (Fig. 6c). However, the SD follows a discrete distribution, which obviously is most conspicuous in the comparisons of small trees (Fig. 6a), whereas, thanks to the use of the bootstrap values, the BSD distribution is continuous (Fig. 7).

Correlation of BSD and SD from the all-against-all tree comparisons of 6901 phylogenetic trees. (a) Trees containing 4–15 species. (b) Trees containing 16–100 species. (c) SD, BSD, and tree size for trees containing between 16 and 100 species

Comparisons of trees with six taxa. Bootstrap values were assigned randomly in each comparison
Figure 7 shows an example of the comparison (all-against-all) of three trees with six species each that differ in one, two, and three splits, resulting in SD values of 0.33, 0.66, and 1, respectively (Fig. 7a). Also, each tree was compared to itself resulting in a SD of 0. Then, bootstrap values were assigned randomly to the trees in order to compare the trees using the BSD method, and this procedure was repeated 1000 times. The resulting plot (Fig. 7b) shows that, for the comparison of trees with SD of 0 and 1, the BSD values ranged from 0 to 0.5 and from 0.5 to 1, respectively, and in principle, could assume all intermediate values. In the case of the comparisons that differed in one split (SD = 0.33), the BSD value was greater than 0.33 in 75% of the comparison, whereas for the comparisons that differed in two splits (SD = 0.67), 25% of the BSD values were greater than 0.67. Thus, the BSD method for tree comparison offers a better resolution than the SD method, especially, for trees with a small number of species.
Figure 8a shows the results of analysis of six simulated alignments with an increasing level of noise (divergence respect to the initial alignment) in each alignment, i.e., from the alignment 0 (without noise and producing trees with bootstrap values of 100) to alignment 5 with the maximum level of noise. For each alignment, a tree was constructed using the UPGMA method from the web server DendroUPGMA (http://genomes.urv.cat/UPGMA). Distances were calculated using the Jaccard coefficient, and bootstraps were generated from 100 replicates. The results of the tree comparison (Fig. 8b) using three different methods, namely, nodal distance (ND), SD, and BSD, show that the BSD method presents a continuous distribution resulting in a better resolution of the distances than the other two methods. Indeed, the SD and ND methods fail to discern the similarity between trees after six changes, whereas the BSD method still reports discernible similarity (Fig. 8b). In order to compare the three tree comparison methods, the distance reported by each method was normalized to the maximum value in each case, i.e., after 46 changes (maximum number of changes in the simulation), the distance to the initial tree is 1.41, 0.30, and 0.42 for ND, SD, and BSD, respectively. All three distance values indicate that the trees are similar far above the random expectation, supporting the robustness of all methods, but the BSD method presents a better resolution in the tree comparison.

Comparison of six trees constructed from alignments with increasing noise levels. (a) Comparison of trees from six simulated alignments. The UPGMA tree from each alignment was reconstructed with the web server DendroUPGMA (http://genomes.urv.cat/UPGMA) using the Jaccard coefficient as the measure of distance and generating 100 bootstraps replicates. Alignment 0 corresponds to the initial alignment without noise that perfectly separates all branches, resulting in a tree with bootstrap values of 100 for all internal nodes. Alignments 1 to 5 correspond to the derivatives of the initial alignment with increasing noise levels at each step. (b) Results of the comparison of each tree [1 to 5] with the initial tree (0). The trees were compared using three methods: split distance (SD), nodal distance (ND), and boot-split distance (BSD). For the purpose of comparison, the results obtained with each of the three methods were normalized to the maximum value in each case
3.1.5 Analysis of Random Trees and the Significance of BSD Results
To assess the significance of the tree comparison by the BSD method, we performed several tree comparisons using random trees containing between 4 and 100 species (Fig. 9). Each test is an all-against-all comparison of 1000 random trees (for complete results see Additional File 2). The results from random tree comparison have to be used to determine whether the detected similarities or differences between trees are significantly different from chance [12]. Figure 9 shows that the distance between random trees monotonically increases with the tree size up to a value of approximately 0.75 for BSD and approximately 0.999 for SD. In other words, although BSD is an extension of the SD method, the results obtained by the two methods are not directly comparable. Therefore, to assess whether the similarity between two trees is better than chance, one must consider the method used for the tree comparison (e.g. SD or BSD) and the size of the tree. For example, consider two trees with 15 species each for which the SD method reports a distance of 0.75. This value is far below randomness (Fig. 9), so the conclusion would be that the two trees are nonrandomly similar. However, if the same distance value (0.75) is reported by the BSD method, the conclusion would be the opposite, namely, that the two trees are no more similar than two random trees of 15 species.

