
Abstract
The 2014 Ebola outbreak in West Africa triggered a global crisis. Nine countries have reported more than 20 000 infection cases in total and nearly 8000 lives have been lost. The actual death toll is likely much higher than this figure; the death rate is as high as 70%, considering confirmed cases.
The Ebola virus launches its destruction by shutting down the host’s innate and adaptive immune systems. The virus then replicates itself out of control and causes a cytokine storm in the host. Consequently, the host’s overdriven immune system attacks its own endothelial cells and this leads to multiple organ hemorrhagic damage, the host dies of septic shock finally. Under current circumstances where no specific interventions have shown effectiveness against the virus, our opinions are justified in applying a non-specific anti-viral approach during the incubation period of virus infection as an essential protection to put the host’s immune system into an alert state and henceforth to slow down the viral replication. When the viral infection proceeds to the terminal stage, the key factor would be applying a non-specific immune modulation approach to suppress the cytokine storm that causes multiple organ failure, in an attempt to open a time window for the host’s immune system to recover.
概要
埃博拉病毒急性感染具有极高的致死率, 其基本特征表现在机体的先天免疫与过继免疫反应被压制, 病毒疯狂复制引发强烈的细胞因子风暴并攻击血管系统, 大面积细胞凋亡导致脓血症和多脏器功能衰竭, 病人最终因体温下降、 血压过低休克致死。 在缺乏特异性治疗药物干预的情况下, 仍有一部分感染者会自然康复, 说明埃博拉感染是一种自限性疾病, 机体自身的免疫系统在非特异性治疗措施帮助下最终能够清除病毒。 埃博拉病毒部分地阻断了I型干扰素的产生和抗病毒信号途径, 但有证据表明I型干扰素仍能够在一定程度上抑制病毒复制, 潜伏期内予以预防性注射I型干扰素可望大幅度降低感染者体内病毒负载; 间充质干细胞具有强大及可塑性的免疫调节作用, 能够压制细胞因子风暴, 支持血管内皮细胞增殖, 抑制细胞凋亡, 具有向炎症组织趋化和参与修复的能力, 对于埃博拉感染终末期患者采用间充质干细胞治疗有望显著降低死亡风险和后遗症的发生率。 当前绝大多数常见的病毒性感染缺乏特异性治疗药物, 在相当长一段时期内, 非特异性治疗手段仍是对抗病毒性疾病的基本策略。
Similar content being viewed by others

The 2014 Ebola outbreak in West Africa triggered a global crisis. Nine countries have reported more than 20 000 infection cases in total and nearly 8000 lives have been lost. The actual death toll is likely much higher than this figure; the death rate is as high as 70%, considering confirmed cases.
The Ebola virus launches its destruction by shutting down the host’s innate and adaptive immune systems. The virus then replicates itself out of control and causes a cytokine storm in the host. Consequently, the host’s overdriven immune system attacks its own endothelial cells and this leads to multiple organ hemorrhagic damage, the host dies of septic shock finally. Under current circumstances where no specific interventions have shown effectiveness against the virus, our opinions are justified in applying a non-specific anti-viral approach during the incubation period of virus infection as an essential protection to put the host’s immune system into an alert state and henceforth to slow down the viral replication. When the viral infection proceeds to the terminal stage, the key factor would be applying a non-specific immune modulation approach to suppress the cytokine storm that causes multiple organ failure, in an attempt to open a time window for the host’s immune system to recover.
1 What makes the Ebola infection so deadly?
The Ebola virus invades antigen presenting cells (APCs) quietly and turns their alarm system off essentially, so that the immune system remains inactivated toward the virus. The virus then grows uncontrollably and invades many organs. Eventually, many premature cells start dying and exploding, and these cells release all their contents, including signal molecules, into the blood. The signals eventually trigger the extreme immune attacks, which cause arteries, veins and capillaries to leak blood and plasma. The host’s body temperature drops and blood pressure falls, causing the host to go into a severe septic shock.
