
Abstract
Background
The discovery and development of anticancer still remain a challenge especially regarding the problem of cancer cell selectivity. Matrix metalloproteinase (MMP) was broadly studied as one of the protein targets to stop cancer angiogenesis as well as its cell migration.
Main text
The MMP degrades extracellular matrix (ECM) such as collagen and gelatin which are important to control the cell migration from one to other sites. In cancer, this cell migration is regarded with metastasis, which is essential for the formation of new blood vessels called angiogenesis. The most common target in MMP, i.e. the catalytic site, is currently reported as being the non-selective target for inhibitor compounds that inhibit all MMPs but is associated with adverse side effects. Hemopexin, especially in MMP9 (PEX9) was found to be different from other domains in the MMP family which could potentially be the next target for anticancer due to the availability of its crystal structure in the Protein Data Bank (PDB).
Conclusion
The PEX9 crystal structure was resolved as a homodimer connected by a hydrophobic area between two blades along the β-propeller which its structure and function for computational drug modelling can be studied.
Similar content being viewed by others


Background
Cancer is the second cause of major health problems worldwide which significantly increases the incidence along with the growth and ageing of the population. And now, the burden has shifted to those who live in developing countries, as reported in 2012 that approximately 57% of all cases of cancer in the world and 65% of all deaths caused by cancer occurred in developing countries [1]. In 2012, cancer killed 8.2 million people all over the world, in which, particularly, lung cancer had the most incidences amongst others with 1.59 million deaths [2]. In the USA, the mortality caused by cancer occurred prevalently in the lungs and bronchi (158,000 deaths), whereas the mortality caused by breast cancer is not so far behind (58,670 deaths) [3]. In general, cancer occurs in 182 people out of 100,000 population, in which this number is higher in males than females. For both of them, the most common cases were lung cancer (16.8% in male and 8.8% in female) as well as colorectal cancer (9.2% in both genders). In particular, males suffered from prostate cancer (15% of all cancers in men), whereas females had a high incidence of breast cancer (25.1%) as reported [2]. As such, it was reported in 2015 that there were 347,792 people in Indonesia suffering from cancer. The most burdened provinces were Central Java with a total of 68,638 people diagnosed with cancer, followed by East Java with 61,230 people diagnosed with cancer. The most common cancer types found in these provinces were cervical and breast cancers [4].
One of the characteristics of cancer is the process of angiogenesis, which proceeds as follows: tumours that have > 2 mm in diameter need more oxygen and nutrition to rapidly grow up. Cells located in the very centre of the tumour will receive less oxygen than the cells on the outer surface. Therefore, hypoxia will induce the tumour cells to carry out angiogenesis, which is an extension of blood vasculature to supply the tumour with more oxygen. In angiogenesis, the endothelial cells of an existing blood vasculature will disconnect and rearrange to form new vasculature, which is in contrast to vasculogenesis wherein new endothelial cells are developed from stem cells [5].
Several studies have analysed the different roles of the many biological signalling molecules (factors) involved in the process of growth and expansion of cancer cells. One of these factors is a family of enzymes called the matrix metalloproteinases (shortened as MMPs) [6]. Humans have more than 20 types of MMPs, which are generally divided into 5 groups based on their substrate specificity, i.e. the components of the extracellular matrix (ECM) [7, 8]. The groups are the gelatinases, collagenases, matrilysins, stromelysins and membrane-type MMPs (MT-MMPs). Structurally, MMPs are zinc-dependent endopeptidases classified as protease enzymes [9]. The main function of these enzymes is to degrade the ECM, which is involved in various physiological processes such as wound healing, inflammation, angiogenesis, vasculogenesis and metastasis [10,11,12,13].