Random BSD and SD depending on the tree size. Results of the tree comparison of random trees (with different sizes ranging from 4 to 100 species) show that the BSD and SD increase up to 0.75 and 0.999, respectively
Another and probably the most important problem of the comparison of phylogenetic trees is how to interpret the results from a biological perspective. To address this issue, we generated random trees containing from 4 to 100 species and performed 1 to 100 permutations (swap of a pair of branches) in each tree. The resulting tree was then compared with the source tree (Fig. 10a, b). The results show the number of permutations required to obtain a particular BSD value for different tree sizes (number of species). For instance, BSD = 0.3 in the comparison of two trees with 20 species indicates that the two trees are separated by one permutation whereas BSD = 0.6 indicates that the trees are separated by approximately 9 permutations (for the complete listing of equivalences between BSD, SD and the number of permutations, see Additional File 3). Considering that each permutation corresponds to an HGT event, the BSD may be construed as the measure of the extent of HGT contributing to the topological difference between the compared trees. Given the discrete distribution of SD values, this measure cannot be used to infer the number of permutations with the same precision as BSD.

The number of permutations and the BSD. (a) BSD depending on the number of permutations and tree size. (b) Mean and standard deviation of the BSD for up to 100 permutations for trees with 20 species
3.2 Analysis of Topological Trends in a Set of Phylogenetic Trees
3.2.1 Calculation of the Tree Inconsistency
A key characteristic of the FOL is the degree of the topological (in)consistency between the constituent trees. To quantify this trend, we introduced the inconsistency score (IS), which is the fraction of the times that the splits from a given tree are found in all N trees that comprise the FOL. Thus, the IS may be naturally taken as a measure of how representative of the entire FOL is the topology of the given tree. The IS is calculated using Eqs. 5–7, where N is the total number of trees, X is the number of splits in the given tree, and Y is the number of times the splits from the given tree are found in all trees of the FOL.
In addition to the calculation of a single value of IS for a given tree by comparing its topology to the topologies of rest of trees in the FOL, IS can be calculated along the depth of the trees, namely, split depth and phylogenetic depth. The split depth was calculated for each unrooted tree according to the number of splits from the tips to the center of the tree. The value of split depth ranged from 1 to 49 ([100 species/2] − 1). The phylogenetic depth was obtained from the branch lengths of a rescaled ultrametric tree, rooted between archaeal and bacterial species, and ranged from 0 to 1. The topology of the ultrametric tree was obtained from the supertree of the 102 NUTs using the CLANN program [67]. The branch lengths from each of the 6901 trees were used to calculate the average distance between each pair of species. The obtained matrix was used to calculate the branch lengths of the supertree of the NUTs. This supertree with branch lengths was then used to construct an ultrametric tree using the program KITSCH from the Phylip package [68] and rescaled to the depth range from 0 to 1. The resulting ultrametric tree was used for the analysis of the dependence of tree inconsistency on phylogenetic depth.
3.2.2 Classical Multidimensional Scaling Analysis
The classical multidimensional scaling (CMDS), also known as principal coordinate analysis, is the multifactorial method best suited to analyze matrices obtained from tree comparison methods like BSD and identify the main trends in a large set of phylogenetic trees. The CMDS embeds n data points implied by a [n × n] distance matrix into an m-dimensional space (m < n) such that, for any k ∈ [1, m], the embedding into the first k dimensions is the best in terms of preserving the original distances between the points [69, 70]. In our analysis, the data points are distances between trees obtained using the BSD method. The choice of the optimal number of clusters is made using the gap statistics algorithm [71]. The number of clusters for which the value of the gap function for cluster k + 1 is not significantly higher than that for cluster k (z-score below 1.96, corresponding to 0.05 significance level) is considered optimal. The CMDS analysis was performed using the K-means function of the R package that implements the K-means algorithm. The CMDS approach has been previously employed by Hillis et al. for phylogenetic tree comparison, with the distances between trees calculated using the Robinson-Foulds distance [72].
3.3 Analysis of Quartets of Species
3.3.1 Definition of Quartets and Mapping Quartets onto Trees
The minimum evolutionary unit in unrooted phylogenetic trees is defined by groups of four species (or quartets), and each quartet may be best represented by the three possible unrooted tree topologies (Fig. 11a). A quartet defined by the set of species A, B, C, and D has three possible unrooted topologies: (1) AB|CD, (2) AC|BD, and (3) AD|BC. To analyze which quartet topology (QT) best represents the relationships among the four species in a quartet, each quartet was compared against the entire set of phylogenetic trees from 100 species (the FOL).

