
Abstract
Single-strand-specific nucleases are used to cleave mismatches in otherwise double-stranded DNA. Assays typically involve PCR amplification followed by a denaturing and annealing step to generate heteroduplexed DNA molecules from PCR products containing nucleotide polymorphisms. This is followed by digestion with nucleases that cleave at the site of the mismatch. The molecular weights of the cleavage products indicate the approximate position of nucleotide polymorphisms in PCR amplicons. Cleaved DNA products are observed by one of several readout platforms such as native gel electrophoresis, denaturing gel electrophoresis, capillary electrophoresis, or denaturing high-performance liquid chromatography (DHPLC). This approach is highly suitable for accurate discovery of natural and induced single-nucleotide polymorphisms (SNPs) and also small insertions and deletions (indels). The use of self-extracted single-strand-specific nucleases, typically prepared from crude extracts of celery (Apium graveolens), is common for reverse-genetics (e.g. Targeting Induced Local Lesions IN Genomes (TILLING)) and studies of natural nucleotide polymorphisms (e.g. Ecotilling). While protocols have been published describing the preparation of single-strand-specific nuclease from plant tissues, many rely on toxic chemicals, dialysis and large-volume preparatory centrifuges and also require steps to be performed at 4 °C. We provide here a streamlined room temperature extraction protocol that uses standard microcentrifuges and eliminates toxic chemicals and traditional dialysis.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Natural and induced mutations are powerful tools for functional genomics and plant breeding. Methods for rapid discovery and genotyping of mutations are important for a variety of applications including marker development, marker-assisted selection, population genetics, and reverse-genetics. Single-nucleotide polymorphisms (SNPs) represent the most common type of natural nucleotide variation in plants. While the spectra of mutations induced by treatment with either chemical or physical mutagens vary depending on the mutagen and dosage of mutagen used, many treatments with chemical mutagens produce predominantly SNP or small indel mutations. For example, data collected from Targeting Induced Local Lesions IN Genomes (TILLING) projects where species were treated with either ethyl methanesulfonate (EMS), sodium azide or a combination of sodium azide, and N-methyl-N-nitrosourea (MNU) show that these mutagens create predominantly single-base-point mutations (Jankowicz-Cieslak et al. 2011). Furthermore, treatment with EMS produces greater than 90 % GC to AT transition mutations for many crop species (Greene et al. 2003; Slade and Knauf 2005; Jankowicz-Cieslak et al. 2011; Jankowicz-Cieslak et al. 2012). There is a broader diversity of effects reported of plants treated with physical mutagens. For example, fast neutron irradiation of plants has been reported to generate genomic deletions ranging in size from less than 56 base pairs to nearly three million base pairs (Li et al. 2001; Rogers et al. 2009; Bolon et al. 2011; Belfield et al. 2012). The most common mutagen used in the development of the ~3200 officially released mutant crop varieties curated in the IAEA’s Mutant Variety Database (http://mvd.iaea.org/) is gamma irradiation. Reports on the type of DNA lesions induced by gamma irradiation are quite variable. In rice, data suggest that >80 % of mutations induced via gamma irradiation are small deletions up to 16 base pairs, with the remainder being large deletions (Sato et al. 2006; Bruce et al. 2009). Yet in maize only deletions greater than 200 kilobases were reported (Yuan et al. 2014). Some caution is needed when trying to draw broad conclusions from these datasets as many technologies employed to discover mutations have strong ascertainment biases. For example, enzymatic mismatch cleavage of PCR products is highly efficient for discovery of SNPs and deletions of up to approximately 50 base pairs, but variations involving a larger number of nucleotides go undetected (Till et al. 2003; Comai et al. 2004). Discovery of large insertions and deletions requires more technically complex and expensive approaches (Bolon et al. 2011; Henry et al. 2014).
