
Abstract
NK cells are able to perform multiple functions, ranging from immunosurveillance to elimination of mutated or damaged cells, through many different cytotoxic mechanisms. Their functions can be very useful for cancer immunotherapy. But to achieve the maximum support from these extraordinary cells it is necessary to know their effector mechanisms and the mechanisms that lead to their suppression. We have briefly summarized some interesting aspect of their role in immunosurveillance of cancer and metastases, the major mechanisms of cell cytotoxicity, in particular their role in antigen dependent cell cytotoxicity, and many promising strategies currently under study to improve the anticancer function of these cells.
Finally, we have taken a closer look at cell therapy in this context, comparing CAR-NK cells and CAR-T cells showing the potential advantages of the former over the latter.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others

Keywords
Introduction
Treatment of head and neck squamous cell carcinomas (SCCHN) rapidly evolved during the last decade, mainly due to the inclusion of immune-checkpoints inhibitors (ICIs) in the routine therapy of relapsed metastatic disease (R/M-SCCHN). However, head and neck cancers remain a major clinical problem and most R/M-SCCHN patients ultimately die of their disease. Nonetheless, the experience matured with ICIs demonstrating that the immune system and their components play a crucial role in the control of R/M-SCCHN.
Natural killer (NK) cells are key-player in cancer immunosurveillance, cancer control and prevention of metastatization. Indeed, in the 1980s, several studies reported a higher incidence of cancers in individuals with defective NK cell function supporting the role of NK cells in immunosurveillance [1, 2].
In human solid tumors, NK cell infiltration is poor in non-small cell lung cancer, colorectal cancer and melanoma, but it is high in breast cancer, kidney cancer and SCCHN. The latter show the highest infiltration of NK cells [3]. The density of infiltrating NK cells correlates with the patient’s prognosis in many solid tumors, including oropharyngeal squamous cell carcinoma (OSCC) [4]. Indeed, Wagner et al. showed a relationship between the prognosis of OSCC patients and NK levels, regardless of HPV status, although higher numbers of CD56 positive (CD56+) cells were found in HPV-positive patients compared to HPV-negative patients. The elevated abundance and activity of cytotoxic NK cells in OSCC patients with HPV driven carcinogenesis might contribute to the favorable outcome in HPV-related OSCC [5].
There is evidence that NK cells are involved in the metastatic spreading: the number of circulating NK cells inversely correlate with circulating tumor cells, and the decline of cytotoxicity and of cytokine production of NK cells after major surgery correlates with the risk of metastases [6].
Indeed, the epithelial-mesenchymal transition (EMT) that is a central step during metastatization, leads to expression of new antigens that reactivate the cytolytic effect of NK cells [7].
Chockley et al. showed that EMT leads to NK cell mediated metastasis specific immuno-surveillance. Indeed, EMT modulates the adhesion molecule CADM1 on the surface of tumor cells, increasing the susceptibility to NK cytotoxicity in lung and breast cancer [8].
NK Cells, Antitumor Effects and Antibody Dependent Cell Cytotoxicity
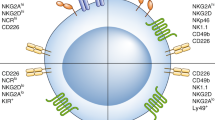
In general, NK cells are extraordinary effective war machines able to kill stressed (infected) or mutated (tumoral) cells through multiple mechanisms (Fig. 5.1). NK cells are divided into two major subsets according to their cell surface expression levels of CD56 and CD16. CD56dim/CD16bright NK cells predominantly mediate natural cytotoxicity, whereas the CD56bright/CD16dim subset plays a role in immune regulation through a high cytokine secretion potential [9].

NK functions
A series of activating and inhibitory receptors on the membrane of NK cells may sense inducible stress molecules and self-proteins. The prevalence of one signal over the other, results in aggression or tolerance. NK cells also bear ligands for death signal receptors expressed on the membrane of the target cells, such as FAS or TRAIL.
Finally, they secrete a high number of cytokines with antitumor activity such as IFN-ɣ.
In addition, NK cells are also the most powerful inducer of antibody dependent cell cytotoxicity (ADCC).
Indeed, FC-gamma receptors (FCɣR), the receptor family linked to ADCC, include both activating and inhibiting receptors, and are expressed on a number of different immune cell lineages. NK cells host only FCɣRIII and FCɣRIIc (CD16 and CD 32c), both activating receptors, which make NK cells the most important lineage able to trigger ADCC.
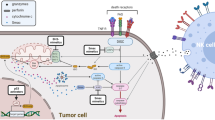
ADCC is a complex but highly efficient mechanism leading to the elimination of damaged, infected or mutated cells. It involves five main actors: (1) the effector cell, (2) the Fcγ Receptor (FcγR), (3) the antibody, (4) the target antigen on the surface of the target cell and (5) the target cell itself [10]. Figure 5.2 reports the five ADCC players.

Main actors of ADCC
As reported above, when we speak about ADCC we consider primarily NK cells.
-
1.
The effector cells.
Cancer cells and many immune cells can damp NK cells, such as tumor associated fibroblast (TAFs), tumor associated macrophages (TAM), T regulatory cells (Tregs) and myeloid derived suppressor cells (MDSC).
They all reduce NK function following NKp44, NKp30 and DNAM1 down-regulation, reduce NK degranulation and IFN-γ production, and inhibit NKG2D expression [11].