1.1 Infection of the Ebola virus
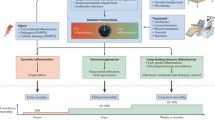
It has been shown that the Ebola virus primarily targets macrophages and dendritic cells (DCs); later, the membrane-associated glycoprotein (GP) in the virus can bind to endothelial cells, causing cell death and vascular permeability. The latter allows the virus to spread to vital organs and execute severe and extensive damage to the body. The symptom of bleeding appears when the virus gains entry into hepatocytes and causes liver failure. Although neutrophils, lymphocytes, and natural killer (NK) cells appear to remain uninfected, those cells undergo ‘bystander apoptosis’, presumably induced by inflammatory mediators and/or the loss of support signals from DCs. Dissemination to regional lymph nodes results in further rounds of virus replication, followed by a spread through the bloodstream to DCs, fixed and mobile macrophages in the liver, spleen, thymus, and other lymphoid tissues, resulting in an extensive tissue necrosis (Mohamadzadeh et al., 2007; Ansari, 2014) (Fig. 1).

System overview of Ebola pathogenesis
This figure was created in reference to Mohamadzadeh et al. (2007) in combination with our own considerations. NK: natural killer; TNF: tumor necrosis factor; IL: interleukin; M-CSF: macrophage colony-stimulating factor; MIP: macrophage inflammatory protein; MCP: macrophage/monocyte chemotactic protein; NO: nitric oxide; sGP: soluble virus glycoprotein
1.2 Impairment of innate and adaptive immunity
The Ebola virus applies multiple physical and biological mechanisms to evade host innate and acquired humoral and cellular immune responses. Interferon (IFN) belongs to the cytokines that are used for communication between cells to trigger the protective defenses of the immune system that help eradicate pathogens. Inhibition of the IFN alarm seems to be one of the most important aspects in the pathogenesis of Ebola. The virus primarily antagonizes both the IFN-α and IFN-β responses in target cells, especially macrophages, monocytes, and DCs, by utilizing the viral protein VP35 to block the phosphorylation of the IFN regulatory factor 3 (IRF3), which acts as a transcription factor for the IFN production (Cárdenas et al., 2006), whereas VP24 to block the IFN-mediated antiviral response (Xu et al., 2014). Macrophages play a critical role in nonspecific defense, and they also help to initiate specific defense mechanisms by recruiting other immune cells such as lymphocytes. Massive infection of macrophages and related cells by the Ebola virus indicates that it is able to block or evade the cells’ innate antiviral mechanisms. The NK cells respond in an antigen-independent manner to viral infections and kill infected cells. NK cell numbers dramatically drop during the course of infection and almost all undergo apoptosis. DCs have a crucial role in both innate and adaptive immunity; however, infected DCs do not produce proinflammatory cytokines or costimulatory molecules, their ability to support T-cell proliferation is thus impaired and the infected DCs undergo anomalous maturation (Hoenen et al., 2006). Impaired APCs function and lymphocyte apoptosis contribute to the failure of specific immune responsiveness. Ebola triggers the systemic dysregulation of immunity that likely results in an uncontrolled virus replication.
1.3 Severe attacks of dysregulated immune system
Macrophage-produced pro-inflammatory cytokines are the key players of the host defense system against pathogens. In most patients, Ebola viral burden elevates by time and triggers an extremely strong immune attack—a phenomenon called ‘cytokine storm’ (Sullivan et al., 2003), during which monocytes and/or macrophages produce a massive amount of pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α) and interleukins (ILs), and chemokines, such as macrophage inflammatory protein 1α/β (MIP-1α/β), macrophage/monocyte chemotactic protein-1 (MCP-1), macrophage colony-stimulating factor (M-CSF), nitric oxide (NO), and eotaxin (Baize et al., 2002). By now, the body response to Ebola infection would be too severe and too late, and the cytokine storm leads to exaggerated inflammatory responses that contribute to lymphoid cell apoptosis and sepsis (Ansari, 2014). The virus eventually disables the vascular system and causes blood leakage combined with massive viremia and intravascular coagulopathy. The terminal stage of the virus infection usually includes diffusive bleeding and hypotensive shock, which would eventually kill the patients.