Enzymatic-based approaches for the discovery and characterization of smaller nucleotide variation date back to the 1970s. A class of enzymes known as single-strand-specific nucleases was first shown to preferentially cleave single-stranded regions of otherwise doubled-stranded DNA (Shenk et al. 1975; Kroeker and Kowalski 1978; Desai and Shankar 2003). By the late 1990s, PCR and capillary Sanger sequencing allowed direct identification of single-nucleotide variation. This remains cost prohibitive for large-scale screening, and assignment of heterozygous indels using this technique is challenging. In 1998, a single-strand-specific nuclease related to S1, named CEL I, was described that could cleave all types of single-base mismatches (Oleykowski et al. 1998). Using this enzyme purified from celery, efficient PCR-based screening approaches were developed for discovery of EMS-induced mutations in TILLING reverse-genetic screens (Colbert et al. 2001). Further studies and optimizations showed that crude enzyme extracts from celery or mung bean sprouts could be prepared that contain a high single-strand-specific nuclease activity and could be used for accurate discovery of both SNP and small indel nucleotide variation (Till et al. 2004). Crude extracts from celery have been commonly used in TILLING assays and also for the detection of natural nucleotide variation (known as Ecotilling), and thousands of nucleotide variants have been discovered (Jankowicz-Cieslak et al. 2011; Till 2014). The use of crude enzyme extracts was shown to result in accurate recovery of DNA variation. In studies of both human samples and highly heterozygous polyploid bananas, false-negative and false-positive discovery errors were reported below 5 % (Till et al. 2006a, b; Till et al. 2010). While many TILLING and Ecotilling projects have utilized fluorescence-based detection of DNA to increase sensitivity and throughput, the development of a more simple and low-cost method employing PCR and standard gel electrophoresis has made mutation discovery suitable for any laboratory equipped for basic molecular biology (Dong et al. 2009; Uauy et al. 2009; Lee et al. 2014).
Assay costs are further reduced by using single-strand-specific nucleases that are self-prepared from plant materials, typically celery. The standard procedure involves two ammonium sulfate cuts carried out at 4 °C in the presence of the toxic protease inhibitor phenylmethylsulfonyl fluoride (PMSF) (Till et al. 2006a, b). Following precipitation, the sample is desalted using 10,000 molecular weight cutoff dialysis membrane. While efficient and low cost, this standard protocol requires all steps to be performed at 4 °C, including large-volume centrifugation. We have simplified this procedure so that nuclease can be prepared at room temperature using a benchtop microcentrifuge common to most molecular biology laboratories. We have also eliminated the use of toxic chemicals. This streamlined approach is diagrammed in Fig. 15.1. Celery is first juiced and buffer added to maintain pH. Particulate matter in the juice is removed by centrifugation. Ammonium sulfate is then added to the clarified juice to a saturation of 25 %. Single-strand-specific nucleases remain in solution at this saturation, but other proteins are precipitated and removed by centrifugation. The supernatant is collected, and ammonium sulfate is added to 85 % saturation, where the desired single-strand-specific nucleases precipitate and are collected by centrifugation. Following removal of the supernatant and suspension of the pellet in buffer, residual salt from the ammonium sulfate precipitation, which is inhibitory to mutation discovery, is removed. This is through the use of specialized filter columns that retain the nuclease. A total of four buffer washes are performed to remove salt. This process also concentrates proteins. The purified enzyme is then collected and activity is tested. The whole procedure can be completed in 1 day. Enzyme for approximately 3000 reactions can be produced from 18 ml of celery juice. The cost of enzyme for one reaction is less than 0.06 euro cents.

Overview of the steps involved in preparation of crude enzyme extracts from celery containing single-strand-specific nuclease activity. Approximate time for each step is indicated
2 Materials
-
1.
Vegetable juicer (e.g. L’Equip) (see Note 1).
-
2.
Celery (see Note 2).
-
3.
Buffer A: 1 M Tris–HCl, pH 7.7.
-
4.
Buffer B: 0.1 M Tris–HCl, pH 7.7, 0.5 M KCl.
-
5.
Ammonium sulfate (NH4)2SO4.
-
6.
Amicon Ultra 0.5 ml 10 K centrifugal filters and collection tubes (EMD Millipore Cat. No. UFC501024).
-
7.
Micropipettes (1000 μl, 200 μl, 20 μl).
-
8.
2.0 ml microcentrifuge tubes.
-
9.
Standard bench microcentrifuge.
-
10.
Magnetic stirrer.
3 Methods
3.1 Enzyme Preparation
-
1.
Rinse celery with water. Remove any leaves and cut off tough tissue at base of stalk. The amount of juice produced depends on the type of juicer or blender used. An efficient juicer will produce ~250 ml from 0.5 kg of celery (see Notes 2 and 3).
-
2.
Juice the celery. Place 18 ml into a clean beaker or tube. Excess juice can be stored at −80 °C for years (see Note 4).
-
3.
Add 2 ml of buffer A to 18 ml of celery juice.
-
4.
Distribute liquid equally into twelve 2.0 ml microcentrifuge tubes.
-
5.
Spin the juice for 20 min at 2,600 × g to pellet debris at room temperature (RT).
-
6.
Pour supernatant into a 25 ml graduated cylinder and record volume.
-
7.