Among the many mechanisms that tumor cells use to impair NK cells, the release of inhibitory soluble ligands such as MIC-A and MIC-B into the tumor microenvironment (TME), the high levels of TGF-β and of other immune-suppressive cytokines are among the best known [12]. Overall, inhibition of NK cells follows the same mechanisms of CD8+ T cell inhibition.
Figure 5.3 summarizes the most important interactions between tumor cells and NK cells and other inhibitory mechanisms.
However, a residual activity of NK cells still exists in the TME, since high NK cell infiltration usually correlates with favourable prognosis in many tumors [4]. In particular, Taylor et al. [13] observed that inducible ADCC was the most predictive marker for clinical outcome in SCCHN. In support of this observation, Lattanzio et al. [14] observed in a series of SCCHN patients treated with cetuximab and radiotherapy, that patients with ADCC activity above the median had a statistic significant benefit in overall survival. Similarly, patients with metastatic, wild-type, colon cancer treated with cetuximab and with ADCC activity above the median had a significant gain in overall survival [15].
-
2.
The FcγR.
The Fc receptor for IgG (FcγR) belongs to the immune globulin superfamily and includes many families (FcγRIa [CD64a], FcγRIIa [CD32a], FcγRIIb [CD32b], FcγRIIc [CD32c], FcγRIIIa [CD16a], FcγRIIIb [CD16b]) which are expressed by many immune cell lineages and link to the Fc fragment of the antibody with different affinity. FcγRIIb is the only inhibitory receptor, links to any IgG subclass with high affinity and is represented in all the immune cells harbouring FcγRs, but not in NK cells.
Therefore, NK cells are the sole immune cells expressing only activator receptors (FcγRIIIa and FcγRIIc) [16]. However, FcγRIIc is expressed only in about 40% of healthy human subjects [17], and is not yet completely understood [18]. Therefore, most attention is devoted to FcγRIIIa and in particular to its polymorphisms. Indeed, experimental models support the positive role of the Valine homozygosis at FcγRIIIa-158 toward an increased ADCC activity [19], although its real impact in clinic is not yet clear [20, 21]. Moreover, Rooney et al. suggested that elevated ADCC activity observed in cancer patients may exceed the value of FcγRIIIa polymorphism as prognosticator [13].
-
3.
The antibody.
IgG includes four classes: 1, 2, 3 and 4. NK-cell dependent ADCC is a Fc segment mediated effector function triggered mainly by IgG1 and IgG3. However, whilst IgG1 seems to be independent from subclasses, IgG3 function is largely allotype-dependent [22]. IgG2 and IgG4 weakly link to FcγRIIIa, but they do not induce ADCC [22, 23]. In conclusion, IgG1 is the most effective IgG class able to trigger ADCC, regardless of allotype.
-
4.
The target antigen (TA).
The TA density on the target cell surface is the fourth actor of ADCC. There is in vitro evidence that the expression of the TA on tumor cell surface is a key factor influencing cytotoxicity [24, 25]. However, the importance of the expression of the TA is not evident in the clinic. For instance, cetuximab has shown clinical activity regardless of EGFR expression. There are many factors that may explain this discrepancy. First, the target effect of cetuximab may mask the immunological effect of the antibody. Second, ADCC depends on the efficiency of additional immunologic variables, such as those we are discussing here.
However, some clinical data supporting the role of the TA density in clinic do exist [26]. Our group observed that in patients treated with cetuximab and radiotherapy, ADCC activity directly correlates with the outcome, but patients with high ADCC and high EGR density (EGFR+++) showed the best outcome [14].
-
5.
The target cells.
The mutational status of the target cell may affect ADCC. For instance, mutation of KRAS leads to constitutive activation of the PI3K/AKT pathway, resulting in direct inhibition of BAD and caspase 9, inhibition of p53 via MDM2 and upregulation of antiapoptotic proteins such as BCL-X, BCL-2 and COX-2. All together these effects confer resistance to apoptosis induced by granzyme B [27, 28].
Therefore, even if all the described actors of ADCC are efficient, the mutational status of the target cell may prevent ADCC induced apoptosis.

Major mechanisms of NK cells stimulation/inhibition
Strategies to Enhance Antitumor NK Cell Function
Since activation or inhibition of immune functions depends on the balance between positive and negative regulators of signaling, the activation, or re-activation, of antitumor NK activity depends upon the upregulation of the former and/or the downregulation of the latter.
A third factor to enhance NK cell activity, is improving their homing into tumor nests, because they are often detained within the stroma surrounding cancer cells.
Reinforcement of NK Cell Activity
Preactivation with cytokines (IL12, IL15, IL18) induces memory-like (cytokine-induced memory-like, CIML) NK cells that show enhanced effector functions lasting for weeks [29].
It is generally accepted that IL-12, IL-15, and IL-18 preactivation induces in NK cells a rapid and prolonged expression of CD25, resulting in a functional up-regulation of high-affinity IL-2 receptor (IL-2Rαβγ) that confers responsiveness to picomolar concentrations of IL-2, favouring NK cells expansion [30].
Cytokine preactivation also induces expression of markers such as the chemotactic receptor CXCR4 necessary for homing of NK cells [31].
Terrèn et al. demonstrated that IL-15 might contribute more than IL12/18 to CIML NK cell-mediated cytotoxicity against target cells, although all the three cytokines are needed to improve activity of NK [32].
