
Abstract
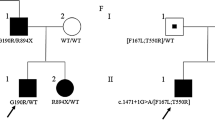
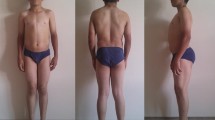
Thomsen’s and Becker’s diseases are the most prevalent nondystrophic myotonias. Their frequency varies, according to different sources, from 1: 100000 to 1: 10000. Thomsen’s myotonia is autosomal dominant, and Becker’s myotonia is autosomal recessive. Both diseases result from mutations of the CLCN1 gene encoding chloride ion channels of skeletal muscles. Molecular genetic analysis of the CLCN1 gene has been performed in patients with diagnoses of nondystrophic Thomsen’s and Becker’s myotonias living in the Russian Federation. A sample of 79 unrelated probands with nondystrophic Thomsen’s and Becker’s myotonias and 44 their relatives has been formed in the Laboratory of DNA Diagnosis of the Medical Genetic Research Center of the Russian Academy of Medical Sciences. Forty CLCN1 gene mutations have been found in a total of 118 chromosomes of 66 probands, including 21 familial and 45 sporadic cases. About half the mutations detected (45%) have been found for the first time; they are not described in the SNP database (ncbi.nlm.nih.gov). The following mutations (substitutions) have been detected in more than one chromosome, accounting for a total of 59.3% of chromosomes with mutations: Gly190Ser (5.9%), c.1437_1450del14 (9.3%), Ala493Glu (5.1%), Thr550Met (3.4%), Tyr686Stop (5.1%), and Arg894Stop (30.5%).
Similar content being viewed by others

References
Colding-Jorgensen, E., Phenotypic Variability in Myotonia Congenita, Muscle Nerve, 2005, vol. 32, no. 1, pp. 19–34.
Meyer-Kleine, C., Stenmeyer, K., and Ricker, K., et al., Spectrum of Mutations in the Major Human Skeletal Muscle Chloride Channel Gene (CLCN1) Leading to Myotonia, Am. J. Hum. Genet., 1995, vol. 57, no. 6, pp. 1325–1334.
Fahlke, C., Durr, C., and George, A.L., Mechanism of Ion Permeation in Skeletal Muscle Chloride Channels, J. Gen. Physiol., 1997, vol. 110, no. 5, pp. 551–64.
Dutzler, R., Campbell, E.B., Cadene, M., et al., X-Ray Structure of a ClC Chloride Channel at 3.0 A Reveals the Molecular Basis of Anion Selectivity, Nature, 2002, vol. 415, no. 6869, pp. 287–94.
Abdalla, J.A., Casley, W.L., Cousin, H.K., et al., Linkage of Thomsen Disease to the T-Cell-Receptor Beta (TCRB) Locus on Chromosome 7q35, Am. J. Hum. Genet., 1992, vol. 51, no. 3, pp. 579–84.
Koch, M.C., Steinmeyer, K., Lorenz, C., et al., The Skeletal Muscle Chloride Channel in Dominant and Recessive Human Myotonia, Science, 1992, vol. 257, no. 5071, pp. 797–800.
Sanger, F., Air, G.M., Barrell, B.G., et al., Nucleotide Sequence of Bacteriophage phi X174 DNA, Nature, 1977, vol. 265, no. 5596, pp. 687–695.
de Diego, C., Gamez, J., Plassart-Schiess, E., et al., Novel Mutations in the Muscle Chloride Channel CLCN1 Gene Causing Myotonia Congenita in Spanish Families, J. Neurol., 1999, vol. 246, no. 9, pp. 825–829.
Kuo, H.C., Hsiao, K.M., Chang, L.I., et al., Novel Mutations at Carboxyl Terminus of CIC-1 Channel in Myotonia Congenita, Acta Neurol. Scand., 2006, vol. 113, no. 5, pp. 342–346.