Bring the supernatant to 25 % (NH4)2SO4 by adding 144 g per liter of solution. Mix gently for 30 min using stir plate and magnetic stir bar (Fig. 15.2). (e.g. if total volume is 18.5 ml, add 2.66 g (NH 4 ) 2 SO 4 ).
-
8.
Distribute liquid into 2.0 ml microcentrifuge tubes. Spin at 15,000 × g at room temperature for 40 min.
-
9.
Pour supernatant into clean graduated cylinder. Record volume. Discard pellet (Fig. 15.3a).
-
10.
Bring the supernatant from 25 to 80 % (NH4)2SO4 by adding 390 g per liter of solution. Mix gently for 30 min in a beaker with magnetic stir bar. (e.g., if total volume is 18 ml, add 7.02 g (NH 4 ) 2 SO 4 ).
-
11.
Distribute liquid into 2.0 ml microcentrifuge tubes. Spin at 15,000 × g for 1.5 h. Save the pellet and discard the supernatant, being careful when decanting the supernatant. The pellet can be stored at −80 °C for months (Fig. 15.3b).
-
12.
Suspend the pellets with buffer B. Add 0.5 ml of buffer to the first tube and pipette until pellet is in solution. Transfer this liquid to the next tube and suspend the pellet. If a large amount of foam is produced, add 0.5 ml of buffer to a new tube. Do not exceed a total of 1.5 ml of buffer. Combine liquid into one tube when all pellets are in the solution (Fig. 15.4a).
-
13.
Desalting. Distribute liquid into four Amicon filter devices, making sure not to exceed 500 μl in any filter (Fig. 15.4b). Attach collection tube and spin at 14,000 × g for 30 min.
-
14.
When complete, remove liquid from collection tube, replace filter onto the same tube, adjust volume to 500 μl with fresh buffer B, pipette up and down 5×, and spin at 14,000 × g for 30 min.
-
15.
Repeat the procedure in step 14 for a total of four times (Fig. 15.5a and b). The removal of salts can be calculated (Table 15.1, see Note 5).
-
16.
To elute sample, invert the filter and place into a new collection tube. Centrifuge at 1000 × g for 2 min.
-
17.
Combine all eluates into a single tube. Approximately 85 μl should be recovered (Fig. 15.5c). This tube contains crude celery juice extract (CJE) consisting of CEL I and other enzymes.
-
18.
Centrifuge for 30 min at 10,000 × g to remove any residual solid material. Transfer to a new tube and perform activity test.

Protein precipitation with (NH4)2SO4

Protein pellets after precipitation with ammonium sulfate. (a) Protein pellet after 25 % (NH4)2SO4 precipitation. At this stage the enzyme remains in the aqueous phase. (b) Protein pellet after 80 % (NH4)2SO4 precipitation. This pellet contains nucleases for mutation discovery

Suspension and desalting of protein precipitate. (a) Approximately 1.5 ml liquid is recovered after resuspension and combination of all pellets (derived from 80 % (NH4)2SO4 precipitation). (b) Liquid is transferred to filter devices for desalting

Stages of enzyme purification and desalting. (a) After first centrifugation (~100 μl liquid is retained in the filter device and ~400 μl flow through). (b) After fourth centrifugation (~35 μl liquid retained in the filter and ~465 μl flow through). (c) Recovered eluates after centrifugation of the inverted Amicon filter devices
3.2 Activity Test
-
1.
Test a range of amounts of extracted enzyme using samples with known mutations. Follow the protocol for enzymatic mismatch cleavage used in Chap. 16 of this book. If you do not have positive control material containing known nucleotide polymorphisms, you can request a kit from the Plant Breeding and Genetics Laboratory that contains genomic DNA, PCR primers, buffers, and nuclease with validated activity (see Note 6).
-
2.
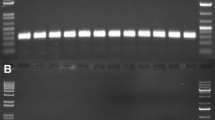
The typical activity of enzyme produced with this protocol is 0.025 μl per reaction (see Note 7). To assay activity, perform a standard titration curve with the outermost points flanking the target range on either side following Table 15.2. Enzyme is diluted with buffer B where indicated. For 1:10 combine 1 μl of enzyme with 9 μl buffer. For 1:100 combine 1 μl enzyme with 99 μl buffer. Mix before use. Perform reactions as in the protocol referenced above by adding 10 μl PCR product with 10 μl of enzyme mix from Table 15.2. Example data are shown in Fig. 15.6.

Enzyme activity tests comparing traditional dialysis-based purification (left) and the benchtop protocol described in this chapter (right). M indicates lanes containing 1 Kb Plus DNA Ladder (Invitrogen). Mutations are easily identified at the 1× concentration
4 Notes
-
1.