Moreover, JAK inhibitors (JAK/STAT pathway, responsible for cytokine regulations) are able to modify NK cell biology in vitro and in vivo.
Schönberg et al. reported that the JAK 1–2 inhibitor ruxolitinib, impairs IL-2 preactivated NK killing ability. Reduced NK cell numbers in ruxolitinib-exposed patients may depend on the inhibition of various important cytokine signals essential for NK maturation (i.e. IL-2 and IL-15) [33].
Ewen et al. found that the activation of NK cells with IL-12/15/18 led to a decreased expression of the inhibitory receptors of the KIR family reinforcing NK effector potential [34].
Moreover IL15-stimulates DCs to activate NK cells in an IL15 dependent manner, indeed, IL-15 DCs, but not IL-4 DCs, promoted NK cell tumoricidal activity towards both NK-sensitive and NK-resistant targets. This effect was found to be mediated by DC surface-bound IL-15 [35].
IL-2 is a well-known growth factor of antigen activated T lymphocytes. IL-2 also stimulates NK cell expansion and activation. However, it also favours Treg expansion through the high affinity sub-unit receptor IL-2Rα (CD25) expressed on these cells. IL-2 variants able to prevent Treg expansion have been generated. Among them, the IL-2 “superkine” with increased affinity to the IL-2Rβ subunit expressed on NK cells and other T effector cells [36].
IL-15 may have stimulating effects similar to IL-2 on NK cells, and also enhances ADCC, without stimulation of Treg expansion [37]. An IL-15 super-agonist, with a long half-life, has been already tested in humans and numerous clinical trials are in progress [38].
There are few preclinical studies investigating cytokine therapy for NK reactivation. However, some data on IL-6 are available. In pancreatic cancer blocking IL 6 not only inhibits tumor growth but also rescue the NK cells from suppression induced by the peripancreatic adipose tissue [39].
Prevent NK Cell Inhibition
Removing the block of NK cells is an emerging, rapidly evolving area and upregulation of some checkpoint molecules (e.g. TIGIT, CD96, PD1, KIRs, NKG2a, IL1R8) represent potential targets for NK-based immunotherapy.
Anti-PD-1
PD-1 is upregulated in several solid tumors, including head and neck cancer [40] and is associated with the inhibition of NK cell activity [41]. It has been demonstrated that PD-1 expression impairs function of intratumoral NK cells. Notably, treatment with PD-1 blockade was able to reverse PD-L1-mediated inhibition of PD-1+ NK cells [42]. Inhibition of PD-1 on NK cells may be important in particular in tumors that poorly express or do not express MHC-I, thereby evading CD-8 T cell attack, but, for the same reason, are a good target for NK cells.
Anti NKG2A
HLA-E is a non-classical MHC-I molecule, frequently up-regulated in SCCHN and is associated with low survival rates. Although MHC-Ia molecules help in cancer cell recognition through the T-cell receptor, HLA-E can be recognized by the inhibitory heterodimeric CD94/NKG2A receptor [43]. This interaction inhibits NK-cell’s cytotoxic functions and prevents autoimmunity, but is also exploited by cytomegalovirus to evade antiviral immunity.
Interactions of HLA-E with CD94/NKG2A significantly impairs IL2 receptor–dependent proliferation of tumor-specific T cells that contributed to reduced cytotoxicity and cytokine production, which improved following antibody-mediated blockade treatment in vitro and ex vivo [44].
Andre P et al. demonstrated the efficacy of anti-NKG2A monalzumab in combination with anti-EGFR.
In a phase II trial of monalizumab combined with cetuximab, responses were observed in 35% of patient who were immunotherapy-naive and 18% in those who received previous chemotherapy. The combination was well tolerated and 93% of adverse events (AE) were of grade 1–2 severity with only 6% of patients experiencing treatment-related grade 3–4 AE. Eight out of 26 patients (31%) achieved a confirmed response (1 complete and 7 partial), 54% had stabilization of disease (SD) [43].
The first patient cohort of the study UPSTREAM (patients not eligible for one of the biomarker-driven cohorts, after platinum progression) treated with single agent monalizumab (10 mg/kg) every 14 days, were reported at ESMO 2019. The sub-study did not meet its primary objective (progressive disease 78%) although 59% of patients had received prior treatment with anti PD1/PD-L1. We hypothesize that blocking the inhibitory axis CD94/NKG2A/HLAE alone might be not sufficient to reverse an immunosuppressive TME.
Anti KIR2
NK cell activation is partially controlled by KIRs upon binding with their ligands. Preclinical hematological studies reported activation of NK through mismatches between KIRs on donor NK cells and recipient MHC class I molecules, with improved relapse-free survival and overall survival [45]. The efficacy and safety of the first-in-class anti-pan-KIR2D agent lirilumab was explored in several clinical trials. Lirilumab can be safely administered but the efficacy in monotherapy is disappointing. Contrary to this, combinations with anti-PD1 antibody and antiCTLA-4 (136 with nivolumab; 22 with ipilimumab) were well-tolerated, with encouraging preliminary results. In SCCHNl the Lirilumab plus nivolumab cohort showed an objective response rate (ORR) of 24%, with durable responses. Notably, increased PD-L1 expression was strongly associated with improved probability of objective response [46].
