
Abstract
Methionine adenosyltransferase (MAT) I/III deficiency can be Inherited as autosomal dominant (AD) or as recessive (AR) traits in which mono- or biallelic MAT1A mutations have been identified, respectively. Although most patients have benign clinical outcomes, some with the AR form have neurological deficits. Here we describe 16 Korean patients with MAT I/III deficiency from 15 unrelated families identified by newborn screening. Ten probands had the AD MAT I/III deficiency, while six had AR MAT I/III deficiency. Plasma methionine (145.7 µmol/L versus 733.2 µmol/L, P < 0.05) and homocysteine levels (12.3 µmol/L versus 18.6 µmol/L, P < 0.05) were lower in the AD type than in AR type. In addition to the only reported AD MAT1A mutation, p.Arg264His, we identified two novel AD mutations, p.Arg249Gln and p.Gly280Arg. In the AR type, four previously reported and two novel mutations, p.Arg163Trp and p.Tyr335*, were identified. No exonic deletions were found by quantitative genomic polymerase chain reaction (PCR). Three-dimensional structural prediction programs indicated that the AD-type mutations were located on the dimer interface or in the substrate binding site, hindering MAT I/III dimerization or substrate binding, respectively, whereas the AR mutations were distant from the interface or substrate binding site. These results indicate that the AD or AR MAT I/III deficiency is correlated with clinical findings, substrate levels and structural features of the mutant proteins, which is important for the neurological management and genetic counseling of the patients.
Similar content being viewed by others

Introduction
Methionine (Met) is the most widely used marker in newborn screening to detect abnormal Met-homocysteine metabolism, especially cystathionine β synthase (CBS) deficiency (MIM #236200), for which patients require urgent medical intervention. However, hypermethioninemia also occurs in other metabolic conditions, including liver disease, tyrosinemia type 1 (MIM #276700), Met adenosyltransferase (MAT) I/III deficiency (MIM #250850), glycine N-methyltransferase (GNMT) deficiency (MIM #606664) and adenosylhomocysteine hydrolase deficiency (MIM #180960) (1,2).
MAT I/III deficiency, the most common cause of persistent hypermethioninemia, is caused by MAT1A mutations (1). MAT1A encodes MAT I, which forms a homotetramer, and MAT III, which forms a homodimer. MAT I/III converts Met to S-adenosylmethionine (SAM). Thus patients with this deficiency develop hypermethioninemia (206–1394 µmol/L; normal plasma range, <50 µmol/L), similar to patients with CBS deficiency (353–1891 µmol/L). However, the plasma homocysteine level is normal or mildly elevated in MAT I/III deficiency (5–26.1 µmol/L; normal plasma range, <15 µmol/L), while it is markedly higher in CBS deficiency (155–471 µmol/L) (1,3).
MAT I/III deficiency can be inherited as autosomal dominant (AD) or as recessive (AR) traits in which mono- or biallelic MAT1A mutations have been identified, respectively (1). The overall incidence of AD and AR MAT I/III deficiency is estimated at 1 in 26,000 ∼ 105,000 newborns (4–6). AD MAT I/III deficiency is more common (58–100% of probands) than the AR type (4–7). In the AD type, p.Arg264His is the only reported mutation to date (8–10), while over 30 mutations have been identified in the AR type (www.hgmd.cf.ac.uk). The AD type has been considered a benign condition, although its long-term prognosis remains unknown (1,4,5,7,11). However, some patients with the AR type may have developmental delay, intellectual disability or brain demyelination (7,11–14). Thus, determination of the inheritance pattern of MAT I/III deficiency is important for the optimal care of the patients as well as for genetic counseling of affected families.
Here, we describe 16 patients from 15 unrelated Korean families with MAT I/III deficiency. Based on the clinical findings, mutation analysis and structural prediction analysis of the mutant protein, the type of MAT I/III deficiency was determined. Notably, two new AD-type mutations were identified, and the differences in the biochemical and molecular structural characteristics were revealed between the AD and AR types.
Materials and Methods
Patients
Newborns with MAT I/III deficiency were identified through neonatal screening from July 2007 to March 2014. At-risk relatives, including siblings and parents of the affected newborns, were screened. Four parents of patients having MAT1A mutation and one sibling with MAT I/III deficiency were identified through family testing. The protocol was approved by the Institutional Review Board of the Asan Medical Center, and written informed consent was obtained from all subjects or their parents.
