
Abstract
Macitentan is a dual endothelin receptor antagonist under phase 3 investigation in pulmonary arterial hypertension. We investigated the effect of cyclosporine (Cs) and rifampin on the pharmacokinetics of macitentan and its metabolites ACT-132577 and ACT-373898 in healthy male subjects. In addition, in vitro studies were performed to investigate interactions between macitentan and its active metabolite ACT-132577 with human organic anion-transporting polypeptides (OATPs). The clinical study (AC-055-111) was conducted as a two-part, one-sequence, crossover study. Ten subjects in each part received multiple-dose macitentan followed by multiple-dose co-administration of Cs (part A) or rifampin (part B). In the presence of Cs, steady-state area under the plasma concentration–time profiles during a dose interval (AUCτ) for macitentan and ACT-373898 increased 10% and 7%, respectively, and decreased 3% for ACT-132577. Steady-state AUCτ of macitentan and ACT-373898 in the presence of rifampin decreased 79% and 64%, respectively. For ACT-132577, no relevant difference in AUCτ between the two treatments was observed. Macitentan co-administered with Cs or rifampin was well tolerated. The complementary in vitro studies demonstrated no marked differences in uptake rates of macitentan and ACT-132577 between the wild-type and OATP over-expressing cells over the concentration range tested. Concomitant treatment with Cs did not have any clinically relevant effect on the exposure to macitentan or its metabolites, at steady-state. Concomitant treatment with rifampin reduced significantly the exposure to macitentan and its metabolite ACT-373898 at steady-state but did not affect the exposure to the active metabolite ACT-132577 to a clinically relevant extent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
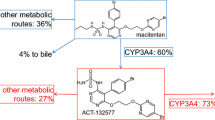
Macitentan (ACT-064992) is an orally active, tissue-targeting dual endothelin ETA and ETB receptor antagonist (ERA) under phase 3 investigation in pulmonary arterial hypertension (1, 2). In humans, macitentan shows a slow absorption; maximum plasma concentrations are achieved approximately 8 h after administration. In addition to macitentan, two metabolites, ACT-132577 (depropylated metabolite) and ACT-373898 (carboxylic acid metabolite), have been identified in plasma with ACT-132577 being pharmacologically active. While the elimination half-life (t 1/2) is approximately 16 h for macitentan, ACT-132577 has a t 1/2 of approximately 48 h. ACT-373898 has a similar pharmacokinetic (PK) profile to macitentan. There are no notable differences in PK parameters of macitentan and its active metabolite between males and females and/or between Caucasian and Japanese subjects (3). In addition, macitentan can be given to subjects irrespective of food intake.
Early phase studies have shown macitentan to have a good safety and tolerability profile. The most frequently reported adverse event (AE) is headache (2,4).
Cyclosporine (Cs) and rifampin were used to investigate the role of metabolism and drug transport in the disposition of macitentan. The immunosuppressant Cs is an inhibitor of CYP3A4, the hepatic uptake transporter organic anion-transporting polypeptide (OATP) (5), and the efflux transporter P-glycoprotein (6). Rifampin, an antibiotic mainly used for the treatment of tuberculosis, is a strong inducer of CYP3A4 and an effective inhibitor of OATP (7–9). As the metabolism of macitentan mainly relies on CYP3A4, the influence of these two drugs on the PK of macitentan is of particular interest (10). In addition, drug–drug interactions (DDIs) at the level of hepatic OATPs have been documented following co-administration of other structurally related ERAs and Cs (11,12). As Cs and rifampin may be used concomitantly with macitentan in clinical practice, we investigated in a two-part, one-sequence, crossover study the effect of multiple-dose Cs (part A) and rifampin (part B) on the PK of macitentan and its metabolites. In addition, in vitro studies were performed to investigate interactions between macitentan and its active metabolite ACT-132577 with human OATP transporters.
MATERIALS AND METHODS
Subjects
It was planned to enroll a total of 20 subjects, 10 in part A and 10 in part B. Healthy male subjects (based on medical history and screening examination) aged between 18 and 45 years, with body mass index (BMI) between 18 and 28 kg/m2, were eligible for this study. In addition, eligible subjects had to have hematology and clinical chemistry results within the normal range and negative results for drug and alcohol use at screening. Subjects were excluded from the study if they had received any drug with immunosuppressive potential within 6 months preceding the screening examination or if they were administered any metabolic enzyme-inducing or enzyme-inhibiting drugs within the last 4 weeks prior to the screening examination.