Anti TIM-3
Interestingly, resistance to anti-PD-1 monoclonal antibodies (mAbs) might depend on up-regulation of alternative immune checkpoints, including TIM-3, LAG3, TIGIT etc. Recent studies showed that T-cell immunoglobulin mucin 3 (TIM3) participates in the regulation of Tregs, and correlates with immunosuppressive microenvironment (Galectin-9, Foxp3, CD68 and CD163) [47].
The increased surface levels of TIM-3 on NK cells in cancers induce NK cell impairments [48], while TIM-3 blockade results in increased NK cell cytotoxicity both in vitro and ex vivo [49]. In SCCHN anti TIM3 reduces Treg activation and decreases CTLA4 and TIGIT.
Currently, therapeutic approaches combining the administration of anti-TIM-3 and anti-PD-1 antibodies showed that the adaptive resistance to PD-1 blockade can be overcome [50].
Several studies are ongoing in phase I both in solid and hematological malignancies as monotherapy or in combination with an anti-PD-1 mAb or anti-LAG3 mAb (NCT03489343, NCT03311412, NCT02817633, NCT03680508, NCT04139902, and NCT03744468).
Anti LAG-3
Lymphocyte activation gene 3 (LAG-3) is an inhibitory receptor on T cells, which increases the effect of Tregs and shows relationship with T cell exhaustion. LAG-3 suppresses immune responses in several tumors, including Hodgkin’s lymphoma, gastric cancer, breast cancer, and other solid tumors. T cells co-expressing both LAG-3 and PD-1 may show a greater degree of exhaustion compared with those expressing LAG-3 alone [51]. Combining anti-LAG-3 mAb and anti-PD-1 mAb synergistically enhances T cell activity, [52] and a phase I/II clinical trial with the combined treatment is ongoing (NCT01968109).
Blockade of LAG-3 pathways has shown to enhance T-cell and NK cell activity, leading to increased antitumor activity and limiting tumor burden in several preclinical studies [52].
In this context, different anti-LAG-3 mAb are currently being used in phase I and phase II clinical trials as monotherapy (NCT03489369 and NCT03250832) or in association with other immune checkpoints inhibitors (NCT04150965, NCT02658981, NCT01968109, NCT03005782, NCT04080804, NCT02676869). A number of additional LAG-3 antibodies are currently in preclinical development.
Anti-TIGIT
T-cell immuno receptor with immunoglobulin and ITIM domains (TIGIT), can suppress T-cell activation and promote T-cell exhaustion. TIGIT and CD96 are co-inhibitory receptors expressed on both T and NK cells and compete with the activating NK cell receptor DNAM-1 for binding to the poliovirus receptor (PVR;CD155) and Nectin2 (CD112) [53]. These receptors participate in a balanced system to control NK cell effector functions. The expression of TIGIT is highly variable among different cancer types and it is highly expressed on tumor-infiltrating NK cells [54].
Notably, the therapeutic effects of anti-TIGIT and anti-PD-L1 monotherapy, or anti-TIGIT and anti-PD-L1 combinations depend on the presence of NK cells [55], indicating the importance of NK cells in checkpoint-targeted immunotherapy. Currently, several ongoing clinical trials (phase I and phase II) focus on testing the feasibility of targeting the TIGIT pathway and improving therapeutic effects through combination with existing immunotherapies, including anti-PD-1 agents (NCT04150965, NCT03119428, NCT04047862, and NCT03563716), mainly in solid tumor patients.
Increase ADCC Through Engineering of Monoclonal Antibodies
Many approved mAbs are of the IgG1 isotype. Fc region in IgG1 includes two N-linked biantennary complex-type oligosaccharides. The Fc region induces ADCC through its interaction with the Fcγ receptor family. However, ADCC activity is influenced by the structure of the Fc region. In physiological conditions, mAb Fc region links to the FcγRIIIa of the effector cells with low affinity, in competition with a specific serum IgG.
The therapeutic mAbs can be engineered to remove fucose residues from the Fc N-glycans. Afucosylated mAbs exhibit strong ADCC activity compared to fucosylated counterpart due to much higher binding affinity to FcγRIIIa. Strong ADCC activity is also maintained at low antigen density, a situation in which the fucosylated mAbs cannot induce detectable ADCC [56, 57].
There is a growing interest in afucosylated mAb and many studies are in progress. Some afucosylated mAb are already approved in clinical practice, such as obinutuzumab (anti CD20), mogamulizumab (anti CCR4) and benralizumab (anti IL5Rα) [58].
Improve NK Cells Trafficking and Homing in the Tumor
In many tumors, NK cells are not in direct contact with tumor cells, but rather, they are restrained in the stroma surrounding tumor nests. It happens even if NK cells express the chemokine receptor CXCR3 and the specific chemokines CXCL9 and CXCL10 are released. Therefore, the endogenous production of chemokines may be insufficient for NK cell recruitment to tumor nests [11].
Chemerin is a super agonist of NK trafficking. It is a chemokine that plays a pivotal role in both immune response, lipid metabolism and in the regulation of programmed cell death, including autophagy and apoptosis. The chemerin chemotactic receptor CMKLR1 is expressed on NK cells, macrophages and subsets of dendritic cells.