Quartets and quartet topologies. (a) Each quartet (q i) is defined by a set of four species (different colors denote species) and may be represented by three possible unrooted tree topologies (q i t i). (b) Quartet topologies (QT). In 100 species, the total number of quartets (Q) is 3,921,225. Each quartet may be represented by three distinct QTs, resulting in a total of 11,763,735 QTs. Each QT was mapped onto the FOL, i.e., for each QT, it was determined which of the three topologies is represented in each phylogenetic tree in the FOL (8.12 × 1010 comparisons). Modified from ref. 61
For 100 species, there are 3,921,225 quartets and, accordingly, 11,763,675 topologies (Fig. 11b). A mapping of quartets onto trees is produced using the SD method [12]. A binary version of this method was employed to compare quartets and trees (a quartet is represented in a tree when SD = 0 and not represented when SD > 0). Figure 12a shows an example of quartet mapping onto a set of ten trees. Here q 1 is a resolved quartet, with the topology q 1 t 1 supported by eight of the ten trees. By contrast, for q 2, three quartet topologies are equally supported, i.e., the topology of this quartet remains unresolved.

Mapping quartets. (a) Mapping quartets onto a set of ten trees. (b) A schematic of the procedure used to reconstruct a species matrix from the map of quartets
To analyze which of the three possible topologies best represents the almost four million quartets in the FOL, each quartet topology was compared with the entire set of 6901 trees, resulting in a total number of 8.12 × 1010 tree comparisons (Fig. 11b), and the number of trees that support each quartet topology was counted for the entire FOL or for the set of 102 NUTs (Fig. 11b).
3.3.2 Distance Matrices and Heat Maps
Using the quartet support values for each quartet, a 100 × 100 between-species distance matrix was calculated as d ij = 1 − S ij/Q ij where d ij is the distance between two species, S ij is the number of trees containing quartets in which the two species are neighbors, and Q ij is the total number of quartets containing the given two species. Then, this distance matrix was used to construct different heat maps using the matrix2png web server ([73], Fig. 12b). In contrast to the BSD method, which is best suited for the analysis of the evolution of individual genes, the distance matrices derived from maps of quartets are used to analyze the evolution of species and to disambiguate treelike evolutionary relationships and “highways” (preferential routes) of HGT.
3.3.3 The Tree-Net Trend (TNT)
The quartet-based between-species distances were used to calculate the Tree-Net Trend (TNT) score. The TNT score is calculated by rescaling each matrix of quartet distances to a 0–1 scale between the supertree-derived matrix (which is taken to represent solely the treelike evolution signal, hence the distance of 0) and the matrix obtained from permuted trees, with distance values around the random expectation of 0.67 (Fig. 13). Two situations may occur in the calculation of the TNT score depending on the relationship between the distance in the supertree matrix (Ds) and the distance in the random matrix (Dr = 0.67). When Ds > Dr (e.g., in comparisons of archaea versus bacteria), S TNT = (d − Dr)/(Ds − Dr), where S TNT is the TNT score and d is the distance between the two compared species in the matrix. When Ds < Dr (in comparisons between closely related species), S TNT = 1 − ((d − Ds)/(Dr − Ds)).

The Tree-Net Trend (TNT). The figure shows a schematic of the TNT calculation and the rescaling procedure. Modified from ref. 61
4 Phylogenetic Concepts in Light of Pervasive Horizontal Gene Transfer
4.1 Patterns in the Phylogenetic Forest of Life
The reconstruction of the evolutionary trends in the FOL is based on the idea that prokaryotes, effectively, share a common gene pool. This gene pool consists of genes with widely different ranges of phyletic spread, from universal to rare ones only present in a few species [74]. Thus, genes, as the elements of this gene pool, have their distinct evolutionary histories blending HGT and vertical inheritance (Fig. 14). In principle, the Forest of Life (FOL) encompasses the complete set of phylogenetic trees for all genes from all genomes. However, a comprehensive analysis of the entire FOL is computationally prohibitive (with over 1000 archaeal and bacterial genomes now available and the computational resources accessible to the authors, estimation of the phylogenetic tree for each gene represented in all these genomes would take weeks of computer time) so a representative subset of the trees needs to be selected and analyzed. Previously [5], we defined such a subset by selecting 100 archaeal and bacterial genomes, which are representative of all major prokaryote groups, and building 6901 maximum likelihood (ML) trees for all genes with a sufficient number of homologs and sufficient level of sequence conservation in this set of genomes; for brevity, we refer to this set of trees as the FOL. In this set of almost 7000 trees, only a very small portion of the forest is represented by nearly universal trees (Fig. 14). Furthermore, bacterial and archaeal universal trees are rare as well, as reflected in Fig. 14 by the small peaks around 41 and 59 species, i.e., all archaea and all bacteria, respectively. The dominant pattern in the major part of the FOL is completely different: the FOL is best represented by numerous small trees, with about 2/3 of the trees including <20 species (Fig. 14).