The ethics committee, Comité de Protection des Personnes Ouest VI, located in Brest (France), approved the study protocol (study number AC-055-111), and all subjects gave written informed consent before any screening procedures were performed. The study was conducted in full conformity with the principles of the Declaration of Helsinki, the EMEA Note for Guidance on Good Clinical Practice (CPMP/ICH/135/95), and International Ethical Guidelines for Biomedical Research Involving Human Subjects (Council for International Organizations of Medical Sciences 2002).
Clinical Study Design
This phase 1, single-center, open-label, two-part, one-sequence crossover study consisted of two parts. Each part comprised two periods: period 1, multiple-dose treatment with macitentan, and period 2, multiple-dose treatment with macitentan and Cs (part A) or multiple-dose treatment with macitentan and rifampin (part B).
An oral dose of 10 mg macitentan once daily was assessed in this study, which corresponds to the maximum dose used in the ongoing phase 3 program. The use of a 30-mg loading dose on day 1 reduced the time to reach steady-state conditions of ACT-132577 (day 4 instead of day 7). In the single-ascending dose study, doses of up to 300 mg had been well tolerated (2). In part A, after 5 days administration of macitentan, subjects were co-administered oral doses of 100 mg of Cs (1 × 100 mg capsule of Neoral®) every 12 h on days 6 through 17. The dose of 2 × 100 mg/day of Cs used in this study corresponds to approximately 2.9 mg/kg/day for a 70-kg man. This is within the recommended range for transplantation indications, i.e., 2 to 6 mg/kg/day, and is below the dose level of 5 mg/kg/day known to potentially increase creatinine and hypertension side effects (13). Both drugs were administered after food.
In part B, after 5 days administration of macitentan, oral doses of 600 mg rifampin (two capsules of 300 mg Rifadin®) were co-administered once daily for 7 days, on days 6 through 12. This dosing regimen is approved for the treatment of tuberculosis and is recommended by the Food and Drug Administration (FDA) for assessment of its enzyme-inducing effect (14,15). Since food intake has a significant effect on the PK of rifampin, drugs were administered under fasted conditions (1 h before a meal).
An End of Study examination, including vital signs, electrocardiogram (ECG), and clinical laboratory tests, took place 2 weeks after the last PK blood sampling, i.e., on day 32 (±2 days) of part A and on day 27 (±2 days) of part B. A telephone safety follow-up for serious adverse events (SAEs) took place 30 days after the last study drug administration.
Trough concentrations of macitentan and its metabolites were determined in blood samples taken from days 1 to 17 (part A) and from days 1 to 12 (part B) prior to daily macitentan administration. Twenty-four-hour concentration–time profiles of macitentan and its metabolites on day 5 (96–120 h) and day 17 (384–408 h) (part A) or day 12 (264–288 h) (part B) were determined in blood samples drawn immediately before and at 1, 3, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 16, and 24 h after macitentan administration.
For each time point, approximately 3 mL of blood was collected from the subject in tubes containing ethylene diamine tetra-acetic acid. Within 30 min after collection, the tubes were centrifuged at approximately 1,500×g for 10 min at 2°C to 8°C. The plasma was stored at −20°C until analysis.
Bioanalytical Methods and PK Assessments
Quantification of macitentan, ACT-132577, and ACT-373898 in plasma was performed by a validated liquid chromatography with tandem mass spectrometry (LCMS/MS) method. Analytes were extracted from plasma by protein precipitation with ethanol (EtOH)/acetonitrile (50:50, v/v) containing the internal standards (polydeuterated analogs of macitentan, ACT-132577, and ACT-373898) and filtered through a protein precipitation plate (0.2 μm pore size), before injection into the high performance liquid chromatography system. The samples were subsequently analyzed by triple-stage quadrupole tandem mass spectrometry (MS/MS). The concentrations of the analytes were determined using the internal standardization method. The square of the correlation coefficient (R 2) was at least 0.9985 for macitentan, 0.9863 for ACT-132577, and 0.9932 for ACT-373898 in all runs. For macitentan, the inter-day precision (coefficient of variation) of control samples was ≤8.6%, and inter-day inaccuracy ranged from −4.3% to 5.3%. For ACT-132577, inter-day precision was ≤7.0%, and inter-day inaccuracy ranged from −5.7% to −2.0%. For ACT-373898, inter-day precision was ≤10.1%, and inter-day inaccuracy was in the range of −9.3% to 1.8%. The lower limit of quantification (LLOQ) was 1 ng/mL for macitentan and ACT-132577, and 0.5 ng/mL for ACT-373898.
