
Abstract
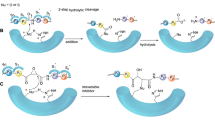
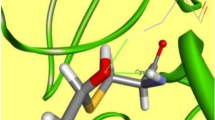
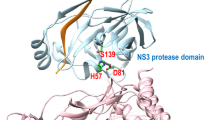
Over the last 2 decades, covalent inhibitors have gained much popularity and is living up to its reputation as a powerful tool in drug discovery. Covalent inhibitors possess many significant advantages including increased biochemical efficiency, prolonged duration and the ability to target shallow, solvent exposed substrate-binding domains. However, rapidly mounting concerns over the potential toxicity, highly reactive nature and general lack of selectivity have negatively impacted covalent inhibitor development. Recently, a great deal of emphasis by the pharmaceutical industry has been placed toward the development of novel approaches to alleviate the major challenges experienced through covalent inhibition. This has unexpectedly led to the emergence of “selective” covalent inhibitors. The purpose of this review is not only to provide an overview from literature but to introduce a technical guidance as to how to initiate a systematic “road map” for the design of selective covalent inhibitors which we believe may assist in the design and development of optimized potential selective covalent HCV NS3/4A viral protease inhibitors.






Similar content being viewed by others
References
Petruzziello A, Marigliano S, Loquercio G et al (2016) Global epidemiology of hepatitis C virus infection: an up-date of the distribution and circulation of hepatitis C virus genotypes. World J Gastroenterol 22:7824–7840. doi:10.3748/wjg.v22.i34.7824
Soumana DI, Ali A, Schiffer CA (2014) Structural analysis of asunaprevir resistance in HCV NS3/4A protease. ACS Chem Biol 9:2485–2490. doi:10.1021/cb5006118
Manns MP, Buti M, Gane E, Pawlotsky JM, Razavi H, Terrault N, Younossi Z (2017) Hepatitis C virus infection. Nat Rev Dis Prim. doi:10.1038/nrdp.2017.6
Villani R, Bellanti F, Serviddio G (2013) Treatment of chronic HCV infection in the era of protease inhibitors. In: Practical management of chronic viral hepatitis. InTech, Croatia, pp 167–184
Kalgutkar AS, Dalvie DK (2012) Drug discovery for a new generation of covalent drugs. Expert Opin Drug Discov 7:561–581. doi:10.1517/17460441.2012.688744
Kumalo HM, Bhakat S, Soliman MES (2015) Theory and applications of covalent docking in drug discovery: merits and pitfalls. Molecules 20:1984–2000. doi:10.3390/molecules20021984
Adeniyi AA, Muthusamy R, Soliman ME (2016) New drug design with covalent modifiers. Expert Opin Drug Discov 11:79–90. doi:10.1517/17460441.2016.1115478
Awoonor-Williams E, Walsh AG, Rowley CN (2017) Modeling covalent-modifier drugs. Biochim Biophys Acta. doi:10.1016/j.bbapap.2017.05.009
Becker D, Kaczmarska Z, Arkona C, Schulz R, Tauber C, Wolber G, Hilgenfeld R, Coll M, Rademann J (2016) Irreversible inhibitors of the 3C protease of Coxsackie virus through templated assembly of protein-binding fragments. Nat Commun 7:12761. doi:10.1038/ncomms12761
Zhu K, Borrelli KW, Greenwood JR et al (2014) Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J Chem Inf Model 54:1932–1940. doi:10.1021/ci500118s
Hagel M, Niu D, St Martin T et al (2011) Selective irreversible inhibition of a protease by targeting a noncatalytic cysteine. Nat Chem Biol 7:22–24. doi:10.1038/nchembio.492
Yao N, Reichert P, Taremi SS et al (1999) Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure 7:1353–1363. doi:10.1016/S0969-2126(00)80025-8
Shiryaev SA, Thomsen ER, Cieplak P et al (2012) New details of HCV NS3/4A proteinase functionality revealed by a high-throughput cleavage assay. PLoS ONE 7:1–12. doi:10.1371/journal.pone.0035759
Hamad HA, Thurston J, Teague T et al (2016) The NS4A cofactor dependent enhancement of HCV NS3 protease activity correlates with a 4D geometrical measure of the catalytic triad region. PLoS ONE 11:e0168002. doi:10.1371/journal.pone.0168002
Chevaliez S, Pawlotsky J-M (2006) HCV genome and life cycle. Hepat C Viruses Genomes Mol Biol 5–47
Kim JL, Morgenstern KA, Lin C et al (1996) Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 87:343–355. doi:10.1016/S0092-8674(00)81351-3
