
Abstract
Despite decades of research and efforts at improving survival, pancreatic ductal adenocarcinoma (PDA) has become the third leading cause of cancer-related deaths in the United States. In fact, by 2020, it is projected to become the second leading cause of cancer-related deaths in the United States. Personalized, or precision, medicine has resulted in improving patient outcomes in other tumor systems. However, for pancreatic cancer patients, there are a limited number of evidence-based targeted therapeutic options that are currently available. Significant advances in DNA sequencing technology have resulted in the identification of a number of genetic mutations and the delineation of core signaling pathways important in PDA. This has subsequently resulted in an advanced understanding of the genetic drivers of the progression of this disease. Facile sequencing technology has moved the field closer to a personalized approach to treating pancreatic cancer. Improvements to the personalized therapy approach will likely result from several factors including the delivery of tumor sequencing results in a clinically relevant timeframe, the development of better targeted drugs, and perhaps a molecular-targeted approach to aspects of PDA biology beyond mutations in the deoxyribonucleic acid (DNA). These advances will allow clinicians to enroll patients in appropriate-matched clinical trials in a timely manner. In this chapter, the opportunities and limitations of a targeted, personalized approach to treating PDA will be discussed.
Similar content being viewed by others

Keywords
1 Introduction
Pancreatic ductal adenocarcinoma (PDA) remains a largely deadly disease with a 5-year survival of only 9% for all stages combined [1]. Currently, it is the third leading cause of cancer-related deaths in the United States and it is on pace to become the second leading cause by 2020 [2]. This mortality rate is due to a number of factors such as aggressive tumor biology; lack of early screening and prevention strategies; and ineffective targeted treatments. Thanks to large-scale high-throughput sequencing studies, our understanding of the molecular driving events in pancreatic tumorigenesis has increased over the past few decades. However, unlike in other cancers, this has not resulted in a similar increase in effective targeted treatment options that are available in the clinic. In fact, the mainstay of pancreatic cancer treatment remains largely conventional and includes surgery for the minority of patients who are diagnosed with resectable disease, and cytotoxic therapy [3,4,5].
The clinical aggressiveness observed in PDA is due, in part, to its cellular complexity and its ability to survive in a harsh tumor microenvironment. These factors likely contribute to resistance to many therapies. First, PDA is associated with a dense stromal reaction. The tumor mass is composed mainly of the tumor microenvironment, and it includes mostly nonneoplastic cells, such as fibroblasts and lymphocytes, and noncellular connective tissue [6, 7]. Additionally, the PDA tumor microenvironment also includes a vasculature, but this cancer is classically hypovascular [8]. This is evident when these tumors are visualized with contrast-enhanced computed tomography imaging, which shows hypoattenuated lesions when compared to the well-enhancing normal surrounding pancreatic parenchyma [9]. PDAs are also genetically complex. Though common driver-mutations are present in essentially all PDAs (i.e., high-frequency mutations, such as KRAS), there are a significant number of low-frequency mutations of which the clinical significance has yet to be determined [10,11,12]. It is this degree of genetic diversity that increases the complexity when considering targeted therapy . For instance, it is unclear which low-frequency mutations contribute to the tumorigenesis in PDA by allowing the tumor to overcome a selective pressure and whether they confer a growth advantage. Some of these low-frequency aberrations may simply be passenger mutations [13]. Additionally, assuming that low-frequency mutations are important in PDA tumorigenesis, it raises the question whether using targeted therapies that will impact a small subset of patients will result in meaningful improvement in overall outcomes in PDA.
This chapter will provide a basic overview of DNA sequencing technology that is available today and how it has contributed to our understanding of dysregulated pathways in PDA. Current targeted therapies and outcomes of precision medicine -based clinical trials in PDA will be reviewed, along with other potential therapeutic strategies that go beyond the targeted approach.
2 Advances in DNA Sequencing and Its Implications in PDA
2.1 Sequencing in PDA
Emerging technologies in sequencing, such as next-generation sequencing (NGS) or whole-exome sequencing (WES) or whole-genome sequencing (WGS) strategies, have been used in PDA to determine its genomic landscape as well as its pathologic progression from precursor lesions into PDA [14, 15]. NGS is a powerful tool that allows for parallel sequencing of multiple genes in one test. Compared to WES, WGS and determination of copy-number alterations (CNAs) provide a more granular view of the genomic landscape of the tumor. WGS and CNAs allow the measurement of alterations in DNA structure (i.e., deletions, amplifications, insertions, and translocations) and result in an improved understanding of the patterns of chromosomal instability that are often observed in PDA [16, 17].
When compared to other tumors, sequencing of PDA is not a simple exercise. In part, this is due to the characteristic desmoplastic stroma that makes analysis of pure tumor epithelial cells difficult. Some of the ways to circumvent this limitation in sequencing PDAs include developing patient-derived cell lines or using laser microdissection, both are methods that enrich the tumor epithelial content [18]. Despite these apparent limitations, over 1300 PDA genomes or exomes have been sequenced, which has added to our understanding of the molecular drivers in PDA (Table 1). More current studies that utilize NGS have focused on WGS and more detailed genomic analyses, combined with ribonucleic acid (RNA) sequencing for a better characterization of PDAs [19, 20]. Despite the increase in utilization of such sophisticated high-throughput studies, novel high-frequency mutations, beyond the key players such as KRAS, have not been identified. However, identification of novel pathways and also subtyping PDA has emerged as a promising deliverable of this work [19, 21].
The first extensive WES analysis of PDA was first published in 2008 by Jones and colleagues [10]. This study used samples from 24 human cell lines and xenografts and utilized Sanger sequencing to sequence 20,661 genes. Genetic alterations that were identified were variable and included point-mutations, deletions, and amplifications. The authors were able to reproducibly identify well-described mutations in KRAS, CDKN2A (p16), TP53, and SMAD4 in PDA genomes. Reproducible alterations in other genes, such as ARID1A, TGFBR2, were identified, but these were found in lower frequencies. Ultimately, the researchers were able to identify 69 genes that were altered in the 24 analyzed samples. Thirty-one of these were further subdivided into 12 core-signaling pathways that were found to be altered in 67–100% of the sequenced samples. Several pathways were found to be genetically altered in 100% of the tumor samples, such as apoptosis and KRAS signaling pathways. This clustering of genetic alterations along with molecular signaling pathways in this first high-throughput analysis provided a practical approach to support this research movement.
Following Jones and colleagues, a number of other studies provided sequencing of PDA samples. A recently published study by Bailey and colleagues would follow Jones and colleagues to provide the next large-scale sequencing data in PDA [19]. Using NGS, the authors performed a whole-genome analysis of 456 PDA samples. They identified 32 mutated genes, which were then grouped into 10 pathways that were consistently dysregulated. Moreover, when expression analyses were performed, they were able to group PDAs into four subtypes: squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine exocrine. Furthermore, these PDA subtypes were correlated with discernable histopathologic characteristics.
A study by Makohon-Moore and colleagues extended these NGS studies. Utilizing very strict inclusion criteria, the authors evaluated primary tumor and metastatic lesion samples by using WGS of patients that were treatment-naïve [31]. The goal was to determine the degree of genetic heterogeneity between primary tumors and metastatic lesions. This is important since it may impact a patient’s response to therapy. For example, if there is significant intratumoral heterogeneity between two different samples in the primary tumor or between the primary tumor and a metastatic lesion, it is likely that a patient would develop early resistance to targeted treatment. However, if the primary tumor is genetically similar to a metastatic lesion, it is plausible that both tumors would be sensitive to the initial therapy [34, 35]. A total of 39 samples were evaluated (26 from metastatic lesions, 3 from different regions of the primary tumor, and normal tissue) in four patients. There was a limited variability of driver mutations in untreated patients with metastatic PDA (mPDA) that were present in the primary tumor and the metastatic lesions. This suggests that in patients with metastatic cancer, there may be a clinical benefit afforded by using targeted therapies geared towards driver mutations in the primary tumor.
Moving forward, the goal is to continue the genetic characterization of PDA, to understand how these genetic aberrations relate to the clinical features of the patient’s disease, and to identify therapeutic targets. Moreover, there will be a continued trend and interest to further continue to characterize dysregulated pathways and subtypes of PDA.
2.2 Current Clinical Use of Next-Generation Sequencing in PDA and Its Implications
There has been an emergence of studies that attempt to link/associate patients’ tumor mutations with currently available targeted therapies. The goal of these studies is to take advantage of the opportunities offered by NGS to characterize genetic pathways that drive a specific PDA and to match it to an available targeted therapy. One such trial has been the Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial from Australia [36]. This was a feasibility trial that aimed to demonstrate the ability to successfully acquire patient samples and to provide quality genomic data for three molecular targets: HER2 amplification, KRAS, and mutations in DNA repair pathways (BRCA1, BRCA2, PALB2, and ATM). The goal was to evaluate whether it would be feasible to provide sequencing results in a clinically relevant timeline. Inclusion criteria included newly diagnosed PDA patients who either received one cycle of gemcitabine for metastatic disease or patients who were treatment-naïve. Patients were randomized in a 1:1 fashion and offered standard therapy (gemcitabine) versus personalized treatment (gemcitabine plus targeted therapy) depending on the patient’s genetic aberrations. At the time that the results of the trial were reported, no patients were successfully treated on the protocol.
The Pancreatic Cancer Action Network (PanCAN) has also launched the Know Your Tumor® Initiative with the goal of providing sequencing data to patients and their oncologists in order to facilitate the use of targeted therapy or clinical trial enrollment. The initial experience resulted in 117 patient sequencing reports, with the identification of an “actionable” finding in approximately 40% of cancers (actionable findings were defined as the availability of a targeted therapy in an identified molecular abnormality in any cancer type or predicted response based on pathway or mechanism-defined for the identified target) [37]. This resulted in 43% of patients being referred to high-priority clinical trials and 53% were recommended in the direction of off-label targeted therapy [38].
A similar multi-institution trial has been designed and implemented in the United States by the authors (MJP and JRB), with the goal of randomizing 60 patients along standard treatment and molecularly targeted therapy (MTT). This trial is also supported by PanCAN and the American Association of Cancer Research. Sequencing of 600 genes and protein expression analyses will be undertaken to further predict the patient’s response to either standard therapy or MTT. The results of this trial are forthcoming. PanCAN has also implemented a multi-institution clinical trial called Precision Promise. Its aim is to promote data-sharing by promoting a number of substudies that investigate different therapies under the same clinical trial umbrella in an effort to expedite the breadth of targeted therapies available to patients with PDA. A similar trial is being implemented in England, called PRECISION-Panc, where multiple subtrials will be carried out under the same umbrella clinical trial. The goal will be to provide molecular profiling of patients’ tumors followed by enrolling patients in clinical trials that utilize the targeted approach [12].