2 What are the differences among fatal, survival, and asymptomatic Ebola infections?
Ebola infection is generally composed of the incubation period, the symptomatic/acute stage, and the convalescent/terminal stage. The virus burden, inflammatory response, and specific antibodies are the main contributors to different outcomes: mortality, survival, or symptomless infection (Fig. 2), suggesting that the appropriate intervention strategy in each stage would accordingly be able to control the Ebola virus.

Abridged general view of different profiles of immune responses in fatal (a), survival (b), and asymptomatic (c) Ebola infections
2.1 Double edges of the inflammatory response
Whether an inflammatory response executes protective or damaging effects depends not only on the specific cytokine profile, but also on a delicate balance between the individual host immune response and the incoming virus (Sullivan et al., 2003).
Some patients have a delayed and prolonged inflammatory response that leads to a cytokine storm characterized by extremely high circulating levels of numerous pro-inflammatory cytokines; these patients generally cannot survive in the viral infection (Misasi and Sullivan, 2014). The average levels of the proinflammatory cytokines in these patients’ blood have been found to be 5–1000 times higher than those in the patients who recovered from the viral infection (Wauquier et al., 2010; Ansari, 2014).
Survivors tend to have a short-lived, balanced pro- and anti-inflammatory responses characterized by the presence of IL-1β, IL-6, and TNF-α in the early clinical course (Bray and Mahanty, 2003), but no IFN-á was detected in either survivors or nonsurvivors (Wauquier et al., 2010).
Some viral infection cases are asymptomatic. An immediate, proper inflammatory response is critical to these cases, which is characterized by transient high levels of IL-1β, IL-6, TNF-α;, MCP-1, and MIP-1α;/β in plasma within 7 d of the first putative infectious contact (Baxter, 2000; Leroy et al., 2001; Baize et al., 2002). It is believed that in these cases, viral replications are suppressed by effective inflammatory responses.
Why some infected patients can develop a proper inflammatory response, whereas the others cannot, remains unclear. Nonetheless, this phenomenon does give clues to the treatment against Ebola virus disease (EVD)—early activated innate immune responses may prevent the viral infection.
2.2 Distinction of specific antibodies
Prompt and appropriate adaptive immune responses to Ebola seem to be important to the resolution of infection. Both virus-specific humoral and cellular mechanisms are required for clearing the viral infection; the humoral immunity is more important at the acute stage for halting viral spread, whereas the cellular immunity is more important for eliminating virus-infected cells that could continuously serve as a source of virus (Ansari, 2014).
Fatal cases have been found to associate with a defective humoral response without specific IgG production, in these cases only low levels of specific IgM were detected in 30% of the patients (Baize et al., 1999; Ksiazek et al., 1999).
Survivors were generally associated more significantly with the early emergence of specific humoral responses and regulated activation of cytotoxic cells that coincided with clearance of viral antigens from the blood. Survivors produced specific IgM as early as in the first 2 d of symptoms onset and specific IgG in 5–8 d (Baize et al., 1999; Ksiazek et al., 1999).
Asymptomatic individuals produced specific IgM and IgG within 2–3 weeks after initial exposure to viral sources, these antibody productions reached only moderate levels one month later (Leroy et al., 2000).
2.3 Diversity of the virus burden
The outcome of an early viral load is unclear as the viral titers in the plasma were undetectable during the incubation period, whereas the virus load was similar between fatalities and survivors during the first days of the symptom stage.
Typically, 10 million or more copies of the Ebola virus per milliliter in plasma can be reached as early as two days after the onset of symptoms in fatal cases. The level of circulating antigens keeps rising until death. Moreover, the virus load at or near the time of death can be 100-fold greater in fatal cases in comparison with nonfatal cases (Schieffelin et al., 2014).
Survivors’ plasma often contained fewer than 100 000 copies of the virus per milliliter of plasma. The level of circulating antigens began declining 3 d after the onset of symptoms and dropped to an undetectable level when the patients recovered (Baize et al., 2002; Hoenen et al., 2006).