The measured individual plasma concentrations of macitentan, ACT-132577, and ACT-373898 were used to calculate PK parameters by model-independent analysis, using the WinNonlin 6.1 or Phoenix software packages (Pharsight Corporation, Mountain View, CA, USA). The measured individual plasma concentrations immediately before the morning dose of macitentan were used to directly obtain trough concentrations (C tr). The area under the plasma concentration–time curve (AUC) was calculated according to the linear trapezoidal rule, using the measured concentration–time values above the LLOQ.
Safety Assessments
Tolerability and safety were evaluated by recording SAEs, AEs, clinical laboratory variables, vital signs, and 12-lead ECG during the study.
Statistical Analysis
The effect of Cs and rifampin on the steady-state area under the plasma concentration time curve during a dose interval (AUCτ) and C tr of macitentan and its metabolites were explored using the ratio of geometric means and its 90% confidence intervals of macitentan + Cs (or macitentan + rifampin) versus macitentan. Tolerability and safety endpoints were analyzed descriptively.
In Vitro Experiments
Chinese Hamster Ovary Cell Culture
Wild-type and Chinese hamster ovary (CHO) cells over-expressing OATP1B1 and OATP1B3 were maintained in Dulbecco's modified eagle's medium supplemented with 10% (v/v) fetal calf serum, penicillin/streptomycin (100 IU/mL), and l-proline (0.05 mg/mL) at 37°C in a humidified atmosphere containing 5% CO2. The culture medium for transfected cells also contained geneticin (500 μg/mL). For transport experiments, cells were seeded 1:10 or 1:20 on 8-cm2 tissue culture dishes (Vitaris, Zug, Switzerland) 3 to 4 days before assaying. One day before the experiments, 5 μM sodium butyrate (Sigma–Aldrich, Buchs, Switzerland) was also added.
Transport Experiments
Transport experiments with wild-type and transfected CHO cells were performed in parallel using two to three 8-cm2 tissue culture dishes for each concentration. After washing the cells three times with a pre-warmed (37°C) buffer (5.3 mM KCl, 1 mM NaH2PO4, 0.8 mM MgSO4, 5.5 mM d-glucose, 20 mM HEPES, 116.4 mM NaCl, pH 7.4 at room temperature), uptake of a test compound was initiated by adding 1 mL of the same buffer containing the test compound at desired concentration (as 14C-labeled alone or as a mixture with non-labeled test compound). The final concentration of organic solvent was 1% (v/v). Uptake of a test compound was stopped after incubation at 37°C for the desired duration by twice adding 2-mL aliquots of ice-cold buffer containing 0.5% (w/v) bovine serum albumin (BSA) (Sigma, Buchs, Switzerland). Cells were then rapidly washed four times with approximately 2 mL of ice-cold buffer and solubilized by the addition of 1 mL of 1% (w/v) Triton X-100. After incubation for at least 20 min, cell lysate (0.5 mL) was mixed with 5 mL of scintillation cocktail IRGA Safe Plus (PerkinElmer, Zurich, Switzerland), and radioactivity was determined using a Tri-Carb 2300 TR liquid scintillation analyzer (Packard Bioscience, Zurich, Switzerland). Aliquots of cell lysates (25 μL) were used to determine the protein content of each sample using the Pierce BCA (bicinchoninic acid) assay (Pierce Science, Lausanne, Switzerland) according to the supplier's protocol. BSA was used as standard.
The inhibitory potential of macitentan and ACT-132577 on the cellular uptake of prototypical OATP substrates was investigated at a single substrate concentration (≤K m) of 1 μM atorvastatin for OATP1B1 and 5 μM estrone-3-sulfate for OATP1B3. After incubation at 37°C for 40 s (OATP1B1) and 90 s (OATP1B3), the uptake was terminated by twice adding 2 mL of ice-cold buffer containing 0.5% (w/v) BSA. Afterwards, sample preparation and analysis were performed as outlined above.