In addition to the trials described, there are a number of currently active trials that are based on identifying genetic aberrations for which targeted therapy is available. These include clinical trials, commonly referred to as basket trials, from the National Cancer institute (NCI) including NCI-Molecular Analysis for Therapy Choice (MATCH) Trial, NCI-Molecular Profiling-Based Assignment of Cancer Therapy (MPACT), and Initiative for Molecular Profiling and Advanced Cancer Therapy (IMPACT 2) [39,40,41]. These studies are not specific for patients with PDA; however, they are likely to recruit patients with PDA based on the trial designs.
3 The Use of Targeted Therapy in the Treatment of PDA
3.1 History of Precision Medicine and its Role in PDA
One of the earliest examples of precision oncology in clinical practice is the use of imatinib in patients with chronic myeloid leukemia (CML) that harbor the Philadelphia chromosome (i.e., the BCR-ABL mutation) [42]. The successful clinical use of mutation-targeted therapies has remained elusive in PDA. Even though the number of US Food and Drug Administration (FDA)-approved mutation-targeted therapies has increased over the years and has resulted in an improvement in outcomes in other cancers, similar results have not been realized in PDA.
In PDA, the use of targeted therapy is limited largely due to the fact that many alterations tend to result in loss-of-function in genes that would traditionally be considered tumor suppressors genes (TSGs). In general, TSGs halt cell proliferation, disrupt the cell cycle, and can initiate apoptosis; therefore, the inactivation of TSGs is a critical event for the progression of tumorigenesis. Since rescue of this genotype would require TSGs to regain function, this limits the use of small molecule compounds or drugs that generally are most effective against inhibiting oncogenes (i.e., in the setting of gain-of-function mutations), and not “turning on” an already “turned off” gene. High-frequency TSG mutations are common in PDAs (e.g., TP53, CDKN2A) and limit the personalized approach. At the present time, perhaps the most effective personalized therapy for PDA is targeting the BRCA pathway, which contains genes that are known TSGs, resulting in synthetic lethality (see next section). Perhaps the resurrection of gene therapy will become a clinical reality. If this happens, one can imagine real precision therapy, wherein specific TSGs can be sequenced in individual tumors and a matched gene therapy option can be utilized (e.g., SMAD4 overexpression for SMAD4 deleted tumors).
3.2 Pathways Dysregulated in PDA and Opportunities for Targeted Therapies
Historically, mutations or genomic alterations of KRAS, CDKN2A (p16), TP53, and SMAD4 have been implicated in the development of pancreatic intraepithelial lesions which ultimately lead to the development of PDA [43, 44]. More generally, multiple pathways are dysregulated in PDA and, in theory, targeted therapies can be used to exploit their specific function. Figure 1 demonstrates commonly altered pathways in PDA and downstream effectors that play a role in tumorigenesis. The remainder of this section will expand on these pathways and provide an overview of therapeutic strategies and options that can be utilized in patients with PDA. Though there have been mixed results with targeting some genetic aberrations, it is the degree and frequency of dysregulation in key cellular processes that make them ideal therapeutic targets in PDA. Therefore, an understanding of the role of individual pathways that are activated or deactivated in PDA will be instrumental to successfully target them in a personalized manner.

Core signaling pathways implicated in PDA, genetic aberrations implicated in tumorigenesis, and potential targeting strategies
3.2.1 Targeting KRAS Signaling and Its Downstream Effectors
The RAS family of oncogenes consists of HRAS, NRAS, and KRAS and one or more isoforms of this gene are mutated in most cancers [45]. The KRAS pathway is one of the best-characterized pathways in cancer and KRAS mutations are frequently observed in PDA, occurring in roughly 95% of tumors [43, 44, 46]. Activity of KRAS is tightly regulated, and under nonpathologic conditions, it exists in an inactive state (i.e., bound to GDP). Extracellular signals, such as growth factors, result in activation of KRAS and the conversion of GDP to GTP and activation of its downstream targets. KRAS mutations are found in PDA precursor lesions, so they are believed to occur early in the progression of PDA. Point mutations in KRAS often occur in codons 12, 13, or 61 resulting in a constitutively active GTPase that is unable to hydrolyze GTP. This results in sustained signaling of a number of downstream KRAS targets that affect cell survival, proliferation, cell cycle progression, apoptosis, and metabolism [45]. The importance of KRAS mutations in the initiation of PDA has been underscored by experiments that utilize genetically engineered mouse models (GEMMs), in which mutant KRAS is driven to be specifically expressed in the pancreas [47,48,49,50].
Targeting KRAS has been difficult to date, and in fact, KRAS is thought to be an “undruggable” target by some [51]. The NCI has started a program that is specifically geared to the development of KRAS inhibitors [52]. The difficulty with developing a small-molecule to target KRAS is, in part, due to the fact that it has a high affinity for GTP. GTP is abundant in the cell, and it effectively blocks access to the active site of the protein by other small molecules. Targeting farnesylation, one of the post-translational modifications of KRAS which affects its localization to the cell membrane, has not resulted in any significant clinical benefits either [53]. Similarly, specifically targeting the localization of KRAS to the cell membrane, which is dependent on PDEδ, with the use of PDEδ inhibitors has shown some success in xenograft models [54]. Targeting mutant KRAS with siRNA has been done in xenograft models, but this has yet to be translated to the clinic [55, 56]. In humans, KRAS siRNA was well tolerated and perhaps even efficacious in patients with locally advanced PDA [57].
Considering the difficulty with targeting KRAS directly, a significant amount of effort has been placed in targeting the downstream effector pathways. KRAS-driven tumors are believed to be dependent on MEK signaling for continued proliferation [58]. Therefore, MEK inhibitors have also been tested in preclinical models with positive results, which have not been reproduced in clinical trials. CI-1040A and AZD6244, two potent MEK inhibitors, have been investigated and were found to be ineffective in patients as second line therapy or as combination therapy with capecitabine in a randomized phase 2 trial [59, 60]. Similarly, trametinib in combination with gemcitabine, when compared to gemcitabine therapy alone, was not found to be superior in a randomized phase 2 trial for patients with treatment-naïve mPDA [61]. These clinical trials underscore the importance of targeting multiple effector pathways simultaneously [62, 63]. For example, concurrent inhibition of MEK and phosphoinositide 3-kinase (PI3K) or AKT may be required to overcome the limitations of targeting and inhibiting a single pathway [64,65,66]. This is due to the fact that there are data to suggest that activation of the PI3K pathway results in resistance to MEK inhibitors [64]. However, despite this, a combination of PI3K and MEK inhibition was not associated with increased survival when compared to modified FOLFOX in patients who failed prior gemcitabine therapy [67]. There have also been studies that have demonstrated synergism with the use of EGFR inhibitors and MEK inhibitors, especially in patients with wild-type KRAS tumors [68, 69]. Erlotinib, an epidermal growth factor receptor (EGFR) inhibitor, is currently FDA-approved for use as a second line therapy for recurrent, mPDA [70]. Moving forward, the combination of many of these therapies along with new targeted agents may be beneficial. In regards to a personalized approach, targeting KRAS mutations would certainly make this an all-inclusive line of therapy (i.e., one size fits all treatment, since the majority of PDAs harbor KRAS mutations). However, some investigators are studying whether specific KRAS amino acid changes, even at the same codon, might be more targetable than others.
3.2.2 Targeting the G1/S Checkpoint
CDKN2A (p16), a TSG, is another high-frequency mutation in PDA, found in over 95% of tumors [10]. It is a cyclin-dependent kinase inhibitor that functions to stop the transition of the cell from entry into S-phase by inhibiting the kinase activity of CDK4 and CDK6 [71,72,73]. In PDA, alterations in p16 expression can be due to promoter hypermethylation, homozygous deletions, or single-allele loss with a concomitant mutation in the second allele [74, 75]. All of these lead to inactivation of p16, which subsequently result in increased phosphorylation of Rb-1. This leads to deactivation of Rb-1 and progression through the G1-S cell cycle checkpoint, resulting in increased cell proliferation [74, 76].
In mutant KRAS-driven cancers, the loss of p16 is common and results in cell cycle dysregulation. Considering this, there is significant interest in recapitulating the function of p16. However, since CDKN2A is a TSG and therapies that result in reinstatement of its expression are limited, there is significant interest in suppressing activity of its targets, CDK4 and CDK6. CDK4/6 inhibitors, such as palbociclib and abemaciclib, have been developed and used in other tumor types and a number of other diseases [77, 78]. In PDA, both in vivo and in vitro studies have shown mixed results with the use of these inhibitors [79,80,81]. In PDA, inhibition of this pathway is currently being investigated. Actively enrolling trials include a phase I clinical trial evaluating the efficacy of palbociclib and gedatolisib, a PI3K/mTOR inhibitor, in patients with a number of solid tumors, including PDA (NCT03065062) and a phase I dose-escalation study evaluating palbociclib in combination with nab-paclitaxel in mPDA (NCT02501902). Another phase I/II clinical trial is evaluating the safety and efficacy of ribociclib in combination with Everolimus in patients with refractory mPDA (NCT02985125). Lastly, another phase Ib dose escalation trial is evaluating the safety of ribociclib in patients with advanced solid tumors and may recruit patients with PDA (NCT02703571). The results of these studies will be forthcoming.
TP53 is a common TSG mutated in most solid tumors and is mutated in 75% of PDAs [82]. TP53 is a transcription factor which modulates the expression of genes that are implicated in cell cycle arrest and apoptosis in the setting of DNA damage or cellular stress [83]. Generally, a mutation accompanied with loss of heterozygosity (LOH) in the second allele leads to its inactivation. Once cells lose TP53 expression, it allows them to bypass the G1-S cell cycle checkpoint, which again, results in increased cell proliferation [76]. Similar to p16, targeting of p53 is difficult since it is also a TSG. Due to this, it has become attractive as a target in tumor immunotherapy. The modified vaccinia virus ankara vaccine expressing p53 (p53MVA) has had some success in preclinical models [84]. Currently, it is being investigated in a clinical trial that includes patients with PDA (NCT02432963), but the success of this therapy is still unknown. A number of preclinical models have attempted to reactivate TP53 with the use of small molecules, such as APR-246 [85, 86]. An ongoing phase II clinical trial is evaluating the efficacy of SGT-53, liposomal nanocomplex tumor-targeting delivery of the wild-type p53 gene, in combination with gemcitabine and nab-paclitaxel in patients with mPDA (NCT02340117). To date, there have not been any clinically relevant therapies that have resulted in recapitulation of TP53 function that have resulted in a clinical benefit for patients with PDA [86, 87]. However, since it is commonly disrupted in PDA, the pursuit of targeting this genetic lesion is a worthy cause [88].