The Forest of Life (FOL). The distribution of the trees in the FOL by the number of species. Modified from ref. 5
4.2 The Nearly Universal Trees (NUTs)
We define the nearly universal trees (NUTs) as trees for those COGs that were represented in more than 90% of the included prokaryotes. This definition yielded 102 NUTs. Not surprisingly, the great majority of the NUTs are genes encoding proteins involved in translation and the core aspects of transcription (Fig. 15). Among the NUTs, only 14 corresponded to COGs that consist of strict 1:1 orthologs (all of them ribosomal proteins), whereas the rest of NUTs included paralogs in some organisms (only the most conserved paralogs were used for tree construction [5]). The 1:1 NUTs were similar to the rest of the NUTs in terms of the connectivity in tree similarity (1-BSD) networks and their positions in the single cluster of NUTs obtained using CMDS.

Distribution of the gene functions among the NUTs. The functional classification of genes was from the COG database [62]
The 102 NUTs were compared to trees produced by analysis of concatenations of universal proteins [49]. The results showed that most of the NUTs were topologically similar to a tree obtained by the concatenation of 31 universal orthologous genes [5]—in other words, the “Universal Tree of Life” constructed by Ciccarelli et al. [49] was statistically indistinguishable from the NUTs and showed properties of a consensus topology. Not surprisingly, the 1:1 ribosomal protein NUTs were even more similar to the universal tree than the rest of the NUTs, in part because these proteins were used for the construction of the universal tree and, in part, presumably because of the low level of HGT among ribosomal proteins.
4.3 The Tree of Life (TOL) as a Central Trend in the FOL
We analyzed the matrix of all-against-all tree comparisons of the NUTs by embedding them into a 30-dimensional tree space using the CMDS procedure [69, 70]. The gap statistics analysis [71] reveals a lack of significant clustering among the NUTs in the tree space. Thus, all the NUTs seem to belong to one unstructured cloud of points scattered around a single centroid. This organization of the tree space is best compatible with individual trees randomly deviating from a single, dominant topology (which may be denoted the TOL), apparently as a result of random HGT (but in part possibly due to random errors in the tree-construction procedure). Therefore, there is an unequivocal general trend among the NUTs. Although the topologies of the NUTs were, for the most part, not identical, so that the NUTs could be separated by their degree of inconsistency (a proxy for the amount of HGT), the overall high consistency level indicated that the NUTs are scattered in the close vicinity of a consensus tree, with HGT events distributed randomly [5].
Thus, the NUTs present a unique and strong signal of unity that seems to reflect the TOL pattern of evolution. The inconsistency score (IS) among the NUTs ranged from 1.4% to 4.3%, whereas the mean IS value for an equivalent set (102) of randomly generated trees with the same number of species was approximately 80%, indicating that the topologies of the NUTs are highly consistent and nonrandom [5].
To further assess the potential contribution of phylogenetic analysis artifacts to observed inconsistencies between the NUTs, we analyzed these trees with different bootstrap support thresholds (i.e., only splits supported by bootstrap values above the respective threshold value were compared). Particularly low IS levels were detected for splits with high bootstrap support, but the inconsistency was never eliminated completely, suggesting that HGT is a significant contributor to the observed inconsistency among the NUTs (IS ranges from 0.3% to 2.1% and 0.3% to 1.8% for splits with a bootstrap value higher than 70 and 90, respectively) [5].
Analysis of the supernetwork built from the 102 NUTs [5] showed that the incongruence among these trees is mainly concentrated at the deepest levels, with a much greater congruence at shallow phylogenetic depths. The major exception is the unambiguous archaeal-bacterial split that is observed despite the apparent substantial interdomain HGT. Evidence of probable HGT between archaea and bacteria was obtained for approximately 44% of the NUTs (13% from archaea to bacteria, 23% from bacteria to archaea, and 8% in both directions), with the implication that HGT is likely to be even more common between the major branches within the archaeal and bacterial domains [5]. These results are compatible with previous reports on the apparently random distribution of HGT events in the history of highly conserved genes, in particular those encoding proteins involved in translation [75, 76], and on the difficulty of resolving the phylogenetic relationships between the major branches of bacteria [77,78,79] and archaea [5, 80, 81]. More specifically, archaeal-bacterial HGT has been inferred for 83% of the genes encoding aminoacyl-tRNA synthetases (compared with the overall 44%), essential components of the translation machinery that are known for their horizontal mobility [42, 82]. In contrast, no HGT has been predicted for any of the ribosomal proteins, which belong to an elaborate molecular complex, the ribosome, and hence appear to be non-exchangeable between the two prokaryotic domains [42, 76]. In addition to the aminoacyl-tRNA synthetases, and in agreement with many previous observations ([83] and references therein), evidence of HGT between archaea and bacteria was seen also for the few metabolic enzymes that belonged to the NUTs, including undecaprenyl pyrophosphate synthase, glyceraldehyde-3-phosphate dehydrogenase, nucleoside diphosphate kinase, thymidylate kinase, and others.
4.4 The NUTs Topologies as the Central Trend and Detection Distinct Evolutionary Patterns in the FOL
Using the BSD method, we compared the topologies of the NUTs to those of the rest of the trees in the FOL. Notably, 2615 trees (~38% of the FOL) showed a greater than 50% similarity (P-value <0.05) to at least one of the NUTs, being the mean similarity of the trees to the NUTs approximately 50% (Fig. 16). For a set of 102 randomized trees of the same size as the NUTs, only about 10% of the trees in the FOL showed the same or greater similarity, indicating that the NUTs were strongly and nonrandomly connected to the rest of the FOL.