Biochemical Analysis
Met was measured by tandem mass spectrometry using dried blood spots, obtained 2 to 3 d after birth, and if the level was high, it was measured quantitatively in plasma amino acid chromatography using a 3200 QTRAP® mass spectrometer (Applied Biosystems, AB Sciex). For subjects with a Met level >50 µmol/L, plasma Met and plasma total homocysteine levels were determined by mass spectrometer and direct competitive chemiluminescent enzyme immunoassay (ADVIA Centaur, Bayer), respectively (15,16).
MAT1A Gene Mutation Analysis
Genomic DNA was extracted from peripheral blood leukocytes using Gentra Puregene Blood kits (Qiagen). All nine MAT1A exons (exon I to IX) and exon-intron boundaries were amplified by polymerase chain reaction (PCR) and directly sequenced using an ABI3130x1 Genetic Analyzer (Applied Biosystems). Real-time PCR was performed to assess MAT1A exonic deletion and/or duplication in the genomic DNA. The genomic dosage was quantified by the MJ Research PTC-200 PCR system (Bio-Rad) with HiPi Real-Time PCR SYBR Green (Elpis Biotech, Inc.) using the comparative Ct method. Microsatellite analysis was carried out using the AmpFSTR COfiler (Applied Biosystems). PCR, followed by capillary electrophoresis, was performed according to the manufacturer’s instructions.
Structural Prediction Analysis of the MAT1A Mutant Proteins
The three-dimensional structure of human MAT1A (PDB code 2OBV) was used to model the mutant proteins using PyMOL software (http://www.pymol.org).
Statistical Analysis
Mann-Whitney U tests were performed to compare Met and homocysteine levels between AD and AR forms in MAT I/III deficiency. SPSS for Windows (version 13, SPSS) was used for all statistical analyses, with P <0.05 considered statistically significant.
Results
Sixteen patients from 15 unrelated Korean families were identified through neonatal screening, which was done 3 d after birth. Ten patients from 10 families had monoallelic MAT1A mutations (62.5%), whereas the other six patients from five families had biallelic MAT1A mutations (37.5%).
MAT I/III Deficiency with a Monoallelic MAT1A Mutation
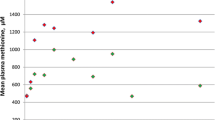
The mean concentrations of plasma Met and homocysteine at diagnosis for the 10 newborns were 145.7 ± 47.1 µmol/L (range 62–207 µmol/L) and 12.3 ± 4.8 µmol/L (6.5–21.7 µmol/L), respectively (Figure 1).

Comparison of plasma methionine and homocysteine levels between the autosomal dominant (AD) form and the autosomal recessive (AR) form in MAT I/III deficiency. (A) The mean of plasma methionine levels at diagnosis and the latest evaluation of the AR form was higher than that of the AD form (P = 0.02, 0.05). (B) The mean of initial and latest homocysteine levels of AR form was higher than that of the AD form (P = 0.04, 0.02). Data represent the mean ± standard deviation. *P < 0.05.
Three different MAT1A mutations, c.746G>A (p.Arg249Gln), c.791G>A (p.Arg264His) or c.838G>A (p.Gly280Arg), were identified in each of the 10 newborns (Table 1). Of these, p.Arg264His was detected in five unrelated newborns and p.Arg249Gln in four, while p.Gly280Arg was found in only one newborn. The variants p.Arg249Gln and p.Gly280Arg were novel and were not found in the 1000 Genomes database (http://browser.1000genomes.org). In silico analysis using the PolyPhen-2 program (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://blocks.fhcrc.org/sift/SIFT.html) predicted both variants as pathogenic. Genomic quantitative PCRs for the nine exons were carried out to rule out a genomic deletion or duplication, and none were detected (subjects 2, 6 and 10) (data not shown).
At the latest evaluation of the 10 newborns at 11.3 ± 5.5 (1–18) months of age, the plasma Met and homocysteine were 126.3 ± 114.5 µmol/L (20–341 µmol/L) and 8.5 ± 3.1 µmol/L (5-11.8 µmol/L), respectively (Figure 1). No patient showed neurologic problems or developmental delay and did not receive any treatment or special diet.
Parental testing was available for three patients (subjects 1, 8 and 9), whereas it was not available in the other seven patients. The paternal inheritance was confirmed in all three families. Met was measured for two fathers (subject 8-1 and 9-1), aged 32 and 38 years, which were 66 and 116 µmol/L, respectively. The fathers had been on normal diets and their intelligence was normal, and all had graduated colleges and were gainfully employed.