The enzymatic activity of MMPs is due to their catalytic site, which employs zinc ions coordinated by a histidine triad and is surrounded by other essential amino acid residues such as glutamic acid and alanine [14]. One type of MMP which is highly expressed during breast cancer is MMP9, which makes it an interesting biological target to control the growth of breast cancer cells. Instead of designing drugs that target the catalytic domain, which has already been done several times and resulted in the non-selective targeting of all MMPs [15], the focus should instead be on another part of the MMP structure. The hemopexin domain is the C-terminal domain of MMPs, and it can potentially be the next target for compounds that selectively inhibit different MMPs due to it having different structures and functionalities for each MMP [16]. This is in contrast to the structure of the catalytic domain which is virtually similar amongst all MMPs. An experiment was carried out in which the catalytic domain of MMP9 was mutated and the histidine triad (401, 405 and 411) has been replaced by alanine. The result was that the mutated MMP9 was still sensitive to the ECM substrate against its inhibitor [17]. Therefore, targeting the MMP9 in the catalytic site could be less favourable in the future.
New strategies for the selective targeting of MMPs have already been reviewed by other authors, in which they discussed about the exosite of the catalytic domain in MMP13, the hemopexin domain in MMP14 and the collagen-binding domain in MMP2 and MMP9 [18]. However, in this review, we will discuss the molecular mechanism of the hemopexin site in MMP9 as the potential protein target and the computational drug design of an MMP9 hemopexin inhibitor.
Matrix metalloproteinases
All classes of MMPs have similarities in their structures which consist of a pro-peptide region, a catalytic domain and many MMPs contain a hemopexin domain, though not all of them do. The pro-peptide region is normally composed of 80 amino acids, whereas the catalytic domain employs approximately 170 amino acid residues with a typical structure in almost all MMPs [19]. On the other hand, the hemopexin domain contains 200 amino acid residues in almost all MMPs except MMP7, MMP23 and MMP26 [20]. As mentioned before, MMPs are divided into classes based on each MMP’s substrate specificity.
The first class of MMPs is the collagenases, which have a role in the degradation of collagen types I, II and III. The MMP types which belong to this class are MMP1, MMP8, MMP13 and MMP18. The next class of MMPs is the stromelysins, which only contain MMP3 and MMP10, whereas the class of the matrilysins only has MMP7 and MMP26. In addition, matrilysins have functions to regulate both tumour necrosis factor α (TNFα) and E-cadherin. The class that contains the most members is the MT-MMPs which include MMP14, MMP15, MMP16, MMP17, MMP24 and MMP25 [21].
Of particular interests are the class of MMPs, known as gelatinases, which consists of MMP2 (also termed as gelatinase A) and MMP9 (termed as gelatinase B). The structure of MMP9 is composed of a NH2- pro-domain having three fibronectin (Fi) repetitions, which is important for collagen or gelatine binding. There are also two zinc ions located at the catalytic domain interacting with cysteine from the pro-peptide domain. The catalytic domain is linked by the O-glycoside domain, and it ends with the hemopexin domain next to it [22].
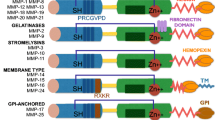
The structure of the hemopexin domain is decorated by four blades of beta-propellers. This facilitates the interaction between MMP9 and other molecules such as a substrate, cell receptors and tissue inhibitors [23]. Interestingly, this hemopexin domain could also act as an antagonist for the activity of the MMP9 itself, which will later be explained in more depth [17]. The natural inhibitors of MMPs are macromolecules called tissue inhibitors of metalloproteinases (TIMPS), where TIMP1, TIMP2 and TIMP3 serve as natural MMP9 inhibitors. TIMP1 has a high affinity towards MMP9 and inhibits this enzyme via interaction through the N-terminal of the catalytic domain. In general, all activities of MMPs could be inhibited by either TIMP1 or TIMP2 [24]. Before MMPs become active enzymes, they are inactive pro-enzymes. This inactivation is caused by the aforementioned interaction between the zinc ion and the cysteine residue, and this phenomenon is called the cysteine switch [20]. The general structure of MMPs is presented in Fig. 1.