The viral load was the lowest in asymptomatic individuals, but the virus RNA was detectable in these subjects for up to three weeks after initial exposure (Leroy et al., 2001; Baize et al., 2002).
2.4 Depletion of relevant cells
NK cells and lymphocytes are not infected by Ebola virus, but they undergo massive ‘bystander apoptosis’ (Hoenen et al., 2006). A strong depletion of both CD4+ and CD8+ lymphocytes and plasma cells was found in fatal cases (Geisbert et al., 2000), followed by extensive intravascular apoptosis, vascular dysfunction, and loss of endothelial barrier function, which kill the patients (Baize et al., 1999).
3 A prospective strategy
In the symptomatic stage, the supportive care is mainly toward aggressive prevention of intravascular volume depletion, correction of profound electrolyte-abnormalities, and prevention of the shock complications (Fowler et al., 2014). There is neither precaution for the incubation period nor effective medication for the terminal stage. We propose that a preventative antiviral intervention for the incubation period may lower the consequent virus burden. Furthermore, we propose that an immunomodulatory strategy for the terminal stage may reduce the damages caused by the cytokine storm, thus prolonging the survival time of the patients, making it possible for the patients’ adoptive immunity to recover and beat the infection.
3.1 Type I IFN intervention during the incubation period may render a beneficial outcome
Type I IFNs are cytokines that are secreted by infected cells. They induce cell-intrinsic antiviral states in infected and neighbor cells, they also modulate innate immune responses in a balanced manner and activate the adaptive immune system (le Bon and Tough, 2002; Ivashkiv and Donlin, 2014) (Fig. 3).

Type I IFN controls innate and adaptive immunity and intracellular antiviral programs
This figure was created in reference to Ivashkiv and Donlin (2014) in combination with our own considerations
Type I IFNs have a broad spectrum of antiviral capability, which is able to fight most virus infections. To give an example, the recombinant IFN-α2b has been shown to have a significant nonspecific inhibition of herpes simplex virus, influenza virus, and severe acute respiratory syndrome (SARS) coronary virus replication (Cao et al., 2011).
The Ebola virus blocks the production of type I IFN by APCs including DCs and macrophages; it also blocks the IFN-mediated antiviral response by virus proteins. It is a crucial mechanism whereby the viruses evade attacks from the host immune system directly. However, recombinant IFN-α2b (200 IU/ml) can suppress Ebola replication by 100-folds in Vero cells in vitro, early treatment of Ebola-infected cynomolgus with recombinant IFN-α2b delayed onset of viremia and death by several days (Jahrling et al., 1999). In addition, IFN-β treatment was associated with reducing the plasma and tissue viral burden; it thus significantly increased survival time in macaques infected with the Ebola virus in vivo (Smith et al., 2013).
Collectively, these results indicate that type I IFN may have therapeutic potential: it is reasonable to postulate direct effects on viral replication as well as adaptive immune response. The observation that the virus titers were not detectable in the incubation period indicates that the vast majority of cells were not infected by the Ebola virus. Based on this fact, we suggest that exogenous administration of type I IFN may induce uninfected cells into an antiviral state. Type I IFN might limit the spread of the Ebola virus and prolong survival if administered immediately after exposure to Ebola viruses.
3.2 Mesenchymal stromal cell therapy during the terminal stage may prevent cytokine storm, massive cell apoptosis, and septic shock
Some patients indeed recovered from the Ebola infection without receiving specific interventions despite Ebola infection’s high mortality rate. This fact suggests that appropriate immune responses can help the body heal itself. The outcome highly depends on two cellular-level competing mechanisms: the apoptosis of endothelial cells executed by autoimmune attacks and vascular regeneration by stem cells. Interestingly, the patients under the age of 21 years had a lower fatality rate of 57%, whereas the fatality rate was up to 94% in those over the age of 45 years, between these two age groups, the fatality rate was 74% (Schieffelin et al., 2014). Stem cells are the essential for the regenerative processes of almost all tissues and organs. The significant differences indicate that patients’ cellular-level healing capacity in different age groups may be relevant to the consumption of age-related changes in the stem cell reservoir. We suggest that treatment with mesenchymal stromal cells (MSCs) during the terminal stage of EVD should be considered.