MAT I/III Deficiency with Biallelic MAT1A Mutations
Six infants, two males and four females, from five families (subjects 11-14, 14-1 and 15) were identified by newborn screening tests, of which two (subjects 14 and 14-1) were sisters. The mean initial plasma Met and homocysteine concentrations in these newborns were 733.2 ± 425.1 µmol/L (205.2–1352.6 µmol/L) and 18.6 ± 6.2 µmol/L (10-24.4 µmol/L), respectively, which were higher than those in patients with monoallelic mutations (P = 0.02 and 0.04, respectively) (Figure 1).
Biallelic MAT1A mutations were found in all five families (Table 1). Six different mutations, c.895C>T (p.Arg299Cys) (3 of 10 alleles), c.274T>C (p.Tyr92His) (2 of 10 alleles), c.487C>T (p.Arg163Trp) (2 of 10 alleles), c.291G>C (p.Lys97Ala) (1 of 10 alleles), c.595C>T (p.Arg199Cys) (1 of 10 alleles) and c.1005C>G (p.Tyr335*) (1 of 10 alleles), were identified. Of these mutations, p.Tyr335* and p.Arg163Trp were novel and were not found in the 1000 Genomes database. In silico analyses using the PolyPhen-2 and SIFT programs predicted p.Arg163Trp as pathogenic. p.Arg163Trp was inherited from the mother of subject 15, but she had normal plasma Met (19.3 µmol/L) and homocysteine (9.7 µmol/L) levels. The mother of subject 14, a carrier of p.Tyr335*, declined plasma Met and homocysteine measurements.
Subject 15 was a homozygote for p.Arg163Trp. Her mother was heterozygous for the mutation, but her father had only wild-type alleles in his leukocytes. Microsatellite analyses revealed biparental inheritance in the genomic DNA of subject 15. Genomic quantitative PCRs for all MAT1A exons also revealed the biallelic presence of MAT1A in the subject (data not shown).
Three patients (subjects 12, 14 and 15) were given low Met formula for 10 months (1–11 months of age), 17 months (1–18 months of age) and 15 months (1–16 months of age), respectively, which decreased plasma Met level transiently.
At the latest evaluation at a mean age of 35.8 ± 25.6 months (2–72 months of age), the mean plasma Met and homocysteine concentrations were 804.2 ± 645.1 µmol/L and 17.7 ± 5.5 µmol/L, respectively, which were significantly higher than those in patients with a monoallelic mutation (P = 0.05, 0.02) (Figure 1). No patient had clinically detectable neurologic problems or developmental delay at the last examination.
Structural Analyses of the Nine MAT1A Mutant Proteins
The three-dimensional structure of MAT I/III revealed that three residues, p.Arg249, p.Arg264 and p.Gly280, derived from a monoallelic mutation, were located near the dimeric interface of MAT I/III (Figure 2A). In particular, p.Arg264 and p.Gly280 appear facing each other on the dimer interface (Figure 2B). The p.Arg264His amino acid substitution from positively charged amino acid arginine to histidine with an imidazole side chain, (Figure 2D) and that of p.Gly280Arg from a small and nonpolar glycine to arginine with a long, positive-charged side chain are expected to hinder dimer formation (Figure 2E). The other monoallelic mutation, p.Arg249Gln, is also located on the substrate or SAM binding site (Figure 2C), and the substitution from the positive-charged arginine to the uncharged glutamine is expected to disrupt the substrate binding site as well as dimer binding (Figure 2F). The other six mutations, including p.Arg163Trp, p.Lys97Ala, p.Tyr92His, p.Arg199Cys, p.Arg299Cys and p.Tyr335*, which were in homozygous or compound heterozygous combinations, were located distant from the dimeric interface or substrate binding site and were located buried in hydrophobic regions or were on the surface of the protein dimer/tetramer (Figure 2A).

Crystal structure of the human MAT IA. (A) Monoallelic mutations (red color) are located on the dimer interface (red dotted circle) or SAM binding site (blue dotted circle), whereas biallelic mutations (blue color) are located distant from the dimer interface. (B) p.Gly280 and p.Arg264 are located on the dimeric interface, facing each other. (C) p.Arg249 is located on the SAM binding pocket as well as on the dimer interface. (D-F) Three missense mutations (p.Arg249Gln, p.Arg264His and p.Gly280Arg) are shown in black color with their structural changes.