Chemerin is released in an inactive form (prochemerin) and is converted into active chemerin in inflamed areas. Chemerin is down regulated in many tumors, but restoring its expression may increase NK infiltration and tumor suppression. Indeed, in a breast cancer model, Pachynski RK et al. forced overexpression of chemerin by tumor cells obtaining significant recruitment of NK cells and T cells within the TME [59]. However, the clinical development of chemerin is hampered by its potential side effects [60].
Lee J et al. recently suggested a novel approach. These authors developed an antibody-based NK-cell-homing protein (NRP-body), namely an antibody able to link to a specific tumor antigen, which drives a cargo domain containing CXCL16, the NK chemoattractant. When the antibody links to the tumor antigen, CXCL16 is released in the TME reaching a very high concentration. In a pancreatic cancer model, the NRP-body increased NK-cell infiltration into tumors. Preclinical results showed promising effects [61].
CAR–NK
Compared to CAR-T cells, CAR-transduced NK cells (CAR-NK) exhibit several advantages, (Table 5.1) such as safety in clinical use, the mechanisms by which they recognize cancer cells, and their abundance in clinical samples. Human primary NK cells and the NK-92 cell line have been successfully transduced to express CARs against several tumors, with most mature results in hematological cancers. Moreover, toxicities concerns appear less serious than CAR-T, cytokine release syndrome (CRS) is not reported although NK cells release IFN-γ, IL-3 and GM-CSF, which may result in a different form of CRS (with few systemic inflammatory response and toxic death).
In the last years to counteract these potentially fatal toxicities, CAR- NK cells are modified with an inducible suicide gene able to be activated pharmacologically to turn off the waterfall [62].
NK-92 cells engineered to CAR-NK was recently approved by FDA for clinical trials, and it was already tested in patients with melanoma, sarcoma, colorectal cancer, renal cell cancer (benefit in 5/11 patients) and NSCLC (benefit in 3/4 pts). To date, the used targets of CAR-NK include different cancer antigens such as CD19, CD20, CD244, HER2, CD38, epithelial cell adhesion molecule (EPCAM), disialoganglioside (GD2), EGFR variant III) [63].
Liu et al. generated cord blood derived CAR-NK by transducing NK cells with a retroviral vector to incorporate the genes for CAR CD19, IL-15 (a cytokine crucial for NK cell persistence), and the caspase-9 suicide gene. CAR.19/IL-15/iC9-NK showed additional activity of CAR-NK compared to CAR-T as NK cells preserve the intrinsic capacity to recognize and target tumor cells. Moreover, as above reported, IL-15 drives NK cell expansion and persistence, as demonstrated by longer persistence and anti-tumor activity compared with CAR.19-transduced NK cells lacking IL-15. Finally, these cells can be easily eliminated by pharmacological activation of Caspase 9 [64].
In acute myeloid leukemia NK cells (NK-92) were transduced with a third generation CAR lentiviral construct containing both CD28 and 4-1BB costimulatory molecules, and were infused after salvage chemotherapy in three patients. CAR NK92 cells were irradiated to prevent both excessive expansion and to treat parenteral cells derived from a lymphoma patient. The study failed to demonstrate clinical efficacy but as the “first in human study” it opens the way to optimize this potentially efficacious treatment [65].
Exciting results in pancreatic cancer models have been reported also with cryopreserved NK cells. Systemic administration of NK cells induced greater in vivo tumor growth suppression when compared with gemcitabine. The potent antitumor effect of NK cells was obtained by increasing infiltration into desmoplastic tumor tissues, apoptosis and IFN-γ, and by inhibition of TGFβ [66].
A promising strategy to increase efficacy of NK is to create NK cell engagers (NKCEs): multifunctional antibodies targeting tumor antigens, NKp46 and CD16. The goal is to increase tumor-cell destruction by bringing tumor cells and NK cells together. The new generation of trifunctional NKCEs targets the two activating receptors, NKp46 and CD16, on NK cells and a tumor antigen on cancer cells. Trifunctional NKCEs were more potent in vitro than clinical therapeutic antibodies targeting the same tumor antigen [67].
Despite the usefulness of NK cells, NK-cell therapy is limited by tumor cell inhibition of NK-cell homing to tumor sites, thereby preventing a sustained antitumor immune response.
Ongoing researches on this topic will hopefully provide new tools to overcame this issue (see above in the: ‘Improve NK Cells Trafficking and Homing to the Tumor’ section).
Conclusions
NK cells represent one of the most important tools of the immune system. They are involved in immune surveillance, control of metastatization, and in the fight against tumor cells inside the tumor.
References
Roder JC, Haliotis T, Klein M, Korec S, Jett JR, Ortaldo J, Heberman RB, Katz P, Fauci AS. A new immunodeficiency disorder in humans involving NK cells. Nature. 1980;284:553–5.
Sullivan JL, Byron KS, Brewster FE, Purtilo DT. Deficient natural killer cell activity in x-linked lymphoproliferative syndrome. Science. 1980;210:543–5.
Mandal R, Şenbabaoğlu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, Lee KW, Ganly I, Hakimi AA, Chan TA, Morris LG. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1(17):e89829.
Habif G, Crinier A, André P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. 2019;16(5):415–22.