MSCs are mesoderm-origin, multipotent cells that exist in many tissues and are capable of differentiating into several different cell types, and the numbers or potential of MSC populations in adult organs decline during aging (Stolzing et al., 2008; Toledano et al., 2012). After exogenous administration, MSCs migrate to injured tissue sites where they can inhibit the release of pro-inflammatory cytokines and promote the survival of damaged cells. MSCs operate through a variety of effector mechanisms on key cells of the innate and adaptive immune systems, mostly through manipulating the cell cycle or inducing maturation arrest without apoptosis (Tyndall and Pistoia, 2009). The therapeutic effects of MSCs may depend largely on the capacity of MSCs to regulate inflammation and tissue homeostasis via an array of immunosuppressive factors, cytokines, growth factors, and differentiation factors. MSCs reduce inflammation by shutting down the TNF-α pathway for immune cell activation and prevent a cytokine storm by inhibiting or disabling T-cell response. MSCs reprogram macrophages, neutrophils, NK cells, DCs, T lymphocytes, and B lymphocytes, all of which counteract sepsis (Plock et al., 2013) (Fig. 4).

Immunological function of MSCs on different cell types of the innate/adaptive immunosystem while MSCs diminish damage and induce repair
This figure was created in reference to Plock et al. (2013) in combination with our own considerations. NK: natural killer; IL: interleukin; PGE2: prostaglandin E2; IDO: indoleamine 2,3-dioxygenase; sHLA-G5: soluble human leukocyte antigen-G5; TNFR: tumor necrosis factor receptor; IFN: interferon; EGF: epidermal growth factor
MSCs can specifically communicate with the inflammatory microenvironment and this immunoregulatory function of MSCs is highly plastic (Wang et al., 2014). While stimulating tissue repair by mitogenic and angiogenic effects, MSCs inhibit ongoing inflammation, apoptosis, and later fibrosis of injured tissue, and support endothelial cell growth and blood vessel repair; this strategy can help avoid the abuse of steroid hormones and various sequelae. It has been reported that graft-versus-host disease (GVHD), systemic lupus erythematosus (SLE), and sepsis can be successfully treated by MSCs (Kebriaei et al., 2009; Sun et al., 2009; Wannemuehler et al., 2012; Pedrazza et al., 2014). It would be interesting to use the available nonhuman primate models of EVD to test such a therapeutic hypothesis.
3.3 Non-specific treatment as an essential strategy against viral diseases in the foreseeable future
It has been almost 40 years since the first Ebola outbreak in 1976. To date, there are no effective therapeutic or prophylactic interventions available to prevent this infection. Several experimental interventions are in early stages of development, and their availability is limited and intermittent (Zhang and Wang, 2014). Positive results were observed in several cases where ZMapp and TKM-Ebola were administrated, but it was unclear whether the positive outcome was due to the drugs or due to better supportive care in Western countries than that of Africa. Clinical trial on an Ebola vaccine developed by Merck and NewLink has been suspended due to unexpected side effects recently. Meanwhile, Médecins Sans Frontières (MSF) has selected three existing interventions for clinical trials, they are Favipiravir approved in Japan for treating influenza, Brincidofivir approved in the USA for virus treatment, and Ebola convalescent serum, and they might all be present less of a supply challenge or are already approved for other purposes. However, preliminary data show that their potency against the Ebola virus is limited. The outlook for the development of Ebola therapeutics is not optimistic.
There are many different kinds of viruses. These viruses mutate their DNA/RNA signatures and evolve rapidly. There has been no specific therapy for even the most common human viral infection such as HBV, HCV, HIV, HPV, and avian influenza. Realistically, the non-specific treatment is an essential strategy against viral diseases in the foreseeable future. When our immune system is given sufficient time for intentional activation when we are exposed to deadly viruses, there is a good chance that it can gear up and eliminate the viruses by itself.