3.2.3 Exploiting BRCAness and DNA Damage Response and Repair Pathways
Genetic alterations in BRCA1/2 and other DNA damage response and repair genes (the DNA damage repair, DDR, pathway) are observed in 5–17% of PDAs [11, 26]. Furthermore, germline mutations in BRCA1 and BRCA2 have been shown to increase a patient’s risk of developing PDA 3.5–10-fold [89], as have mutations in the Fanconi anemia genes (i.e., FANCC, FANCG, and FANCN/PALB2) [90,91,92]. One of the features of tumors that harbor BRCA-related mutations or alterations in the DDR pathways is chromosomal instability [11, 23, 93]. Such mutations have been exploited in ovarian cancer, since tumors that are deficient in DDR have increased susceptibility to platinum-based therapy especially when combined with poly-ADP-ribose polymerase (PARP) inhibitor therapy [94].
This increased susceptibility to platinum-based therapy has been studied in PDA and has shown promising results. Golan and colleagues retrospectively reported on a large cohort of 71 patients with BRCA1 or BRCA2 associated tumors. They found that in patients with stage 3 and 4 disease who received platinum-based therapy (n = 22), when compared to those who received non-platinum based therapy (n = 21), there was improved in median overall survival (22 vs. 9 months, p < 0.039) [95].
As a result of the findings from preclinical models and retrospective studies, prospective trials have investigated the utility of PARP inhibitors in patients with PDA and germline mutations in DDR pathways. PARP inhibitors (PARPi) are a class of drugs that cause an accumulation of single-stranded breaks (SSB) in DNA. Once the replication fork encounters a SSB, it may result in termination or the formation of a double stranded break (DBS); cells that are BRCA-deficient are unable to repair these DSB via homologous recombination leading to cell death through mitotic catastrophe [96, 97]. This is a concept referred to as synthetic lethality [98]. A number of clinical trials have either recently been reported or are currently ongoing in order to investigate the safety and efficacy of PARPi in patients with BRCA1/2 or PALB2 mutations and have shown encouraging results [99,100,101]. At the present time, PARPi are perhaps the most promising avenue that utilizes targeted therapy that may be beneficial to a subset of patients with PDA. Further research will need to show whether tumors that harbor BRCA1/2 mutations are equally as sensitive to PARPi and platinum-based therapy. Moreover, it is important to remember that other genes are commonly mutated in the DDR pathway in PDA. These include ATM, ATR, RAD51, RAD51C, and RPA1. Identification of these targets has raised the possibility of use of ATM and ATR inhibitors in PDA and in other tumors [102]. In fact, there are a number of preclinical models or clinical trials ongoing that are evaluating the use of these therapies in combination with PARP inhibitors and platinum-based chemotherapy [103,104,105]. These studies will address the question of whether mutations in the DDR pathway result in the same cancer phenotype. Lastly, to maximize the benefit afforded with PARPi therapy, both alleles must be inactivated. Therefore, the role of NGS is underscored here where reliable sequencing results must be available to clinicians in order to maximally utilize this targeted therapy.
Chromatin remodeling and mutations in SWItch/sucrose non-fermentable (SWI/SNF) nucleosome complex are common in many tumors [106, 107]. The SWI/SNF nucleosome is a complex that consists of ATP-dependent chromatin remodeling factors that control the transcription of a number of genes by altering the chromatin structure [108, 109]. Loss of ARID1A, one of the components of the SWI/SNF complex, is the most common event (albeit, one that occurs at an overall low-frequency) and it behaves as a TSG in PDA [19, 110, 111]. Mutations in other subunits of the SWI/SNF complex have also been observed, and these include ARID1B, SMRCA4, and SMRCA2 [112]. Recent studies have demonstrated that the use of PARP or ATR inhibitors results in increased sensitivity in tumor cells that are deficient in ARID1A [113, 114]. This preclinical data can be used to expand the use of PARP and ATR inhibitors in patients with PDA who may harbor mutations in the SWI/SNF complex.
3.2.4 Role of SMAD4/TGF-β Signaling
TGF-β signaling has been implicated in pancreatic cancer; a mutation in at least one of the genes in the pathway is present in almost all PDAs [10, 115, 116]. One of the commonly dysregulated genes in this pathway is SMAD4, also known as DPC4, a TSG that is located on chromosome 18q. It encodes for a transcription factor that plays a role in the transforming growth factor beta (TGF-β) signaling pathway [117, 118]. In PDA, aberrations in SMAD4 can occur due to homozygous deletions or LOH, coupled with a point mutation that results in its inactivation. Mutations that result in loss of SMAD4 expression are found in 55% of PDAs. Furthermore, mutations in SMAD4 occur late in the progression of PDA tumorigenesis and are believed to play a role in the metastatic potential of this tumor [15, 119,120,121].
Targeting of this pathway would be clinically useful, considering the frequency with which it is lost along with other elements of this signaling pathway. Inhibition of this pathway can occur by inhibiting the ligand-receptor interaction with the use of TGFβ ligand inhibitors or with the use of TGFβ receptor inhibitors [122, 123]. The use of these compounds is currently being evaluated in a number of other solid tumors. In PDA, LY2157299, a small molecule inhibitor of the TGF-β receptor I kinase, was evaluated in a phase II double-blind clinical trial in combination with gemcitabine in patients with unresectable PDA. This trial showed an improvement in overall survival and progression free survival with the doublet, with an acceptable toxicity profile [124].
Despite the importance of targeting the loss of SMAD4, there have been no synthetic lethal or other targeted therapies that have been used experimentally or clinically to specifically target this molecule. However, due to the pattern of expression of SMAD4, especially in metastatic lesions, it has been proposed to serve as a prognostic marker for poor prognosis [125]. There have been some studies that suggest that in patients with locally advanced PDA that exhibit SMAD4 expression would be suited for chemoradiation, compared to patients with loss of expression of SMAD4 who may not benefit from such intensified local therapy [126, 127].
3.2.5 Targeting the Wnt Signaling Pathway
Alterations in the Wnt signaling pathway are common in many gastrointestinal malignancies. Perhaps the best example of this is mutation of the APC gene and its role in colorectal tumorigenesis [128]. Mutations in the APC gene are relatively uncommon in PDA, especially when compared to other genes within the pathway. These include RNF43, AXIN1/2, and GATA6 [129,130,131]. A number of studies have shown that Wnt signaling is required for the initiation and progression of PDA [131]. Wnt signaling results in expression of β-catenin/TCF4 transcription factor, which in turn results in expression of RNF43. RNF43 encodes an E3 ligase which is responsible for ubiquitination and degradation of Frizzled receptors [132]. Therefore, mutations in RNF43 result in constitutive signaling through the Wnt signaling pathway. The difficulty in targeting genes within the Wnt signaling pathway is reflective of our current limitations in targeting TSGs. However, the use of LGK974, which is an inhibitor of Wnt ligand secretion, has shown promising results [130, 133].
3.2.6 Targeting NOTCH Signaling in PDA Tumorigenesis
The NOTCH signaling pathway is important in a number of malignancies, including PDA [134, 135]. The importance of NOTCH signaling in PDA is further established by GEMMs that demonstrate that, in the setting of oncogenic KRAS, its activation is necessary for the initiation and progression of PanINs [136, 137]. Moreover, NOTCH signaling has been shown to promote “stemness,” epithelial-mesenchymal transition, and chemoresistance [138,139,140]. And aberrations in expression in the NOTCH signaling pathway have been associated with poor clinical outcomes in patients [141, 142].
Though NOTCH mutations are uncommon, studies have shown that other components of the pathway are amplified and result in overexpression [26]. In in vivo and in vitro experiments, there is a strong body of evidence that supports suppression of the NOTCH signaling pathway as therapeutically relevant strategy in PDA [143,144,145,146]. Options of inhibition of NOTCH signaling include inhibitors of gamma-secretase, which is required for transduction of signaling through the pathway. More specifically, interactions with the cell-membrane protein NOTCH by one of its ligands initiate proteolytic cleavage of the protein at both its intra- and extracellular sites. Gamma-secretase is necessary for cleavage of NOTCH in the intracellular space. Once NOTCH has been cleaved, it then translocates to the nucleus and modulates the expression of its target genes [147].
The use of gamma-secretase inhibitors has been explored in clinical trials. A clinical trial to evaluate the safety and efficacy of PF-03084014, a gamma-secretase inhibitor, in PDA has been terminated (NCT02109445). Another trial is currently in place, but not actively recruiting, which will evaluate BMS-906024, another gamma-secretase inhibitor, in solid tumors and may accrue patients with PDA (NCT01292655). Another agent, RO4929097, has been evaluated in patients with previously treated mPDA. Though the study showed that this agent was well tolerated in patients with mPDA, development of this compound has been discontinued by Roche [148].
Another strategy for the targeting of the NOTCH pathway includes the use of monoclonal antibodies. This strategy has shown promising results in xenograft tumors in mice when used in combination with chemotherapy [149]. In clinical trials, however, this therapeutic approach has not been as successful. The use of tarextumab (OMP-59R5), a fully human Notch2/3 monoclonal antibody, has been evaluated in a randomized, placebo-controlled, phase Ib clinical trial in patients with untreated mPDA in combination with gemcitabine and nab-paclitaxel and was shown to be well tolerated, safe, and have some antitumor effects [150]. However, when this combination therapy was studied in a phase 2, nonrandomized, placebo-controlled clinical trial, the results did not reveal any improvement in overall survival in patients with mPDA as a first line therapy [151]. Like many potential targeted therapies, the preclinical data to support targeting of the NOTCH pathway are robust; however, the clinical data thus far have not been as promising. This is highlighted by the importance of this pathway in the tumorigenesis in PDA. Improvements in approaches to target components of the NOTCH signaling pathway may result in promising therapies that can become available in the clinic.
3.2.7 Targeting the Hedgehog Signaling Pathway
In mammals, Hedgehog signaling is important in embryonic development and differentiation gastrointestinal tissue. Beyond the embryonic period, it plays a role in tissue homeostasis and has been implicated in the pathogenesis of a number of diseases [152,153,154]. In PDA, overexpression of Hedgehog is seen early in the development of PanIN-1 s and in preinvasive or invasive epithelium; however, its expression is absent in normal pancreas tissue [155, 156]. Overexpression of hedgehog in abnormal pancreatic tissue depends on expression of oncogenic KRAS, which suggests that Hedgehog is a downstream effector [157]. Yet the question remains whether the role of Hedgehog is dependent on intracellular signaling alone within tumor epithelial cells, or whether it is as a consequence of aberrant ligand signaling in the tumor microenvironment.