Wagner S, Wittekindt C, Reuschenbach M, Hennig B, Thevarajah M, Würdemann N, Prigge ES, von Knebel DM, Dreyer T, Gattenlöhner S, Klussmann JP. CD56-positive lymphocyte infiltration in relation to human papillomavirus association and prognostic significance in oropharyngeal squamous cell carcinoma. Int J Cancer. 2016;138(9):2263–73.
Hoshikawa M, Aoki T, Matsushita H, Karasaki T, Hosoi A, Odaira K, Fujieda N, Kobayashi Y, Kambara K, Ohara O, Arita J, Hasegawa K, Kakimi K, Kokudo N. NK cell and IFN signatures are positive prognostic biomarkers for resectable pancreatic cancer. Biochem Biophys Res Commun. 2018;495(2):2058–65.
López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. 2017;32(2):135–54.
Chockley PJ, Chen J, Chen G, Beer DG, Standiford TJ, Keshamouni VG. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J Clin Invest. 2018;128(4):1384–96.
Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22(11):633–40.
Lo Nigro C, Macagno M, Sangiolo D, Bertolaccini L, Aglietta M, Merlano MC. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann Transl Med. 2019;7(5):105.
Vitale M, et al. Effect of tumor cells and tumor microenvironment on NK-cell function Eur J Immunol. 2014.
Klöß S, Chambron N, Gardlowski T, Arseniev L, Koch J, Esser R, Glienke W, Seitz O, Köhl U. Increased sMICA and TGFβ 1 levels in HNSCC patients impair NKG2D-dependent functionality of activated NK Cells. Oncoimmunology. 2015;4(11):e1055993.
Taylor RJ, Saloura V, Jain A, Goloubeva O, Wong S, Kronsberg S, Nagilla M, Silpino L, de Souza J, Seiwert T, Vokes E, Villaflor V, Cohen EW. Ex Vivo antibody-dependent cellular cytotoxicity inducibility predicts efficacy of cetuximab. Cancer Immunol Res. 2015;3(5):567–74.
Lattanzio L, Denaro N, Vivenza D, Varamo C, Strola G, Fortunato M, Chamorey E, Comino A, Monteverde M, Lo Nigro C, Milano G, Merlano M. Elevated basal antibody-dependent cell-mediated cytotoxicity (ADCC) and high epidermal growth factor receptor (EGFR) expression predict favourable outcome in patients with locally advanced head and neck cancer treated with cetuximab and radiotherapy. Cancer Immunol Immunother. 2017;66(5):573–57.
Lo Nigro C, Ricci V, Vivenza D, Granetto C, Fabozzi T, Miraglio E, Merlano MC. Prognostic and predictive biomarkers in metastatic colorectal cancer anti-EGFR therapy. World J Gastroenterol. 2016;22(30):6944–54.
Nimmerjahn F, Gordan S, Lux A. FcγR dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends Immunol. 2015;36(6):325–36.
Dutertre CA, Bonnin-Gélizé E, Pulford K, Bourel D, Fridman WH, Teillaud JL. A novel subset of NK cells expressing high levels of inhibitory FcgammaRIIB modulating antibody-dependent function. J Leukoc Biol. 2008;84(6):1511–20.
Nagelkerke SQ, Kuijpers TW. Immunomodulation by IVIg and the role of fc-gamma receptors: classic mechanisms of action after all? Front Immunol. 2015;5:674.
Arriga R, Caratelli S, Lanzilli G, Ottaviani A, Cenciarelli C, Sconocchia T, Spagnoli GC, Iezzi G, Roselli M, Lauro D, Coppola A, Dotti G, Ferrone S, Sconocchia G. CD16-158-valine chimeric receptor T cells overcome the resistance of KRAS-mutated colorectal carcinoma cells to cetuximab. Int J Cancer. 2020;146(9):2531–8.
Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie HB, Davidson HC, López-Albaitero A, Gibson SP, Gooding WE, Ferrone S, Ferris RL. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19(7):1858–72. https://doi.org/10.1158/1078-0432.CCR-12-2426.
Trotta AM, Ottaiano A, Romano C, Nasti G, Nappi A, De Divitiis C, Napolitano M, Zanotta S, Casaretti R, D’Alterio C, Avallone A, Califano D, Iaffaioli RV, Scala S. Prospective evaluation of cetuximab-mediated antibody-dependent cell cytotoxicity in metastatic colorectal cancer patients predicts treatment efficacy. Cancer Immunol Res. 2016;4(4):366–74.
de Taeye SW, Bentlage AEH, Mebius MM, Meesters JI, Lissenberg-Thunnissen S, Falck D, Sénard T, Salehi N, Wuhrer M, Schuurman J, Labrijn AF, Rispens T, Vidarsson G. FcγR binding and ADCC activity of human IgG allotypes. Front Immunol. 2020;11:740.
Monteverde M, Milano G, Strola G, Maffi M, Lattanzio L, Vivenza D, Tonissi F, Merlano M, Lo NC. The relevance of ADCC for EGFR targeting: a review of the literature and a clinically-applicable method of assessment in patients. Crit Rev Oncol Hematol. 2015;95(2):179–90.
López-Albaitero A, Lee SC, Morgan S, Grandis JR, Gooding WE, Ferrone S, Ferris RL. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58(11):1853–64.