Compliance with ethics guidelines
Lei ZHANG, Hao WANG, and Yi-qing ZHANG declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Ansari, A.A., 2014. Clinical features and pathobiology of Ebolavirus infection. J. Autoimmun., 55:1–9. [doi:10.1016/j.jaut.2014.09.001]
Baize, S., Leroy, E.M., Georges-Courbot, M.C., et al., 1999. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med., 5(4):423–426. [doi:10.1038/7422]
Baize, S., Leroy, E.M., Georges, A.J., et al., 2002. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol., 128(1):163–168. [doi:10.1046/j.1365-2249.2002.01800.x]
Baxter, A.G., 2000. Symptomless infection with Ebola virus. Lancet, 355(9222):2178–2179. [doi:10.1016/S0140-6736(00)02394-1]
Bray, M., Mahanty, S., 2003. Ebola hemorrhagic fever and septic shock. J. Infect. Dis., 188(11):1613–1617. [doi:10.1086/379727]
Cao, L., Zhang, L., Tian, L., et al., 2011. Antivirus evaluation and clinical evaluation of recombinant human interferon α2b spray. Chin. Med. Biotechnol., 6(5):336–340 (in Chinese). [doi:10.3969/cmba.j.issn.1673-713X.2011.05.004]
Cárdenas, W.B., Loo, Y.M., Gale, M., et al., 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol., 80(11):5168–5178. [doi:10.1128/JVI.02199-05]
Fowler, R.A., Fletcher, T., Fischer, W.A., et al., 2014. Caring for critically ill patients with Ebola virus disease. Perspectives from West Africa. Am. J. Respir. Crit. Care Med., 190(7): 733–737. [doi:10.1164/rccm.201408-1514CP]
Geisbert, T.W., Hensley, L.E., Gibb, T.R., et al., 2000. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest., 80(2):171–186. [doi:10.1038/labinvest.3780021]
Hoenen, T., Groseth, A., Falzarano, D., et al., 2006. Ebola virus: unravelling pathogenesis to combat a deadly disease. Trends Mol. Med., 12(5):206–215. [doi:10.1016/j.molmed.2006.03.006]
Ivashkiv, L.B., Donlin, L.T., 2014. Regulation of type I interferon responses. Nat. Rev. Immunol., 14(1):36–49. [doi:10.1038/nri3581]
Jahrling, P.B., Geisbert, T.W., Geisbert, J.B., et al., 1999. Evaluation of immune globulin and recombinant interferon-α2b for treatment of experimental Ebola virus infections. J. Infect. Dis., 179(s1):S224–S234. [doi:10.1086/514310]
Kebriaei, P., Isola, L., Bahceci, E., 2009. Adult human mesenchymal stem cells added to corticosteroid therapy for the treatment of acute graft-versus-host disease. Biol. Blood Marrow Transpl., 15(7):804–811. [doi:10.1016/j.bbmt.2008.03.012]
Ksiazek, T.G., Rollin, P.E., Williams, A.J., et al., 1999. Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J. Infect. Dis., 179(S1):S177–S187. [doi:10.1086/514321]
le Bon, A., Tough, D.F., 2002. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol., 14(4):432–436. [doi:10.1016/S0952-7915(02)00354-0]
Leroy, E.M., Baize, S., Volchkov, V.E., et al., 2000. Human asymptomatic Ebola infection and strong inflammatory response. Lancet, 355(9222):2210–2215. [doi:10.1016/S0140-6736(00)02405-3]
Leroy, E.M., Baize, S., Debre, P., et al., 2001. Early immune responses accompanying human asymptomatic Ebola infections. Clin. Exp. Immunol., 124(3):453–460. [doi:10.1046/j.1365-2249.2001.01517.x]
Misasi, J., Sullivan, N.J., 2014. Camouflage and misdirection: the full-on assault of Ebola virus disease. Cell, 159(3): 477–486. [doi:10.1016/j.cell.2014.10.006]