Topological similarity between the NUTs and the rest of the FOL. Percentage of trees connected to the NUTs at a different percentage of similarity. Modified from ref. 5
We then analyzed the structure of the FOL by embedding the 3789 COG trees into a 669-dimensional space using the CMDS procedure [69, 70]. A CMDS clustering of the entire set of 6901 trees in the FOL was beyond the capacity of the R software package used for this analysis; however, the set of COG trees included most of the trees with a large number of species for which the topology comparison is most informative. A gap statistics analysis [69, 70] of K-means clustering of these trees in the tree space revealed distinct clusters of trees in the forest. The FOL is optimally partitioned into seven clusters of trees (the smallest number of clusters for which the gap function did not significantly increase with the increase of the number of clusters) (Fig. 17). Clusters 1, 4, 5, and 6 were enriched for bacterial-only trees, all archaeal-only trees belonged to clusters 2 and 3, and cluster 7 consisted entirely of mixed archaeal-bacterial clusters; notably, all the NUTs form a compact group inside cluster 6.

Clusters and patterns in the FOL. The seven clusters identified in the FOL using the CMDS method and the mean similarity values between the 102 NUTs and all trees from each of the seven clusters are shown. Modified from ref. 5
The results of the CMDS clustering (Fig. 17) support the existence of several distinct “attractors” in the FOL. However, we have to emphasize caution in the interpretation of this clustering because trivial separation of the trees by size could be an important contribution. The approaches to the delineation of distinct “groves” within the forest merit further investigation. The most salient observation for the purpose of the present study is that all the NUTs occupy a compact and contiguous region of the tree space and, unlike the complete set of the trees, are not partitioned into distinct clusters by the CMDS procedure. Taken together with the high mean topological similarity between the NUTs and the rest of the FOL, these findings indicate that the NUTs represent a valid central trend in the FOL.
4.5 The Tree and Net Components of Prokaryote Evolution
The TNT map of the NUTs was dominated by the treelike signal (green in Fig. 18a): the mean TNT score for the NUTs was 0.63 (Fig. 19b), so the evolution of the nearly universal genes of prokaryotes appears to be almost “two-third treelike” (i.e., reflects the topology of the supertree). The rest of the FOL stood in a stark contrast to the NUTs, being dominated by the netlike evolution, with the mean TNT value of 0.39 (Fig. 19c) (about “60% netlike”). Remarkably, areas of treelike evolution were interspersed with areas of netlike evolution across different parts of the FOL (Fig. 18b). The major netlike areas observed among the NUTs were retained, but additional ones became apparent including Crenarchaeota that showed a pronounced signal of a non-treelike relationship with diverse bacteria as well as some Euryarchaeota (Fig. 18b). The distribution of the tree and net evolutionary signals among different groups of prokaryotes showed a striking split among the NUTs: among the archaea, the tree signal was heavily dominant (mean TNTNUTs_Archaea = 0.80 ± 0.20), whereas among bacteria the contributions of the tree and net signals were nearly equal (mean TNTNUTs_Bacteria = 0.51 ± 0.38). Among the rest of the trees in the FOL, archaea also showed a stronger tree signal than bacteria, but the difference was much less pronounced than it was among the NUTs (mean TNTFOL_Archaea = 0.47 ± 0.11 and mean TNTFOL_Bacteria = 0.34 ± 0.08). The conclusions on the treelike and netlike components of evolution made here are based on the assumption that the supertree of the NUTs represents the treelike (vertical) signal. We did not perform direct tests of the robustness of these conclusions to the supertree topology. However, observations presented previously [5] suggest that the results are likely to be robust given the coherence of the NUTs topologies as well as the similarity of the supertree topology and the topologies of the individual NUTs to the “Tree of Life” obtained from concatenated sequences of universally conserved ribosomal proteins [49].