Discussion
Here, we have described the clinical and genetic features of 16 patients from 15 unrelated Korean families with MAT I/III deficiency. Analyses of their plasma Met and homocysteine levels, mutation characteristics and parental inheritance patterns indicated that 10 patients from 10 families with monoallelic MAT1A mutations had the AD type of MAT I/III deficiency, whereas the other six patients from five families with biallelic MAT1A mutations had the AR type. Plasma Met and homocysteine levels were lower in the 10 patients with the AD type than in the six patients with AR type, as expected (Figure 1), which is consistent with previous studies. Although the parental studies were limited to a subset of families—three families with the AD type and one family with the AR type—a high Met level was documented in parents carrying the AD mutation, whereas it was normal in a parent who was a carrier for the AR mutation.
Table 2 summarizes the clinical and genetic findings of the previously reported cases that were genetically confirmed with AD or AR MAT I/III deficiencies. Plasma Met (32–666 µmol/L) and homocysteine levels (∼22.8 µmol/L) were mildly elevated in the AD type, in contrast to very high plasma Met (328–2541 µmol/L) and elevated homocysteine (∼59 µmol/L) levels in the AR type (1,3–9,14). The heterozygous carriers in the AR type had normal plasma Met levels. Regarding clinical outcomes, developmental delay and neurological abnormalities, including dyspraxia, fine nystagmus, diadokokinesis, dystonia and epilepsy, were noted in some patients in the AR-type group and exhibited abnormal myelination in a brain imaging study. On the other hand, myelination abnormality was found in only one patient out of 131 patients with AD type, indicating the difference of clinical outcome between the two subtypes.
In silico protein structural modeling analysis also predicted differences in the nature of the mutant proteins causing the AD and AR subtypes. All three mutations identified in the AD type, p.Arg249Gln, p.Arg264His and p.Gly280Arg, are located on the dimer interface and or substrate binding site. The variant p.Arg264His, the only mutation in the AD type reported to date (8–10), is located on the dimer interface. This p.Arg264His impairs the formation of a homodimer with the other mutant allele or a heterodimer with the wild-type allele as a dominant negative effect (8). One novel mutation, p.Gly280Arg, is also located on the dimer interface facing p.Arg264 (Figure 2B) and is expected to significantly alter the protein structure at the dimer interface and inhibit dimer formation, as does p.Arg264His (Figure 2E). Another novel mutation, p.Arg249Gln, is located on the substrate binding site as well as the dimer interface. Importantly, this amino acid change is expected to hinder substrate binding as well as dimer formation (Figure 2F). In addition, the fathers with p.Arg249Gln, subjects 8-1 and 9-1, had high plasma Met levels as well (Table 1).
Therefore, p.Arg249Gln and p.Gly280Arg are suggested as the new mutations causing the AD form of MAT I/III deficiency. Their resultant effect as a dominant mutation would be supported by further studies, such as in vitro experiments measuring enzymatic activity in cells over-expressing the mutant constructs. Previously, other mutations were also reported as monoallelic mutations, including p.Ser247Arg, p.Ala259Val, p.Arg177Trp, p.Arg219Leu, p.Met138Val, p.Arg249Trp, p.Lys289Gln, p.Ile252Thr, p.Gly257Arg and p.Asp258Gly (7,17–20). It was suggested that a mutation in the other allele would exist; however, any mutation in other alleles was not identified by sequence analysis. In the current study, the possibility of exonic deletion was excluded in any patient with the AD form by genomic quantitative PCRs.
On the other hand, all the missense mutations in the AR type involved amino acid substitutions of residues distant from the dimer interface or the substrate (SAM) binding site, including one novel mutation, p.Arg163Trp. These findings indicated that these mutations might not hinder dimerization or substrate binding completely (Figure 2A). It should be noted, however, that a novel nonsense mutation, p.Tyr335*, was also found in the AR type. Notably, the severe types of mutations, including nonsense, frameshift and splicing mutations, have been found exclusively in patients with the AR type (7,11,14,17,21–24). Because MAT I/III functions as a dimer, these types of mutations appear be unable to encode a monomer so that they cannot be involved in dimerization. Thus, the heterozygous carriers with these mutations have normal or slightly increased Met levels, as described in the previous report, supporting the recessive inheritance of these mutations (Table 2) (25). Unfortunately, parental study was unavailable for subjects 14 and 14-1 with p.Tyr335* in our report.