The role of Hedgehog signaling has been extensively studied in mouse models that have helped delineate its mechanism [158,159,160]. Based on GEMMs, the role of Hedgehog ligand was determined to be important in PDA tumorigenesis. In a study by Nolan-Steveaux and colleagues, a mouse model was generated in which SMO-deficient pancreatic progenitor cells (which are insensitive to Hedgehog signaling) were shown to develop PDA at a similar rate as wild-type SMO controls [161]. Moreover, both the SMO-deficient and SMO-wild type mice developed equivalent expression of the Hedgehog ligand and inhibition of GLI1 in both of the groups resulted in increased apoptosis and decreased cell growth [161]. This model suggested that stromal Hedgehog ligand-dependent signaling and noncanonical Gli signaling in tumor epithelial cells are important in KRAS-dependent PDA tumorigenesis [161].
This finding has been further expanded to focus on the Hedgehog ligand, which is produced by tumor epithelial cells, which results in SMO-dependent activation and signaling of adjacent stromal cells (i.e., cancer-associated fibroblasts, CAF) along a canonical signaling pathway [162]. This leads to desmoplasia – one of the hallmarks of PDA. CAFs and cancer-associated stem cells have been implicated in their role in PDA. Co-culture of tumor epithelial cells and CAFs that have been isolated from PDA results in increased proliferation, colony formation, invasion, and resistance to gemcitabine both in vitro and in vivo [162,163,164,165]. Downstream effectors of the Hedgehog signaling pathway, such as SMO or GLI1, are two potential avenues to provide inhibition of this pathway. The SMO-inhibitor, LDE225, has been evaluated in a phase Ib in patients with locally advanced or mPDA in combination with gemcitabine [166]. GDC-0449, also an SMO inhibitor, has shown success in preclinical models [167]. However, when this compound was evaluated in combination with gemcitabine in patients with mPDA, there was no improvement in outcomes when compared to gemcitabine treatment alone [168]. Though there is variability in regards to the success of targeting this pathway, there continues to be much interest in targeting the Hedgehog signaling pathway in PDA. Lastly, targeting of cancer-associated stem cells has also been attempted with the use of monoclonal antibody and is currently being investigated with the use of a “cancer stemness” inhibitor, BBI608 (NCT02231723) [169]. Therapeutic strategies, such as this one, provide a unique way to target vulnerabilities in PDA that go beyond genetic alterations.
3.3 History of Ex Vivo Modeling and the Importance of Preclinical Models in a Personalized Approach to PDA
Molecular and pathologic studies have established a model for progression of PDA, with oncogenic KRAS having an integral role for the inception of tumorigenesis [50]. As discussed in the prior section, a number of genetic aberrations contribute to the tumorigenesis and progression of PDA [47]. The use of genetically engineered mouse models (GEMMs) has been instrumental in our understanding of the initiation and progression of PDA [47]. Moreover, GEMMs have increased our understanding of the role of the tumor microenvironment in PDA and of ligands that are important in dysregulated pathways [161, 170]. Additionally, preclinical models, such as human cell lines, xenograft tumor models, and patient-derived tumor xenografts, have been used to understand the biology of PDA and to identify new therapeutic targets for patients. An exhaustive discussion of ex vivo models is beyond the scope of this chapter, but two new techniques, discussed below, have the potential to significantly propel targeted therapy in PDA: conditionally reprogrammed cells and organoids.
Conditionally reprogrammed cells are a relatively new technique for tumor modeling that allows for quicker regeneration of patient-derived tumor cells that can be used for drug-sensitivity testing [171, 172]. Most recently, three-dimensional culture of patient derived tissue in the form of organoids has been heralded as the next generation ex vivo culture model for PDA [173]. Mouse- and patient-derived organoids have been derived by a number of laboratories around the world and have been genetically modified using CRISPR technology or have been used to test drug sensitivities [174,175,176]. Organoids can be established from surgical specimens and from biopsy specimens. This model allows for the establishment of a pure tumor epithelial population of cells that recapitulates the genomic make-up of the initial tumor specimen [177, 178].
There are a number of preclinical models that are available for translational studies that have attempted to recapitulate the genetic diversity that PDAs exhibit. There are pros and cons that are associated with each model, and at the present time, patient-derived organoids represent perhaps the most promising preclinical model that is available to researchers. There are still many questions that need to be addressed with organoids, including whether the genetic complexity that is seen in the primary tumor is maintained in the organoid. Still, this model can result in an improvement in our understanding of the tumorigenesis and the role or low-frequency mutations in the progression of PDA. This model has already been exploited with intestinal organoids that have been transformed into colorectal carcinoma utilizing genetic engineering [179]. This preclinical model can be used to understand the role of low-frequency mutations by helping delineate those that are truly necessary for tumorigenesis versus those that are just passenger mutations. Having an understanding of the low-frequency mutations that confer survival to PDA tumor cells can then be exploited for targeted drug-development. Ongoing work (including work from JRB’s laboratory) will validate the significance of this model for the pancreatic cancer research community and for the promise of precision therapy.
4 Beyond Genetic Alterations: Finding Alternative Targets
Considering the genetic diversity that is observed in PDA, another option would be approaching the treatment of this devastating disease by utilizing novel therapeutic approaches. For example, in melanoma, the use of immunotherapy has revolutionized the treatment paradigm and has resulted in impressive patient outcomes [180,181,182]. In patients with PDA, the treatment approach would most likely require a combination therapy, in part, due to the genetic diversity that PDA exhibits allowing for compensation to occur along another pathway with targeted blockade. Therefore, other innovative ways of delivering therapy to patients with PDA may be targeting key cellular processes in order to take advantage of a genetic vulnerability, such as the use of PARPi therapy. In this section, alternative strategies to provide “targeted” therapy in PDA in ways that are novel and go beyond genetic alterations that are obtained from tumor sequencing will be discussed.
4.1 Role of Posttranscriptional Modification
Synthesis of messenger RNAs (mRNAs) is one of the essential functions of the cell. Once mRNAs undergo modifications in the nucleus, they are transported to the cytoplasm where they can be involved in a number of functions. Posttranscriptional gene regulation is a key cellular mechanism in which cells are able to modulate gene expression [183]. Regulatory mRNA elements can be present in any portion of the transcript (i.e., 5′-untranslated region (UTR), 3′-UTR, and in some instances even within the coding regions) [184, 185]. These regulatory elements lend themselves to regulation by RNA binding proteins (RBPs) and noncoding RNAs (i.e., microRNAs). Under nonpathologic conditions, posttranscriptional modification and regulation of gene expression are important in many cellular processes. However, there is also increasing evidence that posttranscriptional modification of mRNA transcripts plays an important role in tumor initiation and progression [186, 187]. In the following section, posttranscriptional modification by RBPs and how they can be used as predictors for aggressiveness, response to therapy, or potential therapeutic targets will be explored.
4.1.1 Role of RNA Binding Proteins in PDA Tumorigenesis
RNA-binding proteins (RBPs) are master regulators of mRNA processing and play a role in many vital cellular functions [188]. In cancer, RBPs play a powerful role in driving tumorigenesis, as they are expressed at high frequencies [189].
Perhaps one of the most well-studied RBP is Human Antigen R (HuR) , also known as embryonic lethal, abnormal vision, and Drosophila-like 1 (ELAVL1) [190]. HuR is primarily expressed in the nucleus; however, upon exposure to stress, such as nutrient deprivation, hypoxia, or DNA damage, HuR translocases to the cytoplasm. HuR coordinates a pro-survival network of gene expression by binding to mRNA targets that support cell-survival functions [191, 192]. In vitro, silencing of HuR has been shown to result in decreased tumor growth, impaired migration and invasion, and anchorage-independent growth [193]. Moreover, a number of studies have also demonstrated downstream pro-survival targets of HuR that are important in tumorigenesis [8, 194,195,196]. Finally, a CRISPR knock-out model of HuR in PDA has demonstrated a unique xenograft lethal phenotype in PDA tumor cells [197].
HuR has been shown to be important as both a therapeutic target and a potential biomarker in PDA. Small molecule inhibitors of HuR have been used both in vivo and in vitro [8, 198, 199].
Targeting of HuR by small molecule compounds or siRNA nanoparticle strategies have shown great promise; and there is a hope that these strategies will make it into early phase human trials within the next few years. To date, HuR has also been extensively studied as a biomarker in PDA. In one study, patients with high cytoplasmic HuR have been associated with higher T-stage [200]. And a subsequent study showed that in patients with high cytoplasmic HuR, 5-FU-based therapy as associated with longer disease-free survival when compared to gemcitabine treatment [201]. Additional studies are needed to further elucidate the utility and role of HuR as a biomarker in patients with PDA. HuR may also represent another therapeutic option in PDA, as a drug sensitizer, in order to target a critical drug resistant network in PDA cells, especially in the tumor microenvironment where cells are exposed to low glucose, hypoxic conditions.
4.2 Epigenetic Regulation and its Role in PDA
Epigenetic modifications of DNA, such as histone deacetylation (HDAC) or DNA methylation, have been implicated in tumorigenesis and in metastasis [202]. As an example, in PDA, inactivation of CDKN2A can often times occur due to methylation at its promoter [203]. And this concept, where silencing of TSGs occurs via epigenetic silencing, is not uncommon or unique to PDA. Moreover, epigenetic reprogramming and regulation have also been implicated in metabolic changes in metastatic lesions. This was evaluated in a study by McDonald and colleagues, where matched primary and metastatic samples of PDA were studied in 16 samples from 5 patients [204]. Interestingly, the genetic diversity between the primary tumors and metastatic lesions was unchanged, reaffirming the results from a prior study by Makohon-Moore and colleagues [31]. Yet cells present in metastatic samples had acquired and selectively maintained epigenetic control of a malignant gene expression phenotype in the absence of driver mutations that are metastasis specific.
Targeting of epigenetic regulation has been attempted in PDA with the use of HDAC inhibitors, such as vorinostat, which results in inhibition of tumor growth in vitro and in vivo [205,206,207]. Vorinostat has also been used in clinical trials (NCT00958688), where it was used in combination with 5-FU and radiation in patients with locally advanced PDA; however, the study has been terminated and there are no reported results. A DNA methyltransferase inhibitor, 5-azacytidine, has also been evaluated in patients with advanced PDA in combination with gemcitabine. This study has also been terminated.
Currently enrolling clinical trials that are targeting epigenetic regulation as a therapeutic strategy include a phase II clinical trial in which resected patients with node or margin positive disease who have completed adjuvant therapy go on CC-486 (oral azacytidine) (NCT01845805). Another utilizes decitabine and tetrahydrouridine in patients with mPDA who have failed other therapy (NCT02847000). Therapeutic strategies that aim to target epigenetic modification/reprogramming may be a novel approach for targeted treatment in patients with PDA. This will most likely be further realized as our understanding of the role of epigenetic regulation in metastatic lesions expands, possibly lending itself as a viable therapeutic option in patients with advanced disease.