Data Evaluation
In order to determine uptake rates, cellular uptakes were normalized to total protein content and incubation time. Net OATP-mediated uptake rates were calculated as the difference in the uptake rates of the transfected and wild-type cells for each individual concentration. Half maximal inhibitory concentration (IC50) values for uptake inhibition were determined by plotting the inhibitor concentration (logarithmic scale) against the net uptake rates and nonlinear regression using the equation:
where y is the net uptake rate (picomoles per milligram protein per minute), x is the inhibitor concentration (micromolars), s is the slope at the point of inversion, and Top and Bottom are the maximum and minimum values for cellular uptake rates, respectively. For all graphical data evaluations, the GraphPad Prism software package (version 5.0, GraphPad Software Inc., La Jolla, CA) was used. The fitted parameters are presented as best-fit parameter and standard error (SE).
RESULTS
Demographics and Baseline Characteristics
Ten healthy male subjects in part A (age ± standard deviation [SD], 29 ± 9 years; BMI ± SD, 22.1 ± 2.8 kg/m2) and 10 healthy male subjects in part B (age ± SD, 33 ± 10 years; BMI ± SD, 23.0 ± 1.5 kg/m2) were enrolled. All subjects were Caucasian. All completed the study as per protocol. None of the subjects (in parts A and B) were exposed to any prescribed or over-the-counter drug (including herbal medicines) for 3 months prior to screening.
Pharmacokinetics
Part A
Mean concentration–time profiles of macitentan, ACT-132577, and ACT-373898 in period 1 (multiple-dose macitentan treatment) followed by period 2 (multiple-dose co-administration of Cs) are shown in Fig. 1. In period 1, plasma concentrations of macitentan, ACT-132577, and ACT-373898 approached steady-state on day 5 (96 h) of dosing. In the presence of Cs (period 2), no relevant changes in C tr of macitentan and its metabolites were observed compared to macitentan treatment alone. The mean 24-h plasma concentration–time profiles at steady-state in the two periods, i.e., on day 5 (period 1) and day 17 (period 2), for macitentan and its metabolites were similar (Fig. 2).

Part A: Arithmetic mean ± SD plasma concentration versus time profiles of macitentan, ACT-132577, and ACT-373898 from days 1 to 17. The last day of treatment with macitentan in period 1 (day 5) and last day of treatment with macitentan + Cs in period 2 (day 17) are indicated in the figure

Part A: Arithmetic mean ± SD plasma concentration versus time profiles of macitentan, ACT-132577, and ACT-373898 on day 5 (period 1: macitentan) compared to day 17 (period 2: macitentan + Cs)
Geometric means of steady-state AUCτ for macitentan, ACT-132577, and ACT-373898 in the absence of Cs treatment were 6,876, 16,881, and 530 ng·h/mL, respectively, compared to 7,562, 16,428, and 568 ng·h/mL in the presence of Cs (Table I). The comparison of geometric means of PK variables in part A is shown in Table II. The ratio of geometric means and their 90% confidence limits of AUCτ for ACT-132577 fell entirely within the FDA-recommended bioequivalence range of 0.80–1.25, whereas for macitentan and ACT-373898, at least one of the confidence limits were outside of this range (Table II) (15).
Part B
Mean concentration–time profiles of macitentan and its metabolites ACT-132577 and ACT-373898 in period 1 (multiple-dose macitentan treatment) followed by period 2 (multiple-dose co-administration of rifampin) are shown in Fig. 3. Steady-state conditions were approximately attained on day 5 (96 h) of dosing in the absence of rifampin. In the presence of rifampin treatment, C tr of macitentan decreased from days 6 to 12, and new steady-state conditions were approached on day 12 (264 h). A similar situation was observed for ACT-373898. The start of concomitant treatment of rifampin on day 6 caused an initial increase in C tr of ACT-132577, which gradually decreased with continued dosing and approached steady-state concentrations on day 12 (264 h) which were similar to the original steady-state concentrations. The comparison of 24-h plasma concentration–time profiles at steady-state in the two periods, i.e., on days 5 and 12, showed that concentrations of macitentan and ACT-373898 on day 12 were considerably lower than those on day 5 (Fig. 4). The differences in concentrations of ACT-132577 between days 5 and 12 were much smaller compared to those of macitentan and ACT-373898.