The general structural domain of MMP9 consists of a pro-peptide domain (red tube), catalytic domain (turquoise surface) and three fibronectins (Fi)
MMP9 in breast cancer
It has been reported that MMP9 was expressed in a high concentration during cancers, in which one of them was breast cancer. There are various breast cancer types including luminal A, luminal B, human epidermal growth factor receptor 2 (Her-2)-positive, triple-negative (oestrogen receptor, progesterone and Her-2-negative) [25] and claudin-low. The highest expression of MMP9 was observed during the triple-negative type. Two marketed drugs are available such as tamoxifen and trastuzumab serving as luminal A, luminal B and Her-2-positive breast cancer treatments. In contrast, tamoxifen was reported to induce the expressions of MMP2 and MMP13 in fibroblast at dose 2 mg/100 g food for 5 weeks in mice [26]. However, there is currently no drug designed to treat breast cancer of the triple-negative type [14, 27]. The structures of tamoxifen (1) and trastuzumab (2) are presented in Fig. 2.

The structure of tamoxifen (an oestrogen receptor antagonist = 1), trastuzumab (a Her-2 antagonist = 2), batimastat (3), marimastat (4) and doxycyclin hyclate (5)
When a biopsy of a tumour is obtained and observed under the microscope, the cancer cells are classified into ‘grades’ according to how well-differentiated they are compared to normal cells. The grades indicate how fast they are dividing. Grade 1 represents a cell that is well-differentiated and similar to healthy cells that divide slowly, while grade 3 indicates poor differentiation and the cells divide quickly. Several studies have confirmed that more MMP9 is expressed at grades 2 and 3 than grade 1 [28,29,30,31].
In addition, the progression of breast cancer is classified into ‘stages’ using the TNM system which represents the size of the tumour (T), how many lymph nodes have been infected (N) and whether or not the cancer has metastasised (M). The stages range from stage 0 which indicates that the tumour is still benign, while in stage 4, cancer has already spread over to the organs other than the breast. Stankovic et al. have concluded from their research that the expression of pro-MMP9, and thus MMP9, positively correlates with the progression of the stage of breast cancer [32].
MMP9 can also interact with other biomolecules which have various roles in the progression of cancer. One of them is vascular endothelial growth factor (VEGF) which is an important factor during angiogenesis [33]. Cancer cells produce large amounts of VEGF, but stromal cells in close proximity to the cancer cells can also produce it [34]. When it is secreted, VEGF is bound to the ECM and thus remains inactive. But under hypoxia, the expression of MMP9 is increased which then degrades the ECM; therefore, VEGF can bind to its receptor and trigger angiogenesis. The types of VEGF are VEGF-A, VEGF-B, VEGF-C and VEGF-D which can bind to three types of receptors, i.e. VEGFR-1, VEGFR-2 and VEGFR-3. Amongst them, VEGF-A is one of the most expressed growth factors, which strongly binds to VEGFR-1 rather than VEGFR-2. However, angiogenesis is mainly triggered by the interaction between VEGF-A and VEGFR-2, whereas VEGFR-1 has an important role in regulating the interaction and activation of VEGFR-2 [35].
Another growth factor that has been identified to increase the tumour cell proliferation is the insulin-like growth factor 1 (IGF-1). During the initial phase, the IGF-1 is deactivated by insulin-like growth factor-binding protein 1 (IGFBP-1) by forming a complex structure. MMP9 can degrade this complex to release IGF-1 for tumour cell proliferation [36]. In addition, there is also a chemokine ligand called CXCL12, and its receptor CXCR4, which contributes to cancer metastasis. In people without cancer, the cells of the breast express small amounts of CXCR4, whereas during breast cancer, CXCR4 is expressed in a high concentration. CXCL12 or stromal cell-derived factor 1 (SDF-1) is secreted by a few organs such as the lungs, liver and lymph node. This chemokine serves as a chemoattractive agent for cells expressing CXCR4 to metastasise towards the organs that secrete CXCL12 [37].
It is intriguing to note that while a high expression of MMP9 can activate angiogenesis in cancer, paradoxically, it was also reported that MMP9 also has an antagonistic effect in angiogenesis due to the observation that endostatin, tumstatin and angiostatin (which are the body’s natural angiogenesis inhibitors) were also activated by MMP9 [38]. Endostatin and angiostatin can inhibit the proliferation of endothelial cells. Endostatin is activated via proteolysis of collagen XVII, whereas angiostatin is derived from plasminogen degradation by MMP9. Tumstatin is derived from the degradation of collagen IV, which is able to induce apoptosis of endothelial cells [39]. Another biological macromolecule that blocks tumour development is thrombospondin 1 (TSP-1). It acts by directly inhibiting MMP9, and it has been shown that the levels of MMP9 are reduced in transgenic mice that overexpress TSP-1 [40].