Matthews, E., Fialho, D., Tan, S.V., et al., The Non-Dystrophic Myotonias: Molecular Pathogenesis, Diagnosis and Treatment, Brain J. Neurol., 2010, vol. 133, no. 1, pp. 9–22.
Accardi, A. and Pusch, M., Fast and Slow Gating Relaxations in the Muscle Chloride Channel CLC-1, J. Gen. Physiol., 2000, vol. 116, no. 3, pp. 433–444.
Fialho, D., Schorge, S., Pucovska, U., et al., Chloride Channel Myotonia: Exon 8 Hot-Spot for Dominant-Negative Interactions, Brain J. Neurol., 2007, vol. 130, no. 12, pp. 3265–3274.
Duffield, M., Rychkov, G., Bretag, A., and Roberts, M., Involvement of Helices at the Dimer Interface in ClC-1 Common Gating, J. Gen. Physiol., 2003, vol. 121, no. 2, pp. 149–161.
Steinmeyer, K., Lorenz, C., Push, M., et al., Multimeric Structure of ClC-1 Chloride Channel Revealed by Mutations in Dominant Myotonia Congenita (Thomsen), EMBO J., 1994, vol. 13, no. 4, pp. 737–743.
Shalata, A., Furman, H., Adir, V., et al., Myotonia Congenita in a Large Consanguineous Arab Family: Insight into the Clinical Spectrum of Carriers and Double Heterozygotes of a Novel Mutation in the Chloride Channel CLCN1 Gene, Muscle Nerve, 2009, vol. 41, no. 4, pp. 464–469.
Sun C., Tranebjaerg L., Torbergsen T. et al., Spectrum of CLCN1 mutations in patients with myotonia congenita in Northern Scandinavia, Eur. J. Hum. Genet., 2001, vol. 9, pp. 903–909.
Trip, J., Drost, G., Verbove, D.J., et al., In Tandem Analysis of CLCN1 and SCN4A Greatly Enhances Mutation Detection in Families with Non-Dystrophic Myotonia, Eur. J. Hum. Genet., 2008, vol. 16, no. 8, pp. 921–929.
George, A.L., Crackower, M.A., Abdalla, J.A., et al., Molecular Basis of Thomsen’s Disease (Autosomal Dominant Myotonia Congenita), Nat. Genet., 1993, vol. 3, no. 4, pp. 305–310.
Koty, P.P., Pergoraro, E., Hobson, G., et al., Myotonia and the Muscle Chloride Channel: Dominant Mutations Show Variable Penetrance and Founder Effect, Neurology, 1996, vol. 47, no. 4, pp. 963–968.
Meyer-Kleine, C., Ricker, K., Otto, M., and Koch, M.C., A Recurrent 14 Bp Deletion in the CLCN1 Gene Associated with Generalized Myotonia (Becker), Hum. Mol. Genet., 1994, vol. 3, no. 6, pp. 1015–1016.
Author information
Authors and Affiliations
Corresponding author
Additional information
Original Russian Text © E.A. Ivanova, E.L. Dadali, V.P. Fedotov, S.A. Kurbatov, G.E. Rudenskaya, T.N. Proskokova, A.V. Polyakov, 2012, published in Genetika, 2012, Vol. 48, No. 9, pp. 1113–1123.
According to the current rules of the nomenclature of mutations in the human genome presented at the Human Genome Variation Society website (http://www.hgvs.org/mutnomen/rec-sport.html; den Dunnen, J.T. and Antonarakis, S.E., Hum. Mutat., 2000, vol. 15, pp. 7–12), the names of all mutations mentioned in this paper that have been described at the protein level should begin with p., e.g., p.Gly190Ser instead of Gly190Ser.
Rights and permissions
About this article
Cite this article
Ivanova, E.A., Dadali, E.L., Fedotov, V.P. et al. The spectrum of CLCN1 gene mutations in patients with nondystrophic Thomsen’s and Becker’s myotonias. Russ J Genet 48, 952–961 (2012). https://doi.org/10.1134/S1022795412090049
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1022795412090049