Part B: Arithmetic mean ± SD plasma concentration versus time profiles of macitentan, ACT-132577, and ACT-373898 from days 1 to 12. The last day of treatment with macitentan in period 1 (day 5) and last day of treatment with macitentan + rifampin in period 2 (day 12) are indicated in the figure

Part B: Arithmetic mean ± SD plasma concentration versus time profiles of macitentan, ACT-132577, and ACT-373898 on day 5 (period 1: macitentan) compared to day 12 (period 2: macitentan + rifampin)
Geometric means of steady-state AUCτ for macitentan, ACT-132577, and ACT-373898 from periods 1 to 2 decreased from 5,775 to 1,240 (a 79% decrease), 14,846 to 14,798 (a 0.3% decrease), and 484 to 176 ng·h/mL (a 64% decrease), respectively (Table I). The ratio of geometric means and their 90% confidence limits of AUCτ for ACT-132577 fell entirely within the FDA-recommended bioequivalence range of 0.80–1.25, whereas for macitentan and ACT-373898, they were completely outside of this range (Table II).
Tolerability and Safety
There were no deaths, SAEs, or AEs leading to treatment discontinuation. None of the AEs led to a withdrawal or any change in the conduct of the study. All AEs were of mild or moderate severity and were resolved at the end of the study with no sequelae.
In part A of the study, a total of 12 treatment-emergent AEs (TEAEs) occurred, of which three occurred during period 1, affecting three subjects (30%), and nine affecting six subjects (60%) occurred during period 2. The most frequently reported TEAE was headache, which affected one subject in period 1 and three subjects in period 2.
In part B of the study, a total of eight TEAEs occurred, of which six occurred during period 1, affecting six subjects (60%), and two affecting two subjects (20%) occurred during period 2. The most frequently reported TEAE was headache, which affected five subjects in period 1 and two subjects in period 2.
No clinically relevant treatment-related effects on vital signs, ECG variables, or clinical laboratory variables were detected in either part of the study.
In Vitro Experiments
To assess whether macitentan and its active metabolite ACT-132577 are substrates of human OATPs, time- and concentration-dependence assessments were performed in vitro. Rapid and comparable uptake of macitentan and ACT-132577 was observed in wild-type and OATP1B1 as well as OATP1B3 over-expressing CHO cells (Fig. 5). The uptake of macitentan into the wild-type, OATP1B1, and OATP1B3 over-expressing cells after incubation for 40 s was (mean ± SD) 346 ± 3, 362 ± 6, and 387 ± 11 pmol/mg, respectively, and the uptake of ACT-132577 was 214 ± 11, 237 ± 7, and 288 ± 10 pmol/mg, respectively. No marked difference in uptake rate of macitentan and ACT-132577 between wild-type and OATP over-expressing cells was observed when the effect of different concentrations was tested (Fig. 6).

Time course of cellular uptake of 3 μM macitentan (a) and 3 μM ACT-132577 (b) in wild-type (circles) and human OATP1B1 (squares) and OATP1B3 (triangles) over-expressing CHO cells. Values represent the mean ± SD of one experiment performed in triplicate

Concentration dependence of cellular uptake of macitentan (panels a and b) and ACT-132577 (panels c and d) in wild-type (circles) and human OATP1B1 (a, c) and OATP1B3 (b, d), over-expressing CHO cells (squares). Values represent the mean ± SD of one experiment performed in triplicate
The inhibitory potential of macitentan and ACT-132577 on OATPs was investigated over a wide range of inhibitor concentrations (0.01 to 300 μM). Macitentan inhibited OATP1B1-mediated uptake of atorvastatin and OATP1B3-mediated uptake of estrone-3-sulfate with IC50 ± SE values of 6.3 ± 0.7 and 11.8 ± 5.0 μM, respectively (Fig. 7). For ACT-132577, these values were 19.9 ± 1.4 and 51.6 ± 3.0 μM, respectively (Fig. 7).

Concentration-dependent inhibition of OATP1B1-mediated net uptake of atorvastatin (a, c) and OATP1B3-mediated uptake of estrone-3-sulfate (b, d) by macitentan and ACT-132577. Values represent the mean ± SD of one experiment done in triplicate, and the solid line is best fit to the data
DISCUSSION
The clinical study was conducted in two parts. In both parts, C tr of macitentan and its metabolites were comparable in the absence of Cs or rifampin, and they were consistent with previous results (16).