4.3 Multi-omic Profiling and Its Role in PDA
A new approach in biological analysis is one where data from multiple sources (e.g., omes) are utilized. This includes genomics, proteomics, epigenitome, transcriptome, etc., in order to study biomarkers and therapies [208]. Multi-omic profiling has been explored in PDA by the authors (MJP and JRB) in order to further delineate the relevance of genetic aberrations found in PDA [37, 209]. Multiple platforms exist in order to take advantage of multi-omic profiling, but at the present time, most utilize NGS. With the use of this strategy, phosphoproteomic data have been provided to clinicians successfully and used to guide therapy [38, 210].
Though still in its relative infancy in PDA, the approach to characterizing patients based on multi-omic profiling is powerful and holds a lot of promise. It also integrates a number of important aspects of the patient’s tumor, such as its genetic composition and epigenetic modifications, and offers an opportunity to provide targeted therapy.
4.4 Dysregulation in Axon Guidance Pathways in PDA
Sequencing studies have revealed that in PDA there are aberrations in axon guidance pathways [19, 20]. Other studies have also found epigenetic regulation in SLIT-ROBO, ITGA2, and MET, members of the axon guidance pathway [211]. Under nonpathologic conditions, expression of genes in the axon guidance pathways is important in embryogenesis. However, in cancer, their aberrant expression has been linked to increasing the predisposition of tumor formation and progression [212,213,214]. The exact role of these factors in tumorigenesis is not yet elucidated in PDA and how it may contribute to cell migration, angiogenesis, and cell survival. Considering the degree of dysregulated expression that exists in this gene subset in PDA, additional studies are needed to further elucidate their role. However, these molecules may be potential effective targets in PDA and in other cancers in a personalized manner.
4.5 Targeting the Tumor Microenvironment
One of the hallmarks of PDAs is its pronounced desmoplastic reaction, which makes up the tumor microenvironment (TME) , with a paucity of tumor epithelial cells [6]. As discussed before, in PDA, the tumor microenvironment has a very important role in PDA tumorigenesis and has been shown to interact with the tumor epithelial cells resulting in tumor progression [215,216,217]. Cells that are associated with the tumor stroma include inflammatory, immune, mesenchymal, and endothelial cells [218]. Cancer-associated fibroblasts (CAFs), an example of mesenchymal cells, have also been shown to impose epigenetic and metabolic regulation of tumor epithelial cells [219]. Additionally, activated pancreatic stellate cells, which give rise to CAFs, play an important role in the deposition of extracellular matrix components and the production of cytokines and growth factors [220, 221].
A number of signaling pathways that are dysregulated have been found to be important in the maintenance of the tumor stroma and may be potential therapeutic targets in PDA. TGFβ signaling, as discussed above, is commonly dysregulated in PDA. Ligands produced by the tumor epithelial cells can result in activation of its signaling cascade in stroma cells due to paracrine action, which has been shown to lead to fibroblast proliferation [222, 223]. This interaction is what also makes the use of TGFβ inhibitors a promising therapeutic strategy in PDA. Hedgehog signaling, as discussed in the Targeting NOTCH signaling in PDA tumorigenesis section, is also another attractive targeted therapeutic strategy due to its role in the desmoplastic reaction that’s common in PDA. The tumor stroma has also been evaluated as a prognostic marker. In a study by Bever and colleagues, the density and activity of the stroma was evaluated and high-stromal density was found to be associated with a longer disease-free survival [224]. Other studies have shown that undifferentiated PDA is associated with increased vascularity, raising the potential of VEGF inhibitors as another targeted therapy [225].
Considering the important role of the TME in PDA, especially as mediated by immune cells, a number of compounds have been used to target this specific interaction. In a multicenter, randomized, placebo-controlled, and double-blind clinical trial, ibrutinib, a Burton’s tyrosine kinase (BTK) inhibitor, is being evaluated in combination with nab-paclitaxel and gemcitabine for patients with mPDA as a first line therapy (NCT02436668) [226]. Ibrutinib is also being evaluated in combination with durvalumab, a human IgG1 monoclonal antibody that binds PD-L1 and inhibits its interaction with CD80, in a phase Ib/II multicenter study in patients with relapsed or refractory mPDA (NCT02403271) [227]. Both of these studies have completed enrollment and are ongoing; however, no results have been published as of this writing.
5 Limitations to Precision Therapy in PDA
Molecular profiling has changed the approach to therapy in many cancers, including PDA. NGS and other novel technologies are now becoming routinely incorporated in the care of some patients. However, data on genetic analyses are only useful if patients can take advantage of targeted therapy, whether on- or off-label or in clinical trials, in addition to standard treatment.
It is evident that in PDA, there is a necessity to not only develop better therapies, but to also continue to expand our understanding of the genetic make-up of PDA. Moreover, there is a need for technologies, such as NGS, to provide actionable data in a timely manner so that it becomes routinely incorporated in clinical practice. The results of the IMPaCT trial, which evaluated the feasibility of providing sequencing data to facilitate treatment with targeted therapy, underscore the need to move genomic and molecular information into routine clinical care in order to propel precision medicine as a standard of care treatment strategy in patients with PDA. This will require continued financial support of agencies behind clinical trials that embrace this approach.
With continued improvements in modeling systems, such as patient-derived organoids, there will be an increased understanding of the genetic and nongenetic landscapes of PDAs. The ability to capture the genetic variability that is present in each PDA in these model systems provides a unique research opportunity that could have a significant return in regards to patient treatment. The ultimate goal would be to recapitulate the genetic diversity seen in individual PDAs into the organoid models in an effort to further identify specific drivers of each PDA that will reveal optimal therapeutic opportunities. Model systems, such as the organoids, allow for drug screens, gene editing, and other manipulations that can improve our understanding of the significance of individual gene mutation events. For instance, at the present time, though a number of low-frequency mutations and pathways disrupted in PDA have been identified, the contribution that these mutations have to driving PDA or if they will be susceptible to the current arsenal of available therapies is not yet something that has been elucidated. Understanding the functional implications of these low-frequency pathway disruptions will be integral in guiding drug discovery and efficient clinical trial design. Ultimately, improvements in the preclinical models in PDA will be helpful to study the clinical relevance of targeting dysregulated pathways or genetic mutations and will most likely result in novel insights moving towards precision therapy for PDA.
6 Future Directions
Discoveries that underlie the genetic drivers in PDA have been identified in patient samples and established in GEMMs and ex vivo models. This has been incrementally translated into innovative, successful therapeutic approaches that hope to improve patient outcomes. Though at the present time there is a paucity of FDA approved targeted therapies for patients with PDA, the number of trials that are ongoing that utilize this approach is impressive. NGS has given researchers and clinicians an insight into the genetic diversity of PDA. This technology spans the spectrum – its utilization in research laboratories is increasingly becoming translated to use in the clinic. Though targeting of low-frequency mutations will most likely not yield a significant clinical benefit to many patients with PDA, it will hopefully result in an increased understanding of the tumorigenesis of this disease and, importantly, aid a subset of patients. In fact, the field has accepted that targeting 5–10% of patients at a time might be a logical approach to improving outcomes. This strategy has been descriptively termed as the “pie approach” to treating the disease (i.e., if about 10% of patients are matched with the correct therapeutic strategy, it can lead to significant changes in overall patient outcomes). This can, in turn, be supported by next generation ex vivo models, which will lead to a better understanding of the PDA biology and hopefully will result in higher throughput of drug testing for each patient.
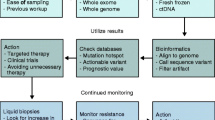
At the present time, in order to make meaningful impact in PDA, researchers, clinicians, and surgeons need to have realistic goals in order to change the current trend in PDA. Ultimately, surgical resection is the only therapeutic option that gives patients a chance for long-term survival. However, since only a minority of patients can benefit from surgery, there need to be improvements in screening, diagnostic, and therapeutic strategies in order to allow more patients an avenue to surgical resection. A schematic for a futuristic clinical trial that employs such a realistic goal for patients in the metastatic setting is presented in Fig. 2. At the time of diagnosis, all patients with metastatic disease should have tumor sampling of both the primary tumor and metastatic lesions. Both tumors should be sequenced and provided to a research laboratory for propagation into organoid cultures. The patient should be offered initial cytotoxic-based therapy followed by targeted therapy based on the sequencing results. Simultaneously, large-scale drug screens should be undertaken for both cytotoxic and targeted therapy while utilizing an organoid-like system. Based on these results from the preclinical model, patients should be advised which therapy they should pursue, whether on- or off-label, or as part of a clinical trial.

Proposed clinical trial schema to optimize the use of precision medicine-based therapies
As a matter of fact, this similar approach can be employed in patients with all stages of PDA. For patients with resectable or locally advanced disease, tumor samples can be obtained, sequenced using whole-genome NGS, and propagated into organoid cultures. The goal should be to have sequencing and organoid drug screening results available to patients and clinicians in a clinically relevant timeline so that this information can be used for better informed clinical decision making. Moreover, utilizing preclinical data for predictive purposes (i.e., high cytoplasmic HuR and drug resistance) will allow clinicians to personalize the treatment approach to each patient. Ultimately, the power of combining NGS, preclinical modeling, such as organoids, and predictive markers will only be fully realized once the use of these technologies become validated.
7 Conclusion
The research community’s understanding of the molecular drivers of PDA has increased over the past decade, with more and more studies delineating the genetic alterations found in this deadly disease. Despite these monumental strides, unlike in other cancers, this progress has been incremental, yet meaningful, in PDA. Our understanding of the implications of genetic aberrations, the role of the tumor microenvironment, metabolic alterations, epigenetic modification, and mechanisms of gene regulation in PDA will continue to increase. However, it is imperative that this is matched with equivalent progress of drug development that results in therapeutic options that can be used in the clinic. Maximizing the results of NGS will require aligning basic research with representative preclinical models. Ex vivo modeling that is done in parallel with NGS at the time of a patient’s diagnosis will help support drug-screening that is based in the fundamental principles of targeted therapy. This strategy will also provide the backbone for well-designed clinical trials in order to produce results that lead to the realization of success in the domain of precision medicine (i.e., better treatment options and improved overall outcomes in patients with PDA).
8 Cross-References
-
EGFR (ErbB) Signaling Pathways in Pancreatic Cancer Pathogenesis
-
Hedgehog Signaling Plays a Dual Role in Pancreatic Carcinogenesis
-
Molecular Pathology of Precursor Lesions of Pancreatic Cancer
-
Notch Signaling in Pancreatic Morphogenesis and Pancreatic Cancer Pathogenesis
-
Smad4/TGF-β Signaling Pathways in Pancreatic Cancer Pathogenesis
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30.