The Tree-Net Trend (TNT) score heatmaps. (a) The 102 NUTs. (b) The FOL without the NUTs (6799 trees). The TNT increases from red (low score, close to random, an indication of netlike evolution) to green (high score, close to the supertree topology, an indication of treelike evolution). The species are ordered according to the topology of the supertree of the 102 NUTs. In (a), the major groups of archaea and bacteria are denoted. Modified from ref. 61

The Tree-Net Trend in the FOL and in the NUTs. (a) A hypothetical equilibrium between the tree and net trends. (b) A schematic representation of the tree tendency in the NUTs. (c) A schematic representation of the net tendency in the FOL
5 Conclusions
The analysis of the phylogenetic FOL is a logical strategy for studying the evolution of prokaryotes because each set of orthologous genes presents its own evolutionary history and no single topology may represent the entire forest. Thus, the methods introduced in this article that compare trees without the use of a preconceived representative topology for the entire FOL may be of wide utility in phylogenomics.
We have shown that, although no single topology may represent the entire FOL and several distinct evolutionary trends are detectable, the NUTs contain a strong treelike signal. Although the treelike signal is quantitatively weaker than the sum total of the signals from HGT, it is the most pronounced single pattern in the entire FOL.
Under the FOL perspective, the traditional TOL concept (a single “true” tree topology) is invalidated and should be replaced by a statistical definition. In other words, the TOL only makes sense as a central trend in the phylogenetic forest.
6 Exercises
-
1.
Calculate the split distance (SD) and boot-split distance (BSD) of the following two trees:
-
(((A,B)61,C)53,D,E);(((A,C)76,B)38,D,E)
-
-
2.
Calculate the inconsistency score of the tree X in the “forest of trees” Y.
-
X = (((A,B),C),D,E)
-
Y = (((A,B),C),D,E); (A,B,(E,D); (((A,C),B),D,E); (A,C,(B,D); (A,B,(C,D); (A,B,(C,E); (A,E,(B,D); (((A,C),D),E,F); (((A,B),D),E,C); (((E,F),A),B,C)
-
Abbreviations
- BSD:
-
Boot-split distance
- CMDS:
-
Classical multidimensional scaling
- COG:
-
Clusters of orthologous genes
- FOL:
-
Forest of Life
- HGT:
-
Horizontal gene transfer
- ND:
-
Nodal distance
- NUTs:
-
Nearly universal trees
- QT:
-
Quartet topology
- SD:
-
Split distance
- TNT:
-
Tree-Net trend
- TOL:
-
Tree of Life
References
Huerta-Cepas J, Dopazo H, Dopazo J, Gabaldon T (2007) The human phylome. Genome Biol 8:R109
Huerta-Cepas J, Bueno A, Dopazo J, Gabaldon T (2008) PhylomeDB: a database for genome-wide collections of gene phylogenies. Nucleic Acids Res 36:D491–D496
Frickey T, Lupas AN (2004) PhyloGenie: automated phylome generation and analysis. Nucleic Acids Res 32:5231–5238
Sicheritz-Ponten T, Andersson SG (2001) A phylogenomic approach to microbial evolution. Nucleic Acids Res 29:545–552
Puigbo P, Wolf YI, Koonin EV (2009) Search for a Tree of Life in the thicket of the phylogenetic forest. J Biol 8:59
Felsenstein J (2004) Inferring phylogenies. Sinauer Associates, Sunderland, MA
Nei M, Kumar S (2001) Molecular evolution and phylogenetics. Oxford University Press, Oxford
Castresana J (2007) Topological variation in single-gene phylogenetic trees. Genome Biol 8:216
Soria-Carrasco V, Castresana J (2008) Estimation of phylogenetic inconsistencies in the three domains of life. Mol Biol Evol 25:2319–2329
Marcet-Houben M, Gabaldon T (2009) The tree versus the forest: the fungal tree of life and the topological diversity within the yeast phylome. PLoS One 4:e4357
Robinson DF, Foulds LR (1981) Comparison of phylogenetic trees. Math Biosci 53:131–147
Puigbo P, Garcia-Vallve S, McInerney JO (2007) TOPD/FMTS: a new software to compare phylogenetic trees. Bioinformatics 23:1556–1558
Steel MA, Penny D (1993) Distribution of tree comparison metrics - some new results. Syst Biol 42:126–141
Bluis J, Shin D-G (2003) Nodal distance algorithm: calculating a phylogenetic tree comparison metric. In: Proceedings of the third IEEE symposium on bioInformatics and bioEngineering, IEEE Computer Society, pp 87–94
Cardona G, Llabres M, Rossello F, Valiente G (2009) Nodal distances for rooted phylogenetic trees. J Math Biol 61(2):253–276
Estabrook GF, McMorris FR, Meachan A (1985) Comparison of undirected phylogenetic trees based on subtree of four evolutionary units. Syst Zool 34:193–200
Allen L, Steel M (2001) Subtree transfer operations and their induced metrics on evolutionary trees. Ann Comb 5:1–15
Waterman MS, Steel M (1978) On the similarity of dendrograms. J Theor Biol 73:789–800