Part A
It has been shown that Cs may affect the PK of numerous compounds through its inhibitory effect on CYP3A4 or OATP transporters (11,12,17–19). The results of a previously performed study investigating the interaction between macitentan and ketoconazole, a strong CYP3A4 inhibitor, showed a small effect of ketoconazole on the exposure to macitentan (approximately two-fold increase of the area under the plasma concentration–time curve from zero to infinity [AUC0–∞]), which was not considered clinically relevant (10). Since Cs is a weaker CYP3A4 inhibitor than ketoconazole, a large effect of Cs on the exposure to macitentan through CYP3A4 inhibition was not expected. However, in this study, the investigation of a potential increase in exposure to macitentan through inhibition of OATP transporters by Cs was of greater interest. The results showed that in the presence of Cs, the exposure to macitentan and its metabolites did not change to a clinically relevant extent, which indicates no significant interaction between these two drugs at the level of metabolizing enzymes and/or transporters such as OATP.
This finding is different from that reported for other ERAs, such as ambrisentan or bosentan, which showed OATP involvement in the interaction of those ERAs with Cs (11,12,19). The significant initial increase in exposure to bosentan seen in the presence of Cs in humans (a 30-fold increase after the first day of co-administration of Cs) was attributed not only to the inhibition of metabolizing enzymes but mainly to the inhibition of OATP by Cs and consequently reduced hepatic uptake (17). Cs may be used concomitantly with pulmonary arterial hypertension therapies, particularly in those patients with connective tissue disease. In addition, calcineurin inhibitors, including Cs, are used to prevent host organ rejection during solid organ transplantation procedures. However, calcineurin inhibitors can limit the success of kidney transplantation due to calcineurin inhibitor-mediated nephrotoxicity. Data from previous studies suggest that some ERAs may attenuate Cs toxicity and may be useful in solid organ transplantation studies (20–22); in contrast to other ERAs, macitentan does not show any interaction with Cs either at the level of CYP enzymes or at the level of OATP transporters. Therefore, macitentan in combination with Cs has potential to be successful in organ transplantation without the need for macitentan dose adjustment.
Part B
In the presence of rifampin, exposure to macitentan decreased markedly. This indicates that the metabolism of macitentan is induced in the presence of rifampin, a strong inducer of CYP3A4. Complex interactions have been observed between rifampin and a number of drugs, including ERAs, which are subject to both CYP3A4-mediated oxidation and OATP-mediated hepatic uptake. These studies have typically revealed a biphasic response characterized by an initial, transient increase in ERA plasma concentration with acute rifampin administration, presumably due to OATP inhibition, followed by reduction in exposure attributed to CYP3A4 induction (23–26). Such biphasic effects of rifampin are not observed with macitentan, which further demonstrates the absence of a contribution from OATP to the disposition of macitentan. The initial increase seen in trough concentrations of ACT-132577 after the first doses of rifampin could be explained by an increase in macitentan metabolism at the level of CYP3A4, resulting in an increased transformation rate to ACT-132577. Rifampin has also been reported to be an inducer of conjugation enzymes, which render drugs more polar and facilitate their excretion (7). In humans, conjugation enzymes are involved in the elimination of ACT-132577, which could in turn be induced by rifampin, accelerating the elimination of ACT-132577. This hypothesis is supported by the observation that the exposure to ACT-132577 after continued dosing (day 12) was very similar to that without rifampin. OATP is unlikely to be involved in the initial increase of exposure to ACT-132577 based on the fact that concomitant treatment with Cs, also a major inhibitor of OATP, did not show the same effect. Since both rifampin and macitentan would be administered on a multiple-dose basis if used together, the exposure at steady-state (day 5 compared to day 12) is considered to be the most relevant comparison between the two treatment conditions.
Macitentan and its metabolite ACT-132577 are both potent dual ERAs, and both contribute to the overall pharmacological effect of macitentan in vivo. ACT-132577 is approximately five-fold less potent than macitentan in vitro. However, systemic exposure in humans to ACT-132577 is 2.5-fold higher than to macitentan. Although the exposure to ACT-132577 at steady-state was not affected by concomitant treatment with rifampin, the approximately four-fold reduction in exposure to macitentan in the presence of rifampin might affect the overall pharmacological effect of the drug. Thus, reduced efficacy of macitentan in the presence of concomitant treatment with rifampin should be considered.
The results from in vitro OATP studies support the observations from the clinical study. High cellular uptake rates of macitentan and ACT-132577 in wild-type cells as well as OATP over-expressing CHO cells suggest that their cellular uptake does not depend on human OATP transporters but is likely to be driven by passive diffusion. A small contribution from active uptake could not be totally excluded, however, although compared to the high passive component, it is considered not to be relevant. The present in vitro data give a possible mechanistic explanation why the concomitant administration of the potent OATP inhibitor Cs did not increase the exposure to macitentan and ACT-132577 to a clinically relevant extent and explain why C tr of macitentan did not show any immediate increase in the presence of rifampin.