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21.
Winter JM, Cameron JL, Campbell KA, Arnold MA, Chang DC, Coleman J, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: a single-institution experience. J Gastrointest Surg. 2006;10(9):1199–210. discussion 210-1
Von Hoff DD, Goldstein D, Renschler MF. Albumin-bound paclitaxel plus gemcitabine in pancreatic cancer. N Engl J Med. 2014;370(5):479–80.
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–25.
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–76.
Chu GC, Kimmelman AC, Hezel AF, DePinho RA. Stromal biology of pancreatic cancer. J Cell Biochem. 2007;101(4):887–907.
Blanco F, Jimbo M, Wulfkuhle J, Gallagher I, Deng J, Enyenihi L, et al. The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene. 2016;35(19):2529–41.
Prokesch RW, Schima W, Chow LC, Jeffrey RB. Multidetector CT of pancreatic adenocarcinoma: diagnostic advances and therapeutic relevance. Eur Radiol. 2003;13(9):2147–54.
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
Dreyer SB, Chang DK, Bailey P, Biankin AV. Pancreatic cancer genomes: implications for clinical management and therapeutic development. Clin Cancer Res. 2017;23(7):1638–46.
Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–24.
Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26(10):1135–45.
Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6(8):2969–72.
Belkadi A, Bolze A, Itan Y, Cobat A, Vincent QB, Antipenko A, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci. 2015;112(17):5473–8.
Pang AW, Macdonald JR, Yuen RK, Hayes VM, Scherer SW. Performance of high-throughput sequencing for the discovery of genetic variation across the complete size spectrum. G3 (Bethesda). 2014;4(1):63–5.
Todd R, Kuo MWLWP. Gene expression profiling using laser capture microdissection. Expert Rev Mol Diagn. 2002;2(5):497–507.
Bailey P, Chang DK, Nones K, Johns AL, Patch A-M, Gingras M-C, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016. https://doi.org/10.1038/nature16965.
Biankin AV, Waddell N, Kassahn KS, Gingras M-C, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405.
Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17(4):500–3.
Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–7.
Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467(7319):1109–13.
Wang L, Tsutsumi S, Kawaguchi T, Nagasaki K, Tatsuno K, Yamamoto S, et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 2012;22(2):208–19.
Jiao Y, Yonescu R, Offerhaus GJA, Klimstra DS, Maitra A, Eshleman JR, et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol. 2014;232(4):428–35.
Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin W-C, Mansour J, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744
Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
Dal Molin M, Zhang M, De Wilde RF, Ottenhof NA, Rezaee N, Wolfgang CL, et al. Very long-term survival following resection for pancreatic cancer is not explained by commonly mutated genes: results of whole-exome sequencing analysis. Clin Cancer Res. 2015;21(8):1944–50.
Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016;6(2):166–75.
Murphy SJ, Hart SN, Halling GC, Johnson SH, Smadbeck JB, Drucker T, et al. Integrated genomic analysis of pancreatic ductal adenocarcinomas reveals genomic rearrangement events as significant drivers of disease. Cancer Res. 2016;76(3):749–61.
Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet. 2017. https://doi.org/10.1038/ng.3764.
Humphris JL, Patch AM, Nones K, Bailey PJ, Johns AL, McKay S, et al. Hypermutation in pancreatic cancer. Gastroenterology. 2017;152(1):68–74.e2.
Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543(7643):65–71.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58.
Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016. https://doi.org/10.1038/nrc.2016.66.
Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, et al. Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin Cancer Res. 2015;21(9):2029–37.
Pishvaian MJ, Brody JR, Matrisian L, Hendifar AE, Engebretson A, Hoos WA, et al. Multi-Omic profiling (MoP) for patients (pts) with pancreatic cancer (PDA): initial results of the Know Your Tumor (KYT) initiative. Proc Am Soc Clin Oncol. 2016. https://doi.org/10.1200/jco.2016.34.4_suppl.282.
Engebretson A, Brody JR, Rahib L, Matrisian L, Hendifar AE, Hoos WA, et al. The Know Your Tumor (KYT) initiative: a national program of multi-omic molecular profiling (MoP) for patients (pts) with pancreatic cancer (PDA). Proc Am Soc Clin Oncol. 2016. https://doi.org/10.1200/jco.2016.34.4_suppl.279.
Mullard A. NCI-MATCH trial pushes cancer umbrella trial paradigm. Nat Rev Drug Discov. 2015;14(8):513–5.
Do K, O’Sullivan Coyne G, Chen AP. An overview of the NCI precision medicine trials – NCI MATCH and MPACT. Chin Clin Oncol. 2015;4(3):31.
Berry DA. The brave new world of clinical cancer research: adaptive biomarker-driven trials integrating clinical practice with clinical research. Mol Oncol. 2015;9(5):951–9.
Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–7.
Maitra A, Adsay NV, Argani P, Iacobuzio-Donahue C, De Marzo A, Cameron JL, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16(9):902–12.
Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–88.
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–74.
McCormick F. KRAS as a Therapeutic Target. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(8):1797–801.
Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–50.
Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302.
Seidler B, Schmidt A, Mayr U, Nakhai H, Schmid RM, Schneider G, et al. A Cre-loxP-based mouse model for conditional somatic gene expression and knockdown in vivo by using avian retroviral vectors. Proc Natl Acad Sci. 2008;105(29):10137–42.
Morris JP, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10(10):683–95.
Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13(11):828–51.
Thompson H. US National Cancer Institute’s new Ras project targets an old foe. Nature medicine. 2013;19(8):949–50.
Van Cutsem E, Van De Velde H, Karasek P, Oettle H, Vervenne W, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol 2004;22(8):1430–1438.
Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, et al. Small molecule inhibition of the KRAS-PDE [delta] interaction impairs oncogenic KRAS signalling. Nature. 2013;497(7451):638.
Khvalevsky EZ, Gabai R, Rachmut IH, Horwitz E, Brunschwig Z, Orbach A, et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci. 2013;110(51):20723–8.
Pecot CV, Wu SY, Bellister S, Filant J, Rupaimoole R, Hisamatsu T, et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol Cancer Ther. 2014;13(12):2876–85.
Golan T, Khvalevsky EZ, Hubert A, Gabai RM, Hen N, Segal A, et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget. 2015;6(27):24560.
Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D, et al. Genetic predictors of MEK dependence in non–small cell lung cancer. Cancer Res. 2008;68(22):9375–83.
Bodoky G, Timcheva C, Spigel DR, La Stella PJ, Ciuleanu TE, Pover G, et al. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig New Drugs. 2012;30(3):1216–23.
Rinehart J, Adjei AA, LoRusso PM, Waterhouse D, Hecht JR, Natale RB, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22(22):4456–62.
Infante JR, Somer BG, Park JO, Li C-P, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50(12):2072–81.
Witkiewicz AK, Borja NA, Franco J, Brody JR, Yeo CJ, Mansour J, et al. Selective impact of CDK4/6 suppression on patient-derived models of pancreatic cancer. Oncotarget. 2015;6(18):15788–801.
Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5(15):6512–25.
Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao Y-M, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69(10):4286–93.
Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, et al. A central role for RAF→ MEK→ ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012;2(8):685–93.
Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res. 2015;21(2):396–404.
Chung V, McDonough S, Philip PA, Cardin D, Wang-Gillam A, Hui L, et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and fluorouracil in patients with metastatic pancreatic cancer after prior therapy: SWOG S1115 study randomized clinical trial. JAMA Oncol. 2017;3(4):516–22.
Diep CH, Munoz RM, Choudhary A, Von Hoff DD, Han H. Synergistic effect between erlotinib and MEK inhibitors in KRAS wild-type human pancreatic cancer cells. Clin Cancer Res. 2011;17(9):2744–56.
Kulke MH, Blaszkowsky LS, Ryan DP, Clark JW, Meyerhardt JA, Zhu AX, et al. Capecitabine plus erlotinib in gemcitabine-refractory advanced pancreatic cancer. J Clin Oncol. 2007;25(30):4787–92.
Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–6.
Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2(10):731–7.
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–46.
Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16ink4a expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011;10(15):2497–503.
Caldas C, Hahn SA, Da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8(1):27–32.
Fukushima N, Sato N, Ueki T, Rosty C, Walter KM, Wilentz RE, et al. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol. 2002;160(5):1573–81.
Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57(15):3126–30.
Cicenas J, Valius M. The CDK inhibitors in cancer research and therapy. J Cancer Res Clin Oncol. 2011;137(10):1409.
Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–38.
Liu F, Korc M. Cdk4/6 inhibition induces epithelial–mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther. 2012;11(10):2138–48.
Heilmann AM, Perera RM, Ecker V, Nicolay BN, Bardeesy N, Benes CH, et al. CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer Res. 2014;74(14):3947–58.
Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5(15):6512–25.
Hruban RH, Iacobuzio-Donahue C, Wilentz RE, Goggins M, Kern SE. Molecular pathology of pancreatic cancer. Cancer J. 2000;7(4):251–8.
Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13(3):217–36.
Song G-Y, Gibson G, Haq W, Huang EC, Srivasta T, Hollstein M, et al. An MVA vaccine overcomes tolerance to human p53 in mice and humans. Cancer Immunol Immunother. 2007;56(8):1193–205.
Lehmann S, Bykov VJ, Ali D, Andrén O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. 2012;30(29):3633–9.
Izetti P, Hautefeuille A, Abujamra AL, de Farias CB, Giacomazzi J, Alemar B, et al. PRIMA-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Investig New Drugs. 2014;32(5):783–94.
Duffy MJ, Synnott NC, McGowan PM, Crown J, O’Connor D, Gallagher WM. p53 as a target for the treatment of cancer. Cancer Treat Rev. 2014;40(10):1153–60.
Yu X, Narayanan S, Vazquez A, Carpizo DR. Small molecule compounds targeting the p53 pathway: are we finally making progress? Apoptosis. 2014;19(7):1055–68.
Hahn SA, Greenhalf B, Ellis I, Sina-Frey M, Rieder H, Korte B, et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst. 2003;95(3):214–21.
van der Heijden MS, Yeo CJ, Hruban RH, Kern SE. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63(10):2585–8.
Van der Heijden MS, Brody JR, Gallmeier E, Cunningham SC, Dezentje DA, Shen D, et al. Functional defects in the fanconi anemia pathway in pancreatic cancer cells. Am J Pathol. 2004;165(2):651–7.
