
Abstract
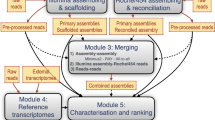
Gene expression analyses of non-model organisms must start with the construction of a high accurate de novo transcriptome as a reference. The best way to determine the suitability of any de novo transcriptome assembling is its comparison with other well-known “reference” transcriptomes. In this study, we took six complete plant transcriptomes (Arabidopsis thaliana, Vitis vinifera, Zea mays, Populus trichocarpa, Triticum aestivum and Oryza sativa) and compared all of them using a series of metrics system for a principal component analysis, resulting that A. thaliana and P. trichocarpa were the best references. This has been automated using AutoFlow. A primary assembly of short reads from Illumina Platform (50 nt, single reads) and long reads from Roche-454 technology from Castanea sativa was performed individually using k-mers from 25 to 35 and different assemblers (Oases v2, SOAPdenovoTrans, RAY, MIRA4 and MINIMUS). The resulting contigs were then reconciled with the aim of obtaining the best transcriptome. Oases and SOAP were used for the assembling of short reads, MIRA and MINIMUS for the assembling of long reads or the reconciliations, and RAY, that can compute de novo transcript assembling from heterogeneous (long and short reads) next-generation sequencing data, was included to avoid the reconciliation step. A total of 90 different assemblies were generated in a single run of the pipeline. A hierarchical clustering on the PCA components (HCPC) was implemented to automatically identify the best assembling strategies based on the shortest distance in HCPC to the two plant reference transcriptomes is selected. In this approach, reconciliation of Roche/454 long reads with Illumina contigs produce more complete and accurate gene reconstructions than other combinations. Surprisingly, reconstructions based only on Illumina and the ones creates with RAY seem to be less accurate. For this specific study, the most complete and accurate transcriptome corresponds to the Illumina contigs obtained with SOAPdenovoTrans and reassembled with 454 long reads using MIRA4. This is only a one example of a transcriptome building. Many other assembling can be performed just changing parameters, k-mers, sequencing technology, assemblers, reference organisms, etc. The pipeline in AutoFlow is easily customizable for those purposes.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others


References
Quintana, J.: Molecular tools to improve chestnut management: El Bierzo as case of study, Ph.D. Thesis (2015)
Seoane, P., Ocaña, S., Carmona, R., Bautista, R., Madrid, E., Torres, A.M., Claros, M.G.: AutoFlow, a versatile workflow engine illustrated by assembling an optimised de novo transcriptome for a non-model species, such as Faba Bean (Vicia faba). Curr. Bioinform. 11(4), 440–450 (2016)
Ocana, S., Seoane, P., Bautista, R., Palomino, C., Claros, G.M., Torres, A.M., Madrid, E.: Large-scale transcriptome analysis in Faba Bean (Vicia Faba L.) under ascochyta fabae infection. PLoS ONE 10(8), 1–17 (2015)
Carmona, R., Zafra, A., Seoane, P., Castro, A., Guerrero-Fernández, D., Castillo-Castillo, T., Medina-García, A., Cánovas, F.M., Aldana-Montes, J.F., Navas-Delgado, I., Alché, J.D., Claros, M.G.: ReprOlive: a database with linked data for the olive tree (Olea europaea L.) reproductive transcriptome. Front. Plant Sci. 6, 625 (2015)
Schulz, M.H., Zerbino, D.R., Vingron, M., Birney, E.: Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092 (2012)
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., He, G., Chen, Y., Pan, Q., Liu, Y.Y.Y.Y., Tang, J., Wu, G., Zhang, H., Shi, Y., Liu, Y.Y.Y.Y., Yu, C., Wang, B., Lu, Y., Han, C., Cheung, D.W., Yiu, S.-M., Peng, S., Xiaoqian, Z., Liu, G., Liao, X., Li, Y., Yang, H., Wang, J.J., Lam, T.-W., Wang, J.J.: SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1(1), 18 (2012)
Boisvert, S., Laviolette, F., Corbeil, J.: Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J. Comput. Biol. 17(11), 1519–1533 (2010)
Pevzner, P.A., Tang, H., Waterman, M.S.: An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. U.S.A. 98, 9748–9753 (2001)
Chevreux, B., Pfisterer, T., Drescher, B., Driesel, A.J., Müller, W.E.G., Wetter, T., Suhai, S.: Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14(6), 1147–1159 (2004)
Huang, X., Madan, A.: CAP3: a DNA sequence assembly program. Genome Res. 9(9), 868–877 (1999)
Sommer, D.D., Delcher, A.L., Salzberg, S.L., Pop, M.: Minimus: a fast, lightweight genome assembler. BMC Bioinform. 8, 64 (2007)
Boisvert, S., Raymond, F., Godzaridis, E., Laviolette, F., Corbeil, J.: Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 13(12), R122 (2012)
Simão, F.A., Waterhouse, R.M., Ioannidis, P., Kriventseva, E.V., Zdobnov, E.M.: BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19), 3210–3212 (2015)
Lê, S., Josse, J., Husson, F.: FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 25(1), 1–18 (2008)
Husson, F., Josse, J., Pagès, J.: Principal component methods - hierarchical clustering - partitional clustering: why would we need to choose for visualizing data? Technical report, pp. 1–17 (2010)
Acknowledgments
This work has been supported by co-funding from the ERDF (European Regional Development Fund) 2014-2020 “Programa Operativo de Crecimiento Inteligente” to the grant RTA2013-00068-C03 of the Spanish INIA and MINECO. The authors also thankfully acknowledge the computer resources and the technical support provided by the Plataforma Andaluza de Bioinformática of the University of Málaga.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper
Cite this paper
Espigares, M., Seoane, P., Bautista, R., Quintana, J., Gómez, L., Claros, M.G. (2017). Obtaining the Most Accurate de novo Transcriptomes for Non-model Organisms: The Case of Castanea sativa . In: Rojas, I., Ortuño, F. (eds) Bioinformatics and Biomedical Engineering. IWBBIO 2017. Lecture Notes in Computer Science(), vol 10209. Springer, Cham. https://doi.org/10.1007/978-3-319-56154-7_44
Download citation
DOI: https://doi.org/10.1007/978-3-319-56154-7_44
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56153-0
Online ISBN: 978-3-319-56154-7
eBook Packages: Computer ScienceComputer Science (R0)