The question whether macitentan and its active metabolite ACT-132577 could act as a perpetrator in DDIs was also assessed in in vitro studies. Macitentan and ACT-132577 exhibited an inhibitory potency on OATP1B1-mediated uptake of atorvastatin, as well as OATP1B3-mediated uptake of estrone-3-sulfate. Comparing the IC50 values and total plasma concentrations of macitentan and ACT-132577 at steady-state in healthy subjects (approximately 0.63 and 1.5 μM, respectively) during a 10-mg once-daily regimen and binding of macitentan and ACT-132577 to human plasma proteins in vitro (99.6% and 99.5%, respectively) indicates that the free steady-state concentration of macitentan and ACT-132577 is around 2.5 and 7.3 nM, respectively. Since free steady-state macitentan and ACT-132577 plasma concentrations are around 2,490-fold lower than the lowest IC50 determined, it appears unlikely that treatment with macitentan will affect the PK of co-administered OATP substrates.
Based on the guidance for industry issued in 2008 by the Center for Drug Evaluation and Research, ACT-373898, identified in the human absorption, distribution, metabolism, and excretion study, was considered as a second major circulating metabolite of macitentan (27,28) and, therefore, was also analyzed in the present study. However, in view of the more recently issued guidance by the International Conference on Harmonization in 2010 (29), ACT-373898 is no longer considered a major metabolite.
Similar to previous phase 1 studies with macitentan, the most frequent TEAE was headache (2,4,10). In part A, no hypertension or AEs relating to renal function, which are associated with Cs treatment, were reported (13). Similarly in part B, no AEs associated with rifampin treatment were reported (14). Overall, tolerability of macitentan when administered concomitantly with Cs or rifampin was good. However, the small number of subjects limits the interpretation of safety data in both parts of the study, and more data in a larger patient population are necessary to evaluate adequately the safety of macitentan in combination with Cs and rifampin.
CONCLUSION
The observed effect of Cs on the exposure to macitentan and its metabolites ACT-132577 and ACT-373898 under steady-state conditions is not considered clinically relevant. Concomitant treatment with rifampin reduced significantly the exposure to macitentan and its metabolite ACT-373898 at steady-state but did not affect the exposure to the active metabolite ACT-132577 to a clinically relevant extent. Reduced efficacy of macitentan in the presence of rifampin should be considered. Hepatic uptake of macitentan and its active metabolite ACT-132577 does not depend on OATP.
REFERENCES
Iglarz M, Binkert C, Morrison K, Fischli W, Gatfield J, Treiber A, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327(3):736–45.
Sidharta PN, van Giersbergen PL, Halabi A, Dingemanse J. Macitentan: entry-into-humans study with a new endothelin receptor antagonist. Eur J Clin Pharmacol. 2011;67(10):977–84.
Bruderer S, Sidharta P, Dingemanse J. Pharmacokinetics, tolerability, and safety of the dual endothelin receptor antagonist macitentan in Japanese and Caucasian healthy subjects. Clin Pharmacol Ther. 2011;89 Suppl 1:83.
Kummer O, Haschke M, Hammann F, Bodmer M, Bruderer S, Regnault Y, et al. Comparison of the dissolution and pharmacokinetic profiles of two galenical formulations of the endothelin receptor antagonist macitentan. Eur J Pharm Sci. 2009;38(4):384–8.
Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest. 2003;33(2):1–5.
International Transporter Consortium. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–36.
Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivistö KT. Pharmacokinetic interactions with rifampin: clinical relevance. Clin Pharmacokinet. 2003;42(9):819–50.
Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampin with organic anion uptake systems of human liver. Hepatology. 2002;36(1):164–72.
Baciewicz AM, Chrisman CR, Finch CK, Self TH. Update on rifampin and rifabutin drug interactions. Am J Sci. 2008;335(2):126–36.
Sidharta PN, Atsmon J, Dingemanse J. Investigation of the effect of ketoconazole on the pharmacokinetics of macitentan in healthy male subjects. Br J Clin Pharmacol. 2010;70:730–1. Abstract.
van Giersbergen PLM, Bodin F, Dingemanse J. Cyclosporin increases the exposure to tezosentan, an intravenous dual endothelin receptor antagonist. Eur J Clin Pharmacol. 2002;58(4):243–5.