Any instrument capable of extracting juice from plant tissues may be used. If using a blender, consider pre-chopping the celery to avoid long fibrous strands that entangle the blender blades.
-
2.
A variety of different plant species and tissues can be used including spinach, mung bean sprouts, and weedy plants growing wild (Oleykowski et al. 1998; Till et al. 2004; Till et al. 2015).
-
3.
No difference in enzyme activity was observed between extraction performed at 4 °C and room temperature.
-
4.
Juice can be stored at −80 °C for at least 2 years. Store juice in aliquots. Activity will reduce due to repeated freeze-thaw cycles.
-
5.
It is important to remove excess salt from the nuclease preparation. Excess salt can cause smearing of gel lanes resulting in higher assay noise and inflated false-negative error rate.
-
6.
A description of the positive controls can be found at http://www-naweb.iaea.org/nafa/pbg/public/manuals-pbg.html. Email Official.Mail@iaea.org with “plant breeding positive controls” in the subject line to request a kit.
-
7.
The optimal amount of nuclease varies depending on the activity of the batch being used. If degradation of the PCR product is not observed at 20×, try 50× and 100× concentrations of enzyme.
References
Belfield EJ, Gan X, Mithani A, Brown C, Jiang C, Franklin K, Alvey E, Wibowo A, Jung M, Bailey K, Kalwani S, Ragoussis J, Mott R, Harberd NP (2012) Genome-wide analysis of mutations in mutant lineages selected following fast-neutron irradiation mutagenesis of Arabidopsis thaliana. Genome Res 22(7):1306–1315
Bolon YT, Haun WJ, Xu WW, Grant D, Stacey MG, Nelson RT, Gerhardt DJ, Jeddeloh JA, Stacey G, Muehlbauer GJ, Orf JH, Naeve SL, Stupar RM, Vance CP (2011) Phenotypic and genomic analyses of a fast neutron mutant population resource in soybean. Plant Physiol 156(1):240–253
Bruce M, Hess A, Bai J, Mauleon R, Diaz MG, Sugiyama N, Bordeos A, Wang GL, Leung H, Leach JE (2009) Detection of genomic deletions in rice using oligonucleotide microarrays. BMC Genomics 10:129
Colbert T, Till BJ, Tompa R, Reynolds S, Steine MN, Yeung AT, McCallum CM, Comai L, Henikoff S (2001) High-throughput screening for induced point mutations. Plant Physiol 126(2):480–484
Comai L, Young K, Till BJ, Reynolds SH, Greene EA, Codomo CA, Enns LC, Johnson JE, Burtner C, Odden AR, Henikoff S (2004) Efficient discovery of DNA polymorphisms in natural populations by ecotilling. Plant J 37(5):778–786
Desai NA, Shankar V (2003) Single-strand-specific nucleases. FEMS Microbiol Rev 26(5):457–491
Dong C, Dalton-Morgan J, Vincent K, Sharp P (2009) A modified TILLING method for wheat breeding. Plant Genome 2(1):39–47
Greene EA, Codomo CA, Taylor NE, Henikoff JG, Till BJ, Reynolds SH, Enns LC, Burtner C, Johnson JE, Odden AR, Comai L, Henikoff S (2003) Spectrum of chemically induced mutations from a large-scale reverse-genetic screen in Arabidopsis. Genetics 164(2):731–740
Henry IM, Nagalakshmi U, Lieberman MC, Ngo KJ, Krasileva KV, Vasquez-Gross H, Akhunova A, Akhunov E, Dubcovsky J, Tai TH, Comai L (2014) Efficient genome-wide detection and cataloging of EMS-induced mutations using exome capture and next-generation sequencing. Plant Cell 26(4):1382–1397
Jankowicz-Cieslak J, Huynh OA, Bado S, Matijevic M, Till BJ (2011) Reverse-genetics by TILLING expands through the plant kingdom. Emirates J Food Agric 23(4):290–300
Jankowicz-Cieslak J, Huynh OA, Brozynska M, Nakitandwe J, Till BJ (2012) Induction, rapid fixation and retention of mutations in vegetatively propagated banana. Plant Biotechnol J 10(9):1056–1066
Kroeker WD, Kowalski D (1978) Gene-sized pieces produced by digestion of linear duplex DNA with mung bean nuclease. Biochemistry 17(16):3236–3243
Lee LS, Till B, Hill H, Huynh O, Jankowicz-Cieslak J (2014) Mutation and mutation screening. In: Henry RJ, Furtado A (eds) Cereal genomics: methods and protocols, vol 1099. Humana Press, New York, pp. 77–95