Lewis GD, Figari I, Fendly B, Wong WL, Carter P, Gorman C, Shepard HM. Differential responses of human tumor cell lines to anti-p185HER2 monoclonal antibodies. Cancer Immunol Immunother. 1993;37(4):255–63. https://doi.org/10.1007/BF01518520.
Seo Y, Ishii Y, Ochiai H, Fukuda K, Akimoto S, Hayashida T, Okabayashi K, Tsuruta M, Hasegawa H, Kitagawa Y. Cetuximab-mediated ADCC activity is correlated with the cell surface expression level of EGFR but not with the KRAS/BRAF mutational status in colorectal cancer. Oncol Rep. 2014;31(5):2115–22.
Zaanan A, Okamoto K, Kawakami H, Khazaie K, Huang S. Sinicrope FA the mutant KRAS gene up-regulates BCL-XL protein via STAT3 to confer apoptosis resistance that is reversed by BIM protein induction and BCL-XL antagonism. J Biol Chem. 2015;290(39):23838–49.
Knickelbein K, Zhang L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015;2(1):4–12.
Uppendahl LD, Felices M, Bendzick L, Ryan C, Kodal B, Hinderlie P, Boylan KLM, Skubitz APN, Miller JS, Geller MA. Cytokine-induced memory-like natural killer cells have enhanced function, proliferation, and in vivo expansion against ovarian cancer cells. Gynecol Oncol. 2019;153(1):149–57. https://doi.org/10.1016/j.ygyno.2019.01.006.
Leong JW, Chase JM, Romee R, Schneider SE, Sullivan RP, Cooper MA, Fehniger TA. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transplant. 2014;20(4):463–73. https://doi.org/10.1016/j.bbmt.2014.01.006.
Levy E, Reger R, Segerberg F, Lambert M, Leijonhufvud C, Baumer Y, Carlsten M, Childs R. Enhanced bone marrow homing of natural killer cells following mRNA transfection with gain of function variant CXCR4r334x. Front Immunol. 2019.
Terrén I, Mikelez I, Odriozola I, Gredilla A, González J, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. Implication of interleukin-12/15/18 and Ruxolitinib in the phenotype, proliferation, and polyfunctionality of human cytokine-preactivated natural killer cells. Front Immunol. 2018;9:737.
Schönberg K, Rudolph J, Wolf D. NK cell modulation by JAK inhibition. Oncoscience. 2015;2(8):677–8.
Ewen E-M, Pahl JHW, Miller M, Watzl C, Cerwenka A. KIR downregulation by IL-12/15/18 unleashes human NK cells from KIR/HLA-I inhibition and enhances killing of tumor cells. Eur J Immunol. 2018;48:355–65. https://doi.org/10.1002/eji.201747128.
Anguille S, Van Acker HH, Van den Bergh J, Willemen Y, Goossens H, Van Tendeloo VF, Smits EL, Berneman ZN, Lion E. Interleukin-15 dendritic cells harness NK cell cytotoxic effector function in a contact- and IL-15-dependent manner. PLoS One. 2015;10(5):e0123340. https://doi.org/10.1371/journal.pone.0123340.
Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, Pande VS, Fathman CG, Boyman O, Garcia KC. Exploiting a natural conformational switch to engineer an interleukin-2 ‘Superkine’. Nature. 2012;484(7395):529–33.
Zhang M, Wen B, Anton OA, Yao Z, Dubois S, Ju W, Sato N, DiLillo DJ, Bamford RN, Ravetch V, Waldmann TA. IL-15 enhanced antibody-dependent cellular cytotoxicity mediated by NK cells and macrophages. PNAS. 2018;115(46):E10915–24.
Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, Weisdorf DJ, Blazar BR, Ustun C, DeFor TE, Vivek S, Peck L, DiPersio JF, Cashen AF, Kyllo R, Musiek A, Schaffer A, Anadkat MJ, Rosman I, Miller D, Egan JO, Jeng EK, Rock A, Wong HC, Fehniger TA, Miller JS. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131(23):2515–27.
Xu Y, Sun J, Sheard MA, Tran HC, Wan Z, Liu WY, Asgharzadeh S, Sposto R, Wu HW, Seeger RC. Lenalidomide overcomes suppression of human natural killer cell anti-tumor functions by neuroblastoma microenvironment-associated IL-6 and TGFβ1. Cancer Immunol Immunother. 2013;62(10):1637–48.
Concha-Benavente F, Kansy B, Moskovitz J, Moy J, Chandran U, Ferris RL. PD-L1 mediates dysfunction in activated PD-1+ NK cells in head and neck cancer patients. Cancer Immunol Res. 2018;6(12):1548–60.
Bi J, Tian Z. NK cell exhaustion. Front Immunol. 2017;8:760.
Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175:1731–43.
Trefny MP, Kaiser M, Stanczak MA, Herzig P, Savic S, Wiese M, Lardinois D, Läubli H, Uhlenbrock F, Zippelius A. PD-1+ natural killer cells in human non-small cell lung cancer can be activated by PD-1/PD-L1 blockade. Cancer Immunol Immunother. 2020. https://doi.org/10.1007/s00262-020-02558-z.
Sarmah N, Baruah MN, Baruah S. Immune modulation in HLA-G expressing head and neck squamous cell carcinoma in relation to human papilloma virus positivity: a study from Northeast India. Front Oncol. 2019;9:58.