Binet I, Wallnöfer A, Weber C, Jones R, Thiel G. Renal hemodynamics and pharmaco-kinetics of bosentan with and without CsA. Kidney Int. 2000;57(1):224–31.
Novartis. Neoral, summary of product characteristics of Cs. 2009.
Sanofi-aventis. Rifadin, summary of product characteristics of rifampin. 2009.
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Draft guidance for industry, drug interaction studies–study design, data analysis, and implications for dosing and labeling. 2006.
Sidharta PN, Dingemanse J. Multiple-dose tolerability, safety, pharmacokinetics, and pharmacodynamics of the dual endothelin receptor antagonist ACT-064992 in healthy human subjects. (Abstract). J Clin Pharmacol. 2008;48:1114.
Treiber A, Schneiter R, Häuser S, Stieger B. Bosentan is a substrate of human OATP1B1 and OATP1B3: inhibition of hepatic uptake as the common mechanism of its interactions with cyclosporin A, rifampin, and sildenafil. Drug Metab Dispos. 2007;35(8):1400–07.
Actelion Pharmaceuticals Ltd. Tracleer (bosentan) tablets. United States Food and Drug Administration Drug Product Label 2009.
Spence R, Mandagere A, Richards DB, Magee MH, Dufton C, Boinpally R. Potential for pharmacokinetic interactions between ambrisentan and cyclosporine. Clin Pharmacol Ther. 2010;88(4):513–20.
Kon V, Sugiura M, Inagami T, Harvie BR, Ichikawa I, Hoover RL. Role of endothelin in cyclosporine-induced glomerular dysfunction. Kidney Int. 1990;37(6):1487–91.
Perico N, Dadan J, Remuzzi G. Endothelin mediates the renal vasoconstriction induced by cyclosporine in the rat. J Am Soc Nephrol. 1990;1(1):76–83.
Fogo A, Hellings SE, Inagami T, Kon V. Endothelin receptor antagonism is protective in in-vivo acute cyclosporine toxicity. Kidney Int. 1992;42(3):770–4.
van Giersbergen PLM, Treiber A, Schneiter R, Dietrich H, Dingemanse J. Inhibitory and inductive effects of rifampin on the pharmacokinetics of bosentan in healthy subjects. Clin Pharmacol Ther. 2007;81(3):414–9.
Harrison B, Magee MH, Mandagere A, Walker G, Dufton C, Henderson LS, Boinpally R. Effects of rifampin (rifampin) on the pharmacokinetics and safety of ambrisentan in healthy subjects: a single-sequence, open-label study. Clin Drug Investig. 2010;30(12):875–85.
Xiong H, Carr RA, Locke CS, Katz DA, Achari R, Doan TT, et al. Dual effects of rifampin on the pharmacokinetics of atrasentan. J Clin Pharmacol. 2007;47(4):423–9.
Zheng HX, Huang Y, Frassetto LA, Benet LZ. Elucidating rifampin's inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin Pharmacol Ther. 2009;85(1):78–85.
Bruderer S, Hopfgartner G, Seiberling M, Wank J, Sidharta P, Treiber A, Dingemanse J. Allschwil: Actelion Pharmaceuticals Ltd. 2011. (Data on file)
US Food and Drug Administration. Guidance for industry 2008, safety testing of drug metabolites. US Department of Health and Human Services. Food and Drug Administration, Center for Drug Evaluation and Research (CDER). February 2008. Pharmacology and Toxicology.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for industry M3(R2) 2010, nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. 2010 (Revision 1).
ACKNOWLEDGMENTS
All authors are employees of Actelion Pharmaceuticals Ltd., except for Dr. Homery who was the principal investigator of the clinical trial and was employee of the contract research organization BIOTRIAL RENNES. BIOTRIAL received financial compensation for the clinical conduct of the study. This study was funded by Actelion Pharmaceuticals Ltd., Allschwil, Switzerland. The authors acknowledge Dr. Urs Simmen (Schötzau & Simmen, Statistische Beratungen, Switzerland) for his assistance in statistical work.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bruderer, S., Äänismaa, P., Homery, MC. et al. Effect of Cyclosporine and Rifampin on the Pharmacokinetics of Macitentan, a Tissue-Targeting Dual Endothelin Receptor Antagonist. AAPS J 14, 68–78 (2012). https://doi.org/10.1208/s12248-011-9316-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-011-9316-3