Li X, Song Y, Century K, Straight S, Ronald P, Dong X, Lassner M, Zhang Y (2001) A fast neutron deletion mutagenesis-based reverse genetics system for plants. Plant J 27(3):235–242
Oleykowski CA, Bronson Mullins CR, Godwin AK, Yeung AT (1998) Mutation detection using a novel plant endonuclease. Nucleic Acids Res 26(20):4597–4602
Rogers C, Wen J, Chen R, Oldroyd G (2009) Deletion based reverse genetics in Medicago truncatula. Plant Physiol 151(3):1077–1086
Sato Y, Shirasawa K, Takahashi Y, Nishimura M, Nishio T (2006) Mutant selection from progeny of gamma-ray-irradiated rice by DNA heteroduplex cleavage using Brassica Petiole extract. Breed Sci 56(2):179–183
Shenk TE, Rhodes C, Rigby PW, Berg P (1975) Biochemical method for mapping mutational alterations in DNA with S1 nuclease: the location of deletions and temperature-sensitive mutations in simian virus 40. Proc Natl Acad Sci USA 72(3):989–993
Slade AJ, Knauf VC (2005) TILLING moves beyond functional genomics into crop improvement. Transgenic Res 14(2):109–115
Till BJ (2014) Mining genetic resources via ecotilling. In: Tuberosa R, Graner A, Frison E (eds) Genomics of plant genetic resources. Springer, Netherlands, pp. 349–365
Till BJ, Reynolds SH, Greene EA, Codomo CA, Enns LC, Johnson JE, Burtner C, Odden AR, Young K, Taylor NE, Henikoff JG, Comai L, Henikoff S (2003) Large-scale discovery of induced point mutations with high-throughput TILLING. Genome Res 13(3):524–530
Till BJ, Burtner C, Comai L, Henikoff S (2004) Mismatch cleavage by single-strand specific nucleases. Nucleic Acids Res 32(8):2632–2641
Till BJ, Zerr T, Bowers E, Greene EA, Comai L, Henikoff S (2006a) High-throughput discovery of rare human nucleotide polymorphisms by ecotilling. Nucleic Acids Res 34(13):e99
Till BJ, Zerr T, Comai L, Henikoff S (2006b) A protocol for TILLING and ecotilling in plants and animals. Nat Protoc 1(5):2465–2477
Till BJ, Jankowicz-Cieslak J, Sagi L, Huynh OA, Utsushi H, Swennen R, Terauchi R, Mba C (2010) Discovery of nucleotide polymorphisms in the Musa gene pool by ecotilling. Theor Appl Genet 121(7):1381–1389
Till BJ, Jankowicz-Cieslak J, Huynh OA, Beshir M, Laport RG, Hofinger BJ (2015) Low-cost methods for molecular characterization of mutant plants. Springer, Switzerland
Uauy C, Paraiso F, Colasuonno P, Tran RK, Tsai H, Berardi S, Comai L, Dubcovsky J (2009) A modified TILLING approach to detect induced mutations in tetraploid and hexaploid wheat. BMC Plant Biol 9:115
Yuan L, Dou Y, Kianian SF, Zhang C, Holding DR (2014) Deletion mutagenesis identifies a haploinsufficient role for gamma-zein in opaque2 endosperm modification. Plant Physiol 164(1):119–130
Acknowledgments
Funding for this work was provided by the Food and Agriculture Organization of the United Nations and the International Atomic Energy Agency through their Joint FAO/IAEA Programme of Nuclear Techniques in Food and Agriculture. This work is part of IAEA Coordinated Research Project D24012.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution-Noncommercial 2.5 License (http://creativecommons.org/licenses/by-nc/2.5/) which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
The images or other third party material in this chapter are included in the work’s Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work’s Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2017 International Atomic Energy Agency
About this chapter
Cite this chapter
Hofinger, B.J., Huynh, O.A., Jankowicz-Cieslak, J., Till, B.J. (2017). A Protocol for Benchtop Extraction of Single-Strand-Specific Nucleases for Mutation Discovery. In: Jankowicz-Cieslak, J., Tai, T., Kumlehn, J., Till, B. (eds) Biotechnologies for Plant Mutation Breeding. Springer, Cham. https://doi.org/10.1007/978-3-319-45021-6_15
Download citation
DOI: https://doi.org/10.1007/978-3-319-45021-6_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45019-3
Online ISBN: 978-3-319-45021-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)