To further complicate the situation, we know of the existence of yet another biological protein involved in carcinogenesis called transforming growth factor β (TGF-β). It has a pleiotropic character, meaning that it can both act as a promoter and inhibitor of cancer. During the early stages of cancer, it acts as a tumour suppressor by arresting cell development and inducing apoptosis. However, during the late stages of cancer, cells become immune to its tumour-suppressing effects and it acts as a tumour promoter. Transforming growth factor β (TGF-β) is secreted as a protein bound to TGF-β-binding protein (TGF-β BP) which is inactive. MMP9 activates TGF-β by releasing it from the binding protein [41,42,43,44].
Hemopexin (PEX)
The hemopexin-binding site in MMP9 (abbreviated as PEX9) has various functions. One important function is to enable MMP9 to bind to gelatine; hence, it is responsible for the substrate specificity of the enzyme [45]. PEX9 also seems to have a role in cell migration. Mantuano et al. proved in an experiment that when PEX9 was inhibited, the migration of Schwann cells (a type of nerve cell) was also inhibited. The migration of Schwann cells is caused by a cell signalling pathway that is activated when MMP9 binds to lipoprotein receptor-related protein (LRP-1) [46].
The mechanism of Schwann cell migration is as follows: when the nervous system is injured, Schwann cells overexpress LRP-1 which interacts with MMP9 to activate extracellular signal-regulated kinase (ERK1/2) and Akt, which leads to cell migration. When the PEX9 domain was inhibited using antibodies, cell migration was blocked [47, 48]. The mechanism of Schwann cell migration blockage related to PEX9 inhibition is illustrated in Fig. 3.

The mechanism of Schwann cell migration blockage related to PEX9 inhibition. a The homodimerisation of MMP9 facilitated by hemopexin domain; b the binding of the homodimer to the LRP-1 which transduces the signal to ERK1/2 leading to the cell migration; c blocking of homodimerisation by small molecule which binds to hemopexin domain, as well as the blocking of the degradation of the ECM by MMP9; d the binding of VEGF that has been released from the ECM by MMP9 to VEGFR which activates angiogenesis. Adapted from Cain [48]
Another study showed that MMP9 promotes the progression of cancer by interacting simultaneously with α4β1 integrin and CD44v at the cell surface. This binding induces Lyn activation, STAT3 phosphorylation and upregulation of MCL-1 and results in cell migration while increasing cell survival [49, 50]. In addition, the researchers also discovered a 17-residue sequence located in blade 4 of the PEX9 that binds to α4β1 integrin, which can be a potential target for the inhibition of cell migration for patients with chronic lymphocytic leukaemia (CLL) [51]. Furthermore, they found synthetic peptides which can disrupt the interaction between PEX9 and CD44 and, as a consequence, also block adhesion and migration of CLL cells [52].
Returning to the topic of breast cancer, Dufour et al. devised an experiment which used MCF-7 cells that were transfected with certain cDNA to prove that the inhibition of PEX9 blocks cell migration in breast cancer. One group of cells was transfected with DNA which made them overexpress pro-MMP9 and TIMP-1, while the other group overexpressed pro-MMP9 and TIMP-2. Cell migration was reduced in the group that expressed TIMP-1, while the other group did not do so. Since it is known that TIMP-1 interacts with PEX9, the conclusion was that the inhibition of PEX9 also retards cell migration [53].
As mentioned earlier, PEX9 may also have activity towards the MMP9 enzyme as an inhibitor. It was found that PEX9 has the capability to strongly bind to gelatin, thus preventing the MMP9 enzyme from binding to gelatin which therefore reduced its activity in the context of tumour invasion and metastasis in colorectal carcinoma [54]. The incorporation of in vitro and in vivo studies by Ezhilarasan et al. showed that enforcing the expression of PEX9 interestingly blocks the activity of MMP9 by decreasing the expression of VEGF and VEGFR2 in human mammary epithelial cells (HMECs). Besides that, it also induced apoptosis via the activation of caspases, as well as inducing the cleavage of poly (ADP-ribose) polymerase (PARP) which also plays a role in apoptosis [55].