One novel mutation, p.Arg163Trp, was found as a homozygote in a patient with the AR form. Although her mother was a carrier for the mutation, her father had only the wild type in peripheral leukocytes. Biparental inheritance and no exonic deletion being demonstrated, it was concluded that this mutation occurred de novo on the paternal allele, or her father may have a germline mosaicism for the mutation.
Despite the results of our study, it should be noted that determining the inheritability of MAT I/III deficiency according to the location of a mutation can also be misleading. For example, p.Arg264Cys, located on the same residue as p.Arg264His, has been found in AR inheritance (13,14). In addition, one of the new AD-type mutations suggested in our study, p.Gly280Arg, was identified only in one patient, in whom parental study was not available. Thus, the inheritance pattern should be determined based on a comprehensive evaluation of the pedigree, degree of Met elevation and the location of a mutation with its predictive effect.
In MAT I/III deficiency, plasma homocysteine levels are usually normal but can be as high as 59 µmol/L in some patients (3,19,24). The causes of hyperhomocysteinemia in MAT I/III deficiency are elusive, but hyperhomocysteinemia might be related to high Met levels that inhibit Met synthase or SAM depletion, as suggested in previous observations (1,3,24). Importantly, SAM is involved in neurotransmitter metabolism, antioxidation and myelination (11,13,26). In addition, a high homocysteine level possibly increases the long-term risk of strokes (27). In our study, all the patients had normal or mildly elevated homocysteine and benign clinical courses without any neurological problems, although long-term outcomes remain to be seen.
Generally, treatment is not indicated for mild or transient hypermethioninemia; monitoring plasma Met and homocysteine levels with developmental assessment may be enough as a follow-up evaluation. However, diet modification such as a low Met diet can be considered for patients with persistent severe hypermethioninemia considering high risk for a neurologic deficit and cardiovascular event (13,17,24). However, there is controversy over Met restriction because it can deplete SAM, affecting neurological outcomes, and only a small number of patients have been treated previously (13,18,21,24). The comprehensive analyses of the overall activities of the enzymes and their cofactors involved in the homocysteine-Met cycle with measurement of their metabolites would give more information regarding the biochemical pathology and treatment options such as SAM or betaine supplementation.
Conclusion
In conclusion, the current study indicates that the mutation spectrum may be more variable than has been known for the AD-type MAT I/III deficiency, which needs to be studied in other ethnic groups as well. Comprehensive analyses, including the measurement of plasma Met and homocysteine, the identification of mono- or biallelic mutations, parental study and macromolecular remodeling help to determine MAT I/III deficiency, either in AD or AR types, which is important for the monitoring of a patient in terms of neurological outcome.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Mudd SH. (2011) Hypermethioninemias of genetic and non-genetic origin: a review. Am. J. Med. Genet. C Semin. Med. Genet. 157C:3–32.
Huemer M, et al. (2015) Newborn screening for homocystinurias and methylation disorders: systematic review and proposed guidelines. J. Inherit. Metab. Dis. 38:1007–19.
Stabler SP, et al. (2002) Elevated plasma total homocysteine in severe methionine adenosyltransferase I/III deficiency. Metabolism. 51:981–8.
Martins E, et al. (2012) Methionine adenosyl-transferase I/III deficiency in Portugal: high frequency of a dominantly inherited form in a small area of Douro high lands. JIMD. Rep. 6:107–12.
Couce ML, et al. (2008) Hypermethioninaemia due to methionine adenosyltransferase I/III (MAT I/III) deficiency: diagnosis in an expanded neonatal screening programme. J. Inherit. Metab. Dis. 31 Suppl 2:S233–9.
Couce ML, et al. (2013) Clinical and metabolic findings in patients with methionine adenosyltransferase I/III deficiency detected by newborn screening. Mol. Genet. Metab. 110:218–21.
Nagao M, Tanaka T, Furujo M. (2013) Spectrum of mutations associated with methionine adenosyltransferase I/III deficiency among individuals identified during newborn screening in Japan. Mol. Genet. Metab. 110:460–4.
Chamberlin ME, Ubagai T, Mudd SH, Levy HL, Chou JY. (1997) Dominant inheritance of isolated hypermethioninemia is associated with a mutation in the human methionine adenosyltransferase 1A gene. Am. J. Hum. Genet. 60:540–6.