Beiko RG, Hamilton N (2006) Phylogenetic identification of lateral genetic transfer events. BMC Evol Biol 6:15
Hickey G, Dehne F, Rau-Chaplin A, Blouin C (2008) SPR distance computation for unrooted trees. Evol Bioinformatics Online 4:17–27
Bogdanowicz D, Giaro K (2017) Comparing phylogenetic trees by matching nodes using the transfer distance between partitions. J Comput Biol 24:422–435
Kubicka E, Kubicki G, McMorris FR (1995) An algorithm to find agreement subtrees. J Classif 12:91–99
Nye TM, Lio P, Gilks WR (2006) A novel algorithm and web-based tool for comparing two alternative phylogenetic trees. Bioinformatics 22:117–119
de Vienne DM, Giraud T, Martin OC (2007) A congruence index for testing topological similarity between trees. Bioinformatics 23:3119–3124
Cotton JA, Page RD (2002) Going nuclear: gene family evolution and vertebrate phylogeny reconciled. Proc Biol Sci 269:1555–1561
Soria-Carrasco V, Talavera G, Igea J, Castresana J (2007) The K tree score: quantification of differences in the relative branch length and topology of phylogenetic trees. Bioinformatics 23:2954–2956
Marcet-Houben M, Gabaldon T (2011) TreeKO: a duplication-aware algorithm for the comparison of phylogenetic trees. Nucleic Acids Res 39:e66
Lu B, Zhang L, Leong HW (2017) A program to compute the soft Robinson-Foulds distance between phylogenetic networks. BMC Genomics 18:111
Koonin EV, Wolf YI, Puigbo P (2009) The phylogenetic forest and the quest for the elusive tree of life. Cold Spring Harb Symp Quant Biol 74:205–213
Zuckerkandl E, Pauling L (1962) Molecular evolution. In: Kasha M, Pullman B (eds) Horizons in biochemistry. Academic, New York, pp 189–225
Woese CR (1987) Bacterial evolution. Microbiol Rev 51:221–271
Bapteste E, O’Malley MA, Beiko RG, Ereshefsky M, Gogarten JP, Franklin-Hall L et al (2009) Prokaryotic evolution and the tree of life are two different things. Biol Direct 4:34
Doolittle WF (2000) Uprooting the tree of life. Sci Am 282:90–95
Doolittle WF, Bapteste E (2007) Pattern pluralism and the Tree of Life hypothesis. Proc Natl Acad Sci U S A 104:2043–2049
Kurland CG, Canback B, Berg OG (2003) Horizontal gene transfer: a critical view. Proc Natl Acad Sci U S A 100:9658–9662
Kurland CG (2005) What tangled web: barriers to rampant horizontal gene transfer. BioEssays 27:741–747
Logsdon JM, Faguy DM (1999) Thermotoga heats up lateral gene transfer. Curr Biol 9:R747–R751
Genereux DP, Logsdon JM Jr (2003) Much ado about bacteria-to-vertebrate lateral gene transfer. Trends Genet 19:191–195
Kunin V, Goldovsky L, Darzentas N, Ouzounis CA (2005) The net of life: reconstructing the microbial phylogenetic network. Genome Res 15:954–959
Daubin V, Moran NA, Ochman H (2003) Phylogenetics and the cohesion of bacterial genomes. Science 301:829–832
Lerat E, Daubin V, Moran NA (2003) From gene trees to organismal phylogeny in prokaryotes: the case of the gamma-proteobacteria. PLoS Biol 1:E19
Woese CR, Olsen GJ, Ibba M, Soll D (2000) Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev 64:202–236
Fitz-Gibbon ST, House CH (1999) Whole genome-based phylogenetic analysis of free-living microorganisms. Nucleic Acids Res 27:4218–4222
Hanage WP, Fraser C, Spratt BG (2006) Sequences, sequence clusters and bacterial species. Philos Trans R Soc Lond B Biol Sci 361:1917–1927
Eisen JA, Fraser CM (2003) Phylogenomics: intersection of evolution and genomics. Science 300:1706–1707
Salzberg SL, White O, Peterson J, Eisen JA (2001) Microbial genes in the human genome: lateral transfer or gene loss? Science 292:1903–1906
Galtier N (2007) A model of horizontal gene transfer and the bacterial phylogeny problem. Syst Biol 56:633–642
Galtier N, Daubin V (2008) Dealing with incongruence in phylogenomic analyses. Philos Trans R Soc Lond B Biol Sci 363:4023–4029
Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P (2006) Toward automatic reconstruction of a highly resolved tree of life. Science 311:1283–1287
Choi IG, Kim SH (2007) Global extent of horizontal gene transfer. Proc Natl Acad Sci U S A 104:4489–4494
Dagan T, Martin W (2009) Getting a better picture of microbial evolution en route to a network of genomes. Philos Trans R Soc Lond B Biol Sci 364:2187–2196
Boucher Y, Douady CJ, Papke RT, Walsh DA, Boudreau ME, Nesbo CL et al (2003) Lateral gene transfer and the origins of prokaryotic groups. Annu Rev Genet 37:283–328
Bucknam J, Boucher Y, Bapteste E (2006) Refuting phylogenetic relationships. Biol Direct 1:26
Schliep K, Lopez P, Lapointe FJ, Bapteste E (2011) Harvesting evolutionary signals in a forest of prokaryotic gene trees. Mol Biol Evol 28:1393–1405