Mohamadzadeh, M., Chen, L., Schmaljohn, A.L., 2007. How Ebola and Marburg viruses battle the immune system. Nat. Rev. Immunol., 7(7):556–567. [doi:10.1038/nri2098]
Pedrazza, L., Lunardelli, A., Luft, C., et al., 2014. Mesenchymal stem cells decrease splenocytes apoptosis in a sepsis experimental model. Inflamm. Res., 63(9):719–729. [doi:10.1007/s00011-014-0745-1]
Plock, J.A., Schnider, J.T., Solari, M.G., et al., 2013. Perspectives on the use of mesenchymal stem cells in vascularized composite allotransplantation. Front. Immunol., 4:175. [doi:10.3389/fimmu.2013.00175]
Schieffelin, J.S., Shaffer, J.G., Goba, A., et al., 2014. Clinical illness and outcomes in patients with Ebola in Sierra Leone. N. Engl. J. Med., 371(22):2092–2100. [doi:10.1056/NEJMoa1411680]
Smith, L.M., Hensley, L.E., Geisbert, T.W., et al., 2013. Interferon-β therapy prolongs survival in rhesus macaque models of Ebola and Marburg hemorrhagic fever. J. Infect. Dis., 208(2):310–318. [doi:10.1093/infdis/jis921]
Stolzing, A., Jones, E., McGonagle, D., et al., 2008. Age-related changes in human bone marrow-derived mesenchymal stem cells: consequences for cell therapies. Mech. Ageing Dev., 129(3):163–173. [doi:10.1016/j.mad.2007.12.002]
Sullivan, N., Yang, Z.Y., Nabel, G.J., 2003. Ebola virus pathogenesis: implications for vaccines and therapies. J. Virol., 77(18):9733–9737. [doi:10.1128/JVI.77.18.9733-9737.2003]
Sun, L., Akiyama, K., Zhang, H., et al., 2009. Mesenchymal stem cell transplantation reverses multiorgan dysfunction in systemic lupus erythematosus mice and humans. Stem Cells, 27(6):1421–1432. [doi:10.1002/stem.68]
Toledano, H., D’Alterio, C., Czech, B., 2012. The let-7-Imp axis regulates ageing of the Drosophila testis stem-cell niche. Nature, 485(7400):605–610. [doi:10.1038/nature 11061]
Tyndall, A., Pistoia, V., 2009. Mesenchymal stem cells combat sepsis. Nat. Med., 15(1):18–20. [doi:10.1038/nm0109-18]
Wang, Y., Chen, X., Cao, W., et al., 2014. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat. Immunol., 15(11): 1009–1016. [doi:10.1038/ni.3002]
Wannemuehler, T.J., Manukyan, M.C., Brewster, B.D., et al., 2012. Advances in mesenchymal stem cell research in sepsis. J. Surg. Res., 173(1):113–126. [doi:10.1016/j.jss.2011.09.053]
Wauquier, N., Becquart, P., Padilla, C., et al., 2010. Human fatal Zaire Ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl. Trop. Dis., 4(10):e837. [doi:10.1371/journal.pntd.0000837]
Xu, W., Edwards, M.R., Borek, D.M., et al., 2014. Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell Host Microbe, 16(2): 187–200. [doi:10.1016/j.chom.2014.07.008]
Zhang, L., Wang, H., 2014. Forty years of the war against Ebola. J. Zhejiang Univ.-Sci. B (Biomed. & Biotechnol.), 15(9):761–765. [doi:10.1631/jzus.B1400222]
Acknowledgements
We thank Paul LEUFKENS of Neurophyxia B.V. (the Netherlands) for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
ORCID: Lei ZHANG, http://orcid.org/0000-0002-0638-6042/0000-0002-0638-6042
Rights and permissions
About this article

Cite this article
Zhang, L., Wang, H. & Zhang, Yq. Against Ebola: type I interferon guard risk and mesenchymal stromal cell combat sepsis. J. Zhejiang Univ. Sci. B 16, 1–9 (2015). https://doi.org/10.1631/jzus.B1400365
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1631/jzus.B1400365