Nagao M, Oyanagi K. (1997) Genetic analysis of isolated persistent hypermethioninemia with dominant inheritance. Acta. Paediatr. Jpn. 39:601–6.
Perez Mato I, et al. (2001) Biochemical basis for the dominant inheritance of hypermethioninemia associated with the R264H mutation of the MAT1A gene: a monomeric methionine adenosyltransferase with tripolyphosphatase activity. J. Biol. Chem. 276:13803–9.
Chamberlin ME, et al. (1996) Demyelination of the brain is associated with methionine adenosyltransferase I/III deficiency. J. Clin. Invest. 98:1021–7.
Tada H, Takanashi J, Barkovich AJ, Yamamoto S, Kohno Y. (2004) Reversible white matter lesion in methionine adenosyltransferase I/III deficiency. Am. J. Neuroradiol. 25:1843–5.
Furujo M, Kinoshita M, Nagao M, Kubo T. (2012) Methionine adenosyltransferase I/III deficiency: neurological manifestations and relevance of S-adenosylmethionine. Mol. Genet. Metab. 107:253–6.
Chamberlin ME, et al. (2000) Methionine adenosyltransferase I/III deficiency: novel mutations and clinical variations. Am. J. Hum. Genet. 66:347–55.
Dietzen DJ, et al. (2008) Rapid comprehensive amino acid analysis by liquid chromatography/tandem mass spectrometry: comparison to cation exchange with post-column ninhydrin detection. Rapid Commun. Mass Spectrom. 22:3481–8.
Demuth K, Ducros V, Michelsohn S, Paul JL. (2004) Evaluation of Advia Centaur automated chemiluminescence immunoassay for determining total homocysteine in plasma. Clin. Chim. Acta. 349:113–20.
Chien YH, Chiang SC, Huang A, Hwu WL. (2005) Spectrum of hypermethioninemia in neonatal screening. Early Hum. Dev. 81:529–33.
Chadwick S, Fitzgerald K, Weiss B, Ficicioglu C. (2014) Thirteen patients with MAT1A mutations detected through newborn screening: 13 years’ experience. JIMD. Rep. 14:71–6.
Fernandez-Irigoyen J, et al. (2010) Enzymatic activity of methionine adenosyltransferase variants identified in patients with persistent hypermethioninemia. Mol. Genet. Metab. 101:172–7.
Shafqat N, et al. (2013) Insight into S-adenosylmethionine biosynthesis from the crystal structures of the human methionine adenosyltransferase catalytic and regulatory subunits. Biochem. J. 452:27–36.
Hirabayashi K, et al. (2013) Neurologically normal development of a patient with severe methionine adenosyltransferase I/III deficiency after continuing dietary methionine restriction. Gene. 530:104–8.
Kim SZ, et al. (2002) Methionine adenosyltransferase I/III deficiency: two Korean compound heterozygous siblings with a novel mutation. J. Inherit. Metab. Dis. 25:661–71.
Hazelwood S, et al. (1998) Normal brain myelination in a patient homozygous for a mutation that encodes a severely truncated methionine adenosyltransferase I/III. Am. J. Med. Genet. 75:395–400.
Chien YH, et al. (2015) Mudd’s disease (MAT I/III deficiency): a survey of data for MAT1A homozygotes and compound heterozygotes. Orphanet. J. Rare. Dis. 10:99.
Mudd SH, et al. (1995) Isolated persistent hypermethioninemia. Am. J. Hum. Genet. 57:882–92.
Bottiglieri T. (2002) S-Adenosyl-L-methionine (SAMe): from the bench to the bedside—molecular basis of a pleiotrophic molecule. Am. J. Clin. Nutr. 76:1151S–7S.
Linnebank M, et al. (2005) Methionine adenosyltransferase (MAT) I/III deficiency with concurrent hyperhomocysteinaemia: two novel cases. J. Inherit. Metab. Dis. 28:1167–8.
Acknowledgments
This study was supported by a grant from the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology (NRF-2011-0019674 and 2015R1D1A1A01058192).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Kim, YM., Kim, J.H., Choi, JH. et al. Determination of Autosomal Dominant or Recessive Methionine Adenosyltransferase I/III Deficiencies Based on Clinical and Molecular Studies. Mol Med 22, 147–155 (2016). https://doi.org/10.2119/molmed.2015.00254
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2015.00254