Beiko RG, Doolittle WF, Charlebois RL (2008) The impact of reticulate evolution on genome phylogeny. Syst Biol 57:844–856
Doolittle WF, Zhaxybayeva O (2009) On the origin of prokaryotic species. Genome Res 19:744–756
Gogarten JP, Townsend JP (2005) Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol 3:679–687
Gogarten JP, Doolittle WF, Lawrence JG (2002) Prokaryotic evolution in light of gene transfer. Mol Biol Evol 19:2226–2238
O’Malley MA, Koonin EV (2011) How stands the Tree of Life a century and a half after The Origin? Biol Direct 6:32
Puigbo P, Wolf YI, Koonin EV (2013) Seeing the Tree of Life behind the phylogenetic forest. BMC Biol 11:46
Puigbo P, Wolf YI, Koonin EV (2010) The tree and net components of prokaryote evolution. Genome Biol Evol 2:745–756
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV et al (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41
Jensen LJ, Julien P, Kuhn M, von Mering C, Muller J, Doerks T et al (2008) eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res 36:D250–D254
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Keane TM, Naughton TJ, McInerney JO (2007) MultiPhyl: a high-throughput phylogenomics webserver using distributed computing. Nucleic Acids Res 35:W33–W37
Creevey CJ, McInerney JO (2005) Clann: investigating phylogenetic information through supertree analyses. Bioinformatics 21:390–392
Felsenstein J (1996) Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol 266:418–427
Torgerson WS (1958) Theory and methods of scaling. Wiley, New York
Gower JC (1966) Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 53:325–328
Tibshirani R, Walther G, Hastie T (2001) Estimating the number of clusters in a data set via the gap statistic. J R Stat Soc B Stat Methodol 63:411–423
Hillis DM, Heath TA, St John K (2005) Analysis and visualization of tree space. Syst Biol 54:471–482
Pavlidis P, Noble WS (2003) Matrix2png: a utility for visualizing matrix data. Bioinformatics 19:295–296
Koonin EV, Wolf YI (2008) Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res 36:6688–6719
Ge F, Wang LS, Kim J (2005) The cobweb of life revealed by genome-scale estimates of horizontal gene transfer. PLoS Biol 3:e316
Brochier C, Bapteste E, Moreira D, Philippe H (2002) Eubacterial phylogeny based on translational apparatus proteins. Trends Genet 18:1–5
Wolf YI, Rogozin IB, Grishin NV, Koonin EV (2002) Genome trees and the tree of life. Trends Genet 18:472–479
Wolf YI, Rogozin IB, Grishin NV, Tatusov RL, Koonin EV (2001) Genome trees constructed using five different approaches suggest new major bacterial clades. BMC Evol Biol 1:8
Creevey CJ, Fitzpatrick DA, Philip GK, Kinsella RJ, O’Connell MJ, Pentony MM et al (2004) Does a tree-like phylogeny only exist at the tips in the prokaryotes? Proc Biol Sci 271:2551–2558
Brochier-Armanet C, Boussau B, Gribaldo S, Forterre P (2008) Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat Rev Microbiol 6:245–252
Elkins JG, Podar M, Graham DE, Makarova KS, Wolf Y, Randau L et al (2008) A korarchaeal genome reveals new insights into the evolution of the Archaea. Proc Natl Acad Sci U S A 105:8102–8107
Wolf YI, Aravind L, Grishin NV, Koonin EV (1999) Evolution of aminoacyl-tRNA synthetases--analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res 9:689–710
Koonin EV (2003) Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat Rev Microbiol 1:127–136
Acknowledgment
The authors’ research is supported by the Department of Health and Human Services intramural program (NIH, National Library of Medicine).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
1 Electronic Supplementary Material
Additional File 1
Results of random analysis: Random test were performed for trees between 4 and 100 species, and each test is the result of 1000 random trees compared all-against-all (PPTX 411 kb)
Additional File 2
Results of the equivalence between topological distance and permutations: Number of permutations required to obtain any distance value (BSD or SD) (XLSX 22354 kb)
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this protocol

Cite this protocol
Puigbò, P., Wolf, Y.I., Koonin, E.V. (2019). Genome-Wide Comparative Analysis of Phylogenetic Trees: The Prokaryotic Forest of Life. In: Anisimova, M. (eds) Evolutionary Genomics. Methods in Molecular Biology, vol 1910. Humana, New York, NY. https://doi.org/10.1007/978-1-4939-9074-0_8
Download citation
DOI: https://doi.org/10.1007/978-1-4939-9074-0_8
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-4939-9073-3
Online ISBN: 978-1-4939-9074-0
eBook Packages: Springer Protocols