Gallmeier E, Calhoun ES, Rago C, Brody JR, Cunningham SC, Hucl T, et al. Targeted disruption of FANCC and FANCG in human cancer provides a preclinical model for specific therapeutic options. Gastroenterology. 2006;130(7):2145–54.
Murphy SJ, Hart SN, Lima JF, Kipp BR, Klebig M, Winters JL, et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology. 2013;145(5):1098–109. e1.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–92.
Golan T, Kanji Z, Epelbaum R, Devaud N, Dagan E, Holter S, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer. 2014;111(6):1132–8.
Ashworth A. A synthetic lethal therapeutic approach: poly (ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26(22):3785–90.
Benafif S, Hall M. An update on PARP inhibitors for the treatment of cancer. Onco Targets Ther. 2015;8:519.
Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689–98.
Villarroel MC, Rajeshkumar N, Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10(1):3–8.
Pishvaian MJ, Wang H, Zhuang T, He AR, Hwang JJ, Hankin A, et al. A phase I/II study of ABT-888 in combination with 5-fluorouracil (5-FU) and oxaliplatin (Ox) in patients with metastatic pancreatic cancer (MPC). Proc Am Soc Clin Oncol. 2013.
Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33(3):244–50.
Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. 2015;149:124–38.
Karnitz LM, Zou L. Molecular pathways: targeting ATR in cancer therapy. Clin Cancer Res. 2015;21(21):4780–5.
Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, et al. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol Ther. 2012;13(11):1072–81.
Bang Y-J, Im S-A, Lee K-W, Cho JY, Song E-K, Lee KH, et al. Randomized, double-blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J Clin Oncol. 2015;33(33):3858–65.
Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601.
Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–92.
Wu C. Chromatin remodeling and the control of gene expression. J Biol Chem. 1997;272(45):28171–4.
Narlikar GJ, Fan H-Y, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108(4):475–87.
Khursheed M, Kolla J, Kotapalli V, Gupta N, Gowrishankar S, Uppin S, et al. ARID1B, a member of the human SWI/SNF chromatin remodeling complex, exhibits tumour-suppressor activities in pancreatic cancer cell lines. Br J Cancer. 2013;108(10):2056–62.
Numata M, Morinaga S, Watanabe T, Tamagawa H, Yamamoto N, Shiozawa M, et al. The clinical significance of SWI/SNF complex in pancreatic cancer. Int J Oncol. 2013;42(2):403–10.
Knudsen ES, O’Reilly EM, Brody JR, Witkiewicz AK. Genetic diversity of pancreatic ductal adenocarcinoma and opportunities for precision medicine. Gastroenterology. 2016;150(1):48–63.
Shen J, Peng Y, Wei L, Zhang W, Yang L, Lan L, et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 2015;5(7):752–67.
Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun. 2016;7:13837.
Truty MJ, Urrutia R. Basics of TGF-ß and pancreatic cancer. Pancreatology. 2007;7(5–6):423–35.
Ijichi H, Ikenoue T, Kato N, Mitsuno Y, Togo G, Kato J, et al. Systematic analysis of the TGF-β-Smad signaling pathway in gastrointestinal cancer cells. Biochem Biophys Res Commun. 2001;289(2):350–7.
Hahn SA, Schutte M, Hoque A. Moskaluk CA. DCP4, a candidate tumor supressor gene at human chromosome 18q21. 1. Science. 1996;271(5247):350.
Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ, et al. Transforming growth factor-β-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem. 2003;278(44):43001–7.
Embuscado EE, Laheru D, Ricci F, Yun KJ, de Boom Witzel S, Seigel A, et al. Immortalizing the complexity of cancer metastasis: genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol Ther. 2005;4(5):548–54.
Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27(11):1806–13.
Winter JM, Tang LH, Klimstra DS, Liu W, Linkov I, Brennan MF, et al. Failure patterns in resected pancreas adenocarcinoma: lack of predicted benefit to SMAD4 expression. Ann Surg. 2013;258(2):331.
Colak S, ten Dijke P. Targeting TGF-β Signaling in cancer. Trends Cancer. 2017;3:56–71.
Neuzillet C, de Gramont A, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, et al. Perspectives of TGF-beta inhibition in pancreatic and hepatocellular carcinomas. Oncotarget. 2014;5(1):78–94.
Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, et al. A phase II, double-blind study of galunisertib+ gemcitabine (GG) vs gemcitabine+ placebo (GP) in patients (pts) with unresectable pancreatic cancer (PC). Proc Am Soc Clin Oncol. 2016.
Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15(14):4674–9.
Tatarian T, Winter JM. Genetics of pancreatic cancer and its implications on therapy. Surg Clin N Am. 2016;96(6):1207–21.
Regine WF, Winter KA, Abrams R, Safran H, Hoffman JP, Konski A, et al. Fluorouracil-based chemoradiation with either gemcitabine or fluorouracil chemotherapy after resection of pancreatic adenocarcinoma: 5-year analysis of the US intergroup/RTOG 9704 phase III trial. Ann Surg Oncol. 2011;18(5):1319–26.
Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7.
White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142(2):219–32.
Jiang X, Hao H-X, Growney JD, Woolfenden S, Bottiglio C, Ng N, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci. 2013;110(31):12649–54.
Zhang Y, Morris JP, Yan W, Schofield HK, Gurney A, Simeone DM, et al. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res. 2013;73(15):4909–22.
Jiang X, Charlat O, Zamponi R, Yang Y, Cong F. Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol Cell. 2015;58(3):522–33.
Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Targeting Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc Natl Acad Sci. 2013;110(50):20224–9.
Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338–51.
Gao J, Long B, Wang Z. Role of notch signaling pathway in pancreatic cancer. Am J Cancer Res. 2017;7(2):173–86.
De La OJ, Murtaugh LC. Notch and Kras in pancreatic cancer: at the crossroads of mutation, differentiation and signaling. Cell Cycle. 2009;8(12):1860–4.
Thomas MM, Zhang Y, Mathew E, Kane KT, Maillard I, Pasca di Magliano M. Epithelial notch signaling is a limiting step for pancreatic carcinogenesis. BMC Cancer. 2014;14:862.
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69(6):2400–7.
Kabashima-Niibe A, Higuchi H, Takaishi H, Masugi Y, Matsuzaki Y, Mabuchi Y, et al. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci. 2013;104(2):157–64.
Du X, Zhao YP, Zhang TP, Zhou L, Chen G, Wang TX, et al. Alteration of the intrinsic apoptosis pathway is involved in notch-induced chemoresistance to gemcitabine in pancreatic cancer. Arch Med Res. 2014;45(1):15–20.
Doucas H, Mann CD, Sutton CD, Garcea G, Neal CP, Berry DP, et al. Expression of nuclear Notch3 in pancreatic adenocarcinomas is associated with adverse clinical features, and correlates with the expression of STAT3 and phosphorylated Akt. J Surg Oncol. 2008;97(1):63–8.
Mann CD, Bastianpillai C, Neal CP, Masood MM, Jones DJ, Teichert F, et al. Notch3 and HEY-1 as prognostic biomarkers in pancreatic adenocarcinoma. PLoS One. 2012;7(12):e51119.
Mizuma M, Rasheed ZA, Yabuuchi S, Omura N, Campbell NR, de Wilde RF, et al. The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol Cancer Ther. 2012;11(9):1999–2009.
Palagani V, Bozko P, El Khatib M, Belahmer H, Giese N, Sipos B, et al. Combined inhibition of notch and JAK/STAT is superior to monotherapies and impairs pancreatic cancer progression. Carcinogenesis. 2014;35(4):859–66.
Yabuuchi S, Pai SG, Campbell NR, de Wilde RF, De Oliveira E, Korangath P, et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013;335(1):41–51.
Cook N, Frese KK, Bapiro TE, Jacobetz MA, Gopinathan A, Miller JL, et al. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med. 2012;209(3):437–44.
Shih Ie M, Wang TL. Notch signaling, gamma-secretase inhibitors, and cancer therapy. Cancer Res. 2007;67(5):1879–82.
De Jesus-Acosta A, Laheru D, Maitra A, Arcaroli J, Rudek MA, Dasari A, et al. A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Investig New Drugs. 2014;32(4):739–45.
Yen W-C, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res. 2015;21(9):2084–95.
O’Reilly EM, Smith LS, Bendell JC, Strickler JH, Zalupski M, Gluck W, et al. Final results of phase Ib of anticancer stem cell antibody tarextumab (OMP-59R5, TRXT, anti-Notch 2/3) in combination with nab-paclitaxel and gemcitabine (Nab-P+ Gem) in patients (pts) with untreated metastatic pancreatic cancer (mPC). Proc Am Soc Clin Oncol. 2015. https://doi.org/10.1200/jco.2015.33.3_suppl.278.
O’REILLY EM, Sahai V, Bendell JC, BULLOCK A, LOCONTE N, Hatoum H, et al. Results of a randomized phase 2 trial of an anti-notch 2/3, Tarextumab (OMP-59R5, TRXT, anti-notch 2/3), in combination with Nab-paclitaxel and Gemcitabine (Nab-P+ Gem) in patients (pts) with untreated metastatic pancreatic cancer (mPC). Proc Am Soc Clin Oncol. 2017. https://doi.org/10.1200/JCO.2017.35.4_suppl.279.
Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22(18):2454–72.
Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15(6):801–12.
Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010;29(4):469–81.
Nakashima H, Nakamura M, Yamaguchi H, Yamanaka N, Akiyoshi T, Koga K, et al. Nuclear factor-kappaB contributes to hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. 2006;66(14):7041–9.
Kasperczyk H, Baumann B, Debatin KM, Fulda S. Characterization of sonic hedgehog as a novel NF-kappaB target gene that promotes NF-kappaB-mediated apoptosis resistance and tumor growth in vivo. FASEB J. 2009;23(1):21–33.
Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(1):105–20.
Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–6.
Apelqvist Å, Ahlgren U, Edlund H. Sonic hedgehog directs specialised mesoderm differentiation in the intestine and pancreas. Curr Biol. 1997;7(10):801–4.
di Magliano MP, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006;20(22):3161–73.
Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernández-Zapico ME, et al. GLI1 is regulated through smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23(1):24–36.
Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene. 2009;28(40):3513–25.
Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14(19):5995–6004.
Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67(5):2187–96.
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–61.
Macarulla T, Tabernero J, Palmer DH, Sharma S, KH Y, Sellami DB, et al. A phase Ib dose escalation, safety, and tolerability study of sonidegib in combination with gemcitabine in patients with locally advanced or metastatic pancreatic adenocarcinoma. Proc Am Soc Clin Oncol. 2016. https://doi.org/10.1200/jco.2016.34.4_suppl.371.
Singh BN, Fu J, Srivastava RK, Shankar S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLoS One. 2011;6(11):e27306.
Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N, et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res. 2014. https://doi.org/10.1158/1078-0432.CCR-14-1269.
Hidalgo M, Cooray P, Jameson MB, Carrato A, Parnis F, Jeffery M, et al. A phase Ib study of the anti-cancer stem cell agent demcizumab (DEM) & gemcitabine (GEM)+/−paclitaxel protein bound particles (nab-paclitaxel) in pts with pancreatic cancer. Proc Am Soc Clin Oncol. 2015.
Feldmann G, Habbe N, Dhara S, Bisht S, Alvarez H, Fendrich V, et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut. 2008;57(10):1420–30.
Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A, et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 2017;12(2):439–51.
Beglyarova N, Banina E, Zhou Y, Mukhamadeeva R, Andrianov G, Bobrov E, et al. Screening of conditionally reprogrammed patient-derived carcinoma cells identifies ERCC3–MYC interactions as a target in pancreatic cancer. Clin Cancer Res. 2016;22(24):6153–63.
Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van Den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141(5):1762–72.
van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161(4):933–45.
Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015;21(3):256–62.
Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell-and patient-derived tumor organoids. Nat Med. 2015;21(11):1364–71.
Boj SF, Hwang C-I, Baker LA, Chio IIC, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1):324–38.
Baker LA, Tiriac H, Clevers H, Tuveson DA. Modeling pancreatic cancer with organoids. Trends Cancer. 2016;2(4):176–90.
Drost J, Van Jaarsveld RH, Ponsioen B, Zimberlin C, Van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521(7550):43–7.
Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616–22.
Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti–PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44.
Riley JL. Combination checkpoint blockade–taking melanoma immunotherapy to the next level. The New England journal of medicine. 2013;369(2):187–9.
Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet. 2007;8(7):533–43.
Day D, Tuite MF. Post-transcriptional gene regulatory mechanisms in eukaryotes: an overview. J Endocrinol. 1998;157(3):361–71.
Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582(14):1977–86.
Audic Y, Hartley RS. Post-transcriptional regulation in cancer. Biol Cell. 2004;96(7):479–98.
Jewer M, Findlay SD, Postovit L-M. Post-transcriptional regulation in cancer progression. J Cell Comm Signal. 2012;6(4):233–48.
Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3(3):195–205.
Kechavarzi B, Janga SC. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol. 2014;15(1):R14.
Srikantan S, Gorospe M. HuR function in disease. Front Biosci. 2012;17:189.
Brennan CM, Gallouzi I-E, Steitz JA. Protein ligands to HuR modulate its interaction with target mRNAs in vivo. J Cell Biol. 2000;151(1):1–14.
Kim HH, Abdelmohsen K, Gorospe M. Regulation of HuR by DNA damage response kinases. J Nucleic Acids. 2010;2010:981487.
Jimbo M, Blanco FF, Huang Y-H, Telonis AG, Screnci BA, Cosma GL, et al. Targeting the mRNA-binding protein HuR impairs malignant characteristics of pancreatic ductal adenocarcinoma cells. Oncotarget. 2015;6(29):27312.
Lal S, Burkhart RA, Beeharry N, Bhattacharjee V, Londin ER, Cozzitorto JA, et al. HuR posttranscriptionally regulates WEE1: implications for the DNA damage response in pancreatic cancer cells. Cancer Res. 2014;74(4):1128–40.
Pineda DM, Rittenhouse DW, Valley CC, Cozzitorto JA, Burkhart RA, Leiby B, et al. HuR’s post-transcriptional regulation of death receptor 5 in pancreatic cancer cells. Cancer Biol Ther. 2012;13(10):946–55.
Burkhart RA, Pineda DM, Chand SN, Romeo C, Londin ER, Karoly ED, et al. HuR is a post-transcriptional regulator of core metabolic enzymes in pancreatic cancer. RNA Biol. 2013;10(8):1312–23.
Lal S, Cheung EC, Zarei M, Preet R, Chand SN, Mambelli-Lisboa NC, et al. CRISPR knockout of the HuR gene causes a xenograft lethal phenotype. Mol Cancer Res. 2017;15:696–707.
Blanco FF, Preet R, Aguado A, Vishwakarma V, Stevens LE, Vyas A, et al. Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis. Oncotarget. 2016;7:74043–58.
Kaur K, Wu X, Fields JK, Johnson DK, Lan L, Pratt M, et al. The fungal natural product azaphilone-9 binds to HuR and inhibits HuR-RNA interaction in vitro. PLoS One. 2017;12(4):e0175471.
Richards NG, Rittenhouse DW, Freydin B, Cozzitorto JA, Grenda D, Rui H, et al. HuR status is a powerful marker for prognosis and response to gemcitabine-based chemotherapy for resected pancreatic ductal adenocarcinoma patients. Ann Surg. 2010;252(3):499–506.
Tatarian T, JiangW, Leiby BE, Grigoli A, Jimbo M, Dabbish N, et al. Cytoplasmic HuR Status Predicts Disease-free Survival in Resected Pancreatic Cancer: A Post-hoc Analysis From the International Phase III ESPAC-3 Clinical Trial. Ann Surg. 2017. https://doi.org/10.1097/SLA.0000000000002088
Suvà ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339(6127):1567–70.
Deer EL, González-Hernández J, Coursen JD, Shea JE, Ngatia J, Scaife CL, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39(4):425.
McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nature genetics. 2017;49(3):367–76.
Millward M, Price T, Townsend A, Sweeney C, Spencer A, Sukumaran S, et al. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Investig New Drugs. 2012;30(6):2303–17.
Kumagai T, Wakimoto N, Yin D, Gery S, Kawamata N, Takai N, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int J Cancer. 2007;121(3):656–65.
Lee HS, Park SB, Kim SA, Kwon SK, Cha H, Lee DY, et al. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci Rep. 2017;7:41615.
Kristensen VN, Lingjærde OC, Russnes HG, Vollan HKM, Frigessi A, Børresen-Dale A-L. Principles and methods of integrative genomic analyses in cancer. Nat Rev Cancer. 2014;14(5):299–313.
Pishvaian MJ, Matrisian L, Hendifar AE, Engebretson A, Rahib L, Hoos WA, et al. Preliminary observations of blood-based (BB) molecular testing in a subset of patients with pancreatic cancer (PDA) participating in the Know Your Tumor (KYT) initiative. Proc Am Soc Clin Oncol. 2016;34:268.
Bender RJ, Halverson D, Mason K, Luo L, Brody JR, Rahib L, et al. Molecular biomarkers as predictors of patient survival in pancreatic adenocarcinoma (PDA): An analysis of the Know Your Tumor initiative (KYT). Journal of Clinical Oncology. 2017;35(4_suppl):278.
Nones K, Waddell N, Song S, Patch AM, Miller D, Johns A, et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer. 2014;135(5):1110–8.
Narayan G, Goparaju C, Arias-Pulido H, Kaufmann AM, Schneider A, Dürst M, et al. Promoter hypermethylation-mediated inactivation of multiple Slit-Robo pathway genes in cervical cancer progression. Mol Cancer. 2006;5(1):16.
Xian J, Clark KJ, Fordham R, Pannell R, Rabbitts TH, Rabbitts PH. Inadequate lung development and bronchial hyperplasia in mice with a targeted deletion in the Dutt1/Robo1 gene. Proc Natl Acad Sci. 2001;98(26):15062–6.
Dumartin L, Quemener C, Laklai H, Herbert J, Bicknell R, Bousquet C, et al. Netrin-1 mediates early events in pancreatic adenocarcinoma progression, acting on tumor and endothelial cells. Gastroenterology. 2010;138(4):1595–606. e8.
Ricci F, Kern SE, Hruban RH, Iacobuzio-Donahue CA. Stromal responses to carcinomas of the pancreas: juxtatumoral gene expression conforms to the infiltrating pattern and not the biologic subtype. Cancer Biol Ther. 2005;4(3):302–7.
Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68(3):918–26.
Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6(4):1186–97.
Kong X, Li L, Li Z, Xie K. Targeted destruction of the orchestration of the pancreatic stroma and tumor cells in pancreatic cancer cases: molecular basis for therapeutic implications. Cytokine Growth Factor Rev. 2012;23(6):343–56.
Sherman MH, Ruth TY, Tseng TW, Sousa CM, Liu S, Truitt ML, et al. Stromal cues regulate the pancreatic cancer epigenome and metabolome. Proc Natl Acad Sci. 2017;114:1129–34.
Apte M, Pirola RC, Wilson JS. Pancreatic stellate cell: physiologic role, role in fibrosis and cancer. Curr Opin Gastroenterol. 2015;31(5):416–23.
Sherman MH, Ruth TY, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159(1):80–93.
Vogelmann R, Ruf D, Wagner M, Adler G, Menke A. Effects of fibrogenic mediators on the development of pancreatic fibrosis in a TGF-β1 transgenic mouse model. Am J Physiol –Gastrointest Liver Physiol. 2001;280(1):G164–G72.
Löhr M, Schmidt C, Ringel J, Kluth M, Müller P, Nizze H, et al. Transforming growth factor-β1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001;61(2):550–5.
Bever KM, Sugar EA, Bigelow E, Sharma R, Laheru D, Wolfgang CL, et al. The prognostic value of stroma in pancreatic cancer in patients receiving adjuvant therapy. HPB. 2015;17(4):292–8.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–47.
Tempero MA, Coussens LM, Fong L, Manges R, Singh P, Li Y, et al. A randomized, multicenter, double-blind, placebo-controlled study of the Bruton tyrosine kinase inhibitor, ibrutinib, versus placebo in combination with nab-paclitaxel and gemcitabine in the first-line treatment of patients with metastatic pancreatic adenocarcinoma (RESOLVE). Journal of Clinical Oncology. 2016;34(4_suppl):TPS483-TPS.
Borazanci EH, Hong DS, Gutierrez M, Rasco DW, Reid TR, Veeder MH, et al. Ibrutinib + durvalumab (MEDI4736) in patients (pts) with relapsed or refractory (R/R) pancreatic adenocarcinoma (PAC): A phase Ib/II multicenter study. Journal of Clinical Oncology. 2016;34(4_suppl):TPS484-TPS.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media LLC
About this entry

Cite this entry
Dukleska, K., Yeo, C.J., Pishvaian, M.J., Brody, J.R. (2018). Precision Medicine Based on Next Generation Sequencing and Master Controllers. In: Neoptolemos, J., Urrutia, R., Abbruzzese, J., Büchler, M. (eds) Pancreatic Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-6631-8_71-1
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6631-8_71-1
Received:
Accepted:
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-6631-8
Online ISBN: 978-1-4939-6631-8
eBook Packages: Springer Reference Biomedicine and Life SciencesReference Module Biomedical and Life Sciences