Ruggeri L, Capanni M, Casucci M, Volpi I, Tosti A, Perruccio K, Urbani E, Negrin RS, Martelli MF, Velardi A. Role of natural killer cell alloreactivity in HLA-mismatched hematopoietic stem cell transplantation. Blood. 1999;94:333–9.
Vey N, Karlin L, Sadot-Lebouvier S, Broussais F, Berton-Rigaud D, Rey J, et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget. 2018;9:17675–88.
Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113:3821–30. https://doi.org/10.1182/blood-2008-10-185884.
Gallois A, Silva I, Osman I, Bhardwaj N. Reversal of natural killer cell exhaustion by TIM-3 blockade. Onco Targets Ther. 2014;3:e946365. https://doi.org/10.4161/21624011.2014.946365.
Ju Y, Hou N, Meng J, Wang X, Zhang X, Zhao D, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol. 2010;52:322–9. https://doi.org/10.1016/j.jhep.2009.12.005.
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.
Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogenlike protein 1 is a major immune inhibitory ligand of LAG-3. Cell. 2019;176:334–47 e12. https://doi.org/10.1016/j.cell.2018.11.010.
Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. https://doi.org/10.1038/ni.1679.
Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. 2015;15:243–54. https://doi.org/10.1038/nri3799.
Nold-Petry CA, Lo CY, Rudloff I, Elgass KD, Li S, Gantier MP, et al. IL-37 requires the receptors IL-18Ralpha and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat Immunol. 2015;16:354–65. https://doi.org/10.1038/ni.3103.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, Wang Z, Wu Q, Peng H, Wei H, Sun R, Tian Z. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–32.
Niwa R, Sakurada M, Kobayashi Y, Uehara A, Matsushima K, Ueda R, Nakamura K, Shitara K. Enhanced natural killer cell binding and activation by low-fucose IgG1 antibody results in potent antibody-dependent cellular cytotoxicity induction at lower antigen density. Clin Cancer Res. 2005;11(6):2327–36.
Yamane-Ohnuki N, Satoh M. Production of therapeutic antibodies with controlled fucosylation. MAbs. 2009;1(3):230–6.
Pereira NA, Chan KF, Lin PC, Song Z. The “less-is-more” in therapeutic antibodies: afucosylated anti-cancer antibodies with enhanced antibody-dependent cellular cytotoxicity. MAbs. 2018;10(5):693–711.
Pachynski RK, Wang P, Salazar N, Zheng Y, Nease L, Rosalez J, Leong WI, Virdi G, Rennier K, Shin WJ, Nguyen V, Butcher EC, Zabel BA. Chemerin suppresses breast cancer growth by recruiting immune effector cells into the tumor microenvironment. Front Immunol. 2019;10:983.
Helfer G, Wu QF. Chemerin: a multifaceted adipokine involved in metabolic disorders. J Endocrinol. 2018;238(2):R79–94.
Lee J, Kang TH, Yoo W, Choi H, Jo S, Kong K, Lee SR, Kim SU, Kim JS, Cho D, Kim J, Kim JY, Kwon ES, Kim S. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol Res. 2019;7(2):219–29. https://doi.org/10.1158/2326-6066.CIR-18-0317.
van der Ploeg K, Chang C, Ivarsson MA, Moffett A, Wills MR, Trowsdale J. Modulation of human leukocyte antigen-C by human cytomegalovirus stimulates KIR2DS1 recognition by natural killer cells. Front Immunol. 2017;8:298.
Nowakowska P, Romanski A, Miller N, Odendahl M, Bonig H, Zhang C, Seifried E, Wels WS, Tonn T. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol Immunother. 2018;67(1):25–38.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, Gagea M, Banerjee P, Cai R, Bdaiwi MH, Basar R, Muftuoglu M, Li L, Marin D, Wierda W, Keating M, Champlin R, Shpall E, Rezvani K. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–31.
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, Yin J, You F, Zhu M, Shen W, Chen G, Zhu X, Wu D, Yu J. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–9.
Oh E, Min B, Li Y, Lian C, Hong J, Park GM, Yang B, Cho SY, Hwang YK, Yun CO. Cryopreserved human natural killer cells exhibit potent antitumor efficacy against orthotopic pancreatic cancer through efficient tumor-homing and cytolytic ability (Running Title: Cryopreserved NK Cells Exhibit Antitumor Effect). Cancers (Basel). 2019;11(7):E966. https://doi.org/10.3390/cancers11070966.
Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, Trichard S, Cesari C, Sapet M, Bosco F, Rispaud-Blanc H, Guillot F, Cornen S, Roussel A, Amigues B, Habif G, Caraguel F, Arrufat S, Remark R, Romagné F, Morel Y, Narni-Mancinelli E, Vivier E. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. 2019;177(7):1701–1713.e16.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this paper

Cite this paper
Nerina, D., Merlano, M.C. (2021). NK Cells in Immunotherapy: How Important Are They?. In: Vermorken, J.B., Budach, V., Leemans, C.R., Machiels, JP., Nicolai, P., O’Sullivan, B. (eds) Critical Issues in Head and Neck Oncology. Springer, Cham. https://doi.org/10.1007/978-3-030-63234-2_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-63234-2_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-63233-5
Online ISBN: 978-3-030-63234-2
eBook Packages: MedicineMedicine (R0)