Tanoury GJ, Eastham S, Harrison CL et al (2016) Telaprevir: from drug discovery to the manufacture of drug substance. In: Comprehensive accounts of pharmaceutical research and development: from discovery to late-stage process development, vol 1. American Chemical Society, Washington, DC. doi:10.1021/bk-2016-1239.ch012
Steinkühler C, Biasiol G, Brunetti M et al (1998) Product inhibition of the hepatitis C virus NS3 protease. BioChemistry 37:8899–8905. doi:10.1021/bi980313v
Lamarre D, Anderson PC, Bailey M et al (2003) An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. doi:10.1038/nature02099
Chen KX, Njoroge FG (2011) NS3 protease covalent Inhibitors. Hepatitis C Antiviral Drug Discov Dev
Baillie TA (2016) Targeted covalent inhibitors for drug design. Angew Chem Int Ed 55:13408–13421. doi:10.1002/anie.201601091
Swinney DC (2004) Opinion: biochemical mechanisms of drug action: what does it take for success?. Nat Rev Drug Discov 3:801–808. doi:10.1038/nrd1500
Johnson DS, Weerapana E, Cravatt BF (2010) Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med Chem 2:949–964. doi:10.4155/fmc.10.21
Johansson MH (2012) Reversible michael additions: covalent inhibitors and prodrugs. Mini Rev Med Chem 12:1330–1344. doi:10.2174/13895575112091330
Hallenbeck KK, Turner DM, Renslo AR, Arkin MR (2017) Targeting non-catalytic cysteine residues through structure-guided drug discovery. Curr Top Med Chem 17:4–15
Blake L, Soliman MES (2014) Identification of irreversible protein splicing inhibitors as potential anti-TB drugs: insight from hybrid non-covalent/covalent docking virtual screening and molecular dynamics simulations. Med Chem Res 23:2312–2323. doi:10.1007/s00044-013-0822-y
Rose PW, Bi C, Bluhm WF et al (2013) The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res 41:D475–D482. doi:10.1093/nar/gks1200
Sievers F, Wilm A, Dineen D et al (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi:10.1038/msb.2011.75
Edgar RC (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi:10.1093/nar/gkh340
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi:10.1093/nar/gkf436
Papadopoulos JS, Agarwala R (2007) COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 23:1073–1079. doi:10.1093/bioinformatics/btm076
Di Tommaso P, Moretti S, Xenarios I et al (2011) T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. doi:10.1093/nar/gkr245
Källberg M, Wang H, Wang S et al (2012) Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7:1511–1522. doi:10.1038/nprot.2012.085
Fiser A, Šali A (2003) MODELLER: generation and refinement of homology-based protein structure models. Methods Enzymol 374:461–491. doi:10.1016/S0076-6879(03)74020-8
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. doi:10.1093/bioinformatics/bti770
Huang B (2009) MetaPocket: a meta approach to improve protein ligand binding site prediction. Omi A J Integr Biol 13:325–330. doi:10.1089/omi.2009.0045
Hernandez M, Ghersi D, Sanchez R (2009) SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic Acids Res 37:W413–W416. doi:10.1093/nar/gkp281
Wass MN, Kelley LA, Sternberg MJE (2010) 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res 38:gkq406. doi:10.1093/nar/gkq406
Dundas J, Ouyang Z, Tseng J et al (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 34:116–118. doi:10.1093/nar/gkl282
Weisel M, Proschak E, Schneider G (2007) PocketPicker: analysis of ligand binding-sites with shape descriptors. Chem Cent J 1:7. doi:10.1186/1752-153X-1-7
Dessailly BH, Lensink MF, Orengo CA, Wodak SJ (2008) LigASite: a database of biologically relevant binding sites in proteins with known apo-structures. Nucleic Acids Res. doi:10.1093/nar/gkm839
Brylinski M, Skolnick J (2008) A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc Natl Acad Sci USA 105:129–134. doi:10.1073/pnas.0707684105
Pettersen EF, Goddard TD, Huang CC et al (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi:10.1002/jcc.20084
Hanwell MD, Curtis DE, Lonie DC et al (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 4:17. doi:10.1186/1758-2946-4-17
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38. doi:10.1016/0263-7855(96)00018-5
DeLano WL (2014) The PyMOL molecular graphics system, Version 1.8. Schrödinger LLC, New York. http://www.pymol.org. doi:10.1038/hr.2014.17
Irwin JJ, Sterling T, Mysinger MM et al (2012) ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52:1757–1768. doi:10.1021/ci3001277
Xie X-QS (2010) Exploiting PubChem for virtual screening. Expert Opin Drug Discov 5:1205–1220. doi:10.1517/17460441.2010.524924
Koes DR, Camacho CJ (2012) ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res. doi:10.1093/nar/gks378
Pence HE, Williams A (2010) Chemspider: an online chemical information resource. J Chem Educ 87:1123–1124. doi:10.1021/ed100697w
Wishart DS (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34:D668–D672. doi:10.1093/nar/gkj067
Gaulton A, Bellis LJ, Bento AP et al (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. doi:10.1093/nar/gkr777
Cosconati S, Forli S, Perryman AL et al (2010) Virtual screening with AutoDock: theory and practice. Expert Opin Drug Discov 5:597–607. doi:10.1517/17460441.2010.484460
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461. doi:10.1002/jcc.21334
Ouyang X, Zhou S, Su CTT et al (2013) CovalentDock: automated covalent docking with parameterized covalent linkage energy estimation and molecular geometry constraints. J Comput Chem 34:326–336. doi:10.1002/jcc.23136
Verdonk ML, Cole JC, Hartshorn MJ et al (2003) Improved protein-ligand docking using GOLD. Proteins Struct Funct Genet 52:609–623. doi:10.1002/prot.10465
Jones G, Willett P, Glen RC et al (1997) Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267:727–748. doi:10.1006/jmbi.1996.0897
Friesner RA, Murphy RB, Repasky MP et al (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49:6177–6196. doi:10.1021/jm051256o
Schmidt B (2014) DOCKTITE: a highly versatile step-by-step workflow for covalent docking and virtual screening in MOE. J Chem Inf Model. doi:10.1021/ci500681r
Götz AW, Clark MA, Walker RC (2014) An extensible interface for QM/MM molecular dynamics simulations with AMBER. J Comput Chem 35:95–108. doi:10.1002/jcc.23444
Salomon-Ferrer R, Case DA, Walker RC (2013) An overview of the Amber biomolecular simulation package. Wiley Interdiscip Rev Comput Mol Sci 3:198–210. doi:10.1002/wcms.1121
Van Der Spoel D, Lindahl E, Hess B et al (2005) GROMACS: fast, flexible, and free. J Comput Chem 26:1701–1718. doi:10.1002/jcc.20291
Velde GTE, Bickelhaupt FM, Baerends EJ et al (2001) Chemistry with ADF. J Comput Chem 22:931–967. doi:10.1002/jcc.1056
Valiev M, Bylaska EJ, Govind N et al (2010) NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput Phys Commun 181:1477–1489. doi:10.1016/j.cpc.2010.04.018
Woodcock HL, Hodošček M, Gilbert ATB et al (2007) Interfacing Q-Chem and CHARMM to perform QM/MM reaction path calculations. J Comput Chem 28:1485–1502. doi:10.1002/jcc.20587
Metz S, Kästner J, Sokol AA et al (2014) ChemShell-a modular software package for QM/MM simulations. Wiley Interdiscip Rev Comput Mol Sci 4:101–110. doi:10.1002/wcms.1163
Ward RA, Colclough N, Challinor M et al (2015) Structure-guided design of highly selective and potent covalent inhibitors of ERK1/2. J Med Chem 58:4790–4801. doi:10.1021/acs.jmedchem.5b00466
Xiao C, Zhang Y (2007) Design-atom approach for the quantum mechanical/molecular mechanical covalent boundary: a design-carbon atom with five valence electrons. J Chem Phys 127:1–25. doi:10.1063/1.2774980
Allen M (2004) Introduction to molecular dynamics simulation. Comput Soft Matter 23:1–28. doi:10.1016/j.cplett.2006.06.020
Groenhof G (2013) Introduction to QM/MM simulations. In: Biomolecular simulations: methods and protocols. Springer, New York, pp 43–66
Senn HM, Thiel W (2009) QM/MM methods for biomolecular systems. Angew Chemie Int Ed 48:1198–1229
Karplus M, McCammon JA (2002) Molecular dynamics simulations of biomolecules. Nat Struct Mol Biol 9:646–652. doi:10.1038/nsb0902-646
Acknowledgements
The authors acknowledge the National Research Foundation for their financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare no potential conflicts of interest.
Rights and permissions
About this article

Cite this article
Shunmugam, L., Ramharack, P. & Soliman, M.E.S. Road Map for the Structure-Based Design of Selective Covalent HCV NS3/4A Protease Inhibitors. Protein J 36, 397–406 (2017). https://doi.org/10.1007/s10930-017-9736-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-017-9736-8