So far, the evidence seems to indicate that the inhibition of PEX9 can indeed inhibit the progression of certain aspects of cancer. However, the main issue is whether a selective PEX9 inhibitor can be designed. A study had been conducted to compare the structure of MMP9 from 12 different vertebrates to analyse their structural similarities based on the natural selection hypothesis. It showed that amino acids in the middle region were homologous, while the amino acids in the C-terminal region (particularly in exon 13) showed the least similarities. This exon is located in the hemopexin domain, which means that it may be feasible to create a drug that can selectively bind to this site [56]. The similarity of amino acid sequences in the hemopexin domain amongst all types of MMPs was only 25–33%; thus, it has been concluded that this allosteric site can control the function and specificity of MMPs due to their molecular nature and modular domain organisation, as reviewed [57, 58].
Structurally, the complete MMP9 enzyme is interesting because it contains a 30-A-long O-glycosylated (OG) domain which links the enzyme with its hemopexin domain. This OG domain takes on a flexible conformation which facilitates independent movements of the terminal domains [59]. Almost all the MMPs have linker domains, but MMP9 has the longest linker in the form of an OG domain. This domain contains many glycine and proline residues, and despite the domain being heavily glycosylated, its length allows it to flex, fold and form compact conformations [60, 61].
It is proposed that the activity of the catalytic site of MMP9 could be induced by long-range conformational transitions, wherein the activator proteins or ligand is bound to an allosteric area [62,63,64]. Therefore, PEX9 as the domain connected by the linker (OG domain) to the catalytic domain is a possible device to be exploited as an allosteric controller of MMP9 activity. Since it has been shown that the formation of the PEX9 dimer leads to a biological activity, inhibiting the formation of the said dimer could be the first step in inhibiting the activity [65, 66]. As another example, the allosteric function of PEX9 can be seen in the interaction between MMP9 and CD44. The interaction may facilitate the binding of the enzyme and the substrate, which increases the probability of proteolysis [23].
Although, it has been confirmed that the formation of homodimers of PEX9 is what leads to biological activity [67, 68]. Later reports showed that when MMP9 was still in its inactive pro-enzyme form, it was bound to TIMP-1 and forms a circular homotrimer. This was confirmed by electrophoresis, atomic force microscopy and transmission electron microscopy [69]. It was remarked that the pro-MMP9 trimer demonstrated a higher affinity towards TIMP-1 than its monomer with a 50-fold difference in magnitude.
Cha et al. [70] have crystallised a single PEX9 domain, providing the model for structure-based drug design. This crystal consists of four-bladed β-propeller structures and four antiparallel β-strands for each blade. A peptide loop connected the blades with each other within those four antiparallel β-strands. A disulfide bond between Cys516 and Cys704 connected them to maintain the integrity of the structure. A highly conserved disulfide bond was present amongst all previous PEX domains as shown in both monomeric and dimeric states. It is to be noted that the homodimerisation of PEX9 is reduction-sensitive, which was understood at that time to mean that dimerisation was linked by disulfide bonds. However, later investigations showed that the link between the monomers did not belong to the disulfide bond variety, but rather a hydrophobic interaction between blades 4 for each sub-unit. Despite this, there is an important disulfide bond between blade 1 and blade 4 (the aforementioned bond between Cys516 and Cys70) that functions to maintain the structure of the β-propellers for each individual monomer. This disulfide bond is easily cleaved by reducing agents, leading to dimer dissociation and explaining the sensitivity of the pro-MMP9 dimer.
The crystallised PEX9 structure has no inhibitor bound within any pocket; however, there are two sulfate ions bound at blades 1 and 3. The sulfate was bound to this site with a strong interaction to the Arg105 residue measured to be 3.03 Å long via electrostatic interactions. Considering that there are no other PEX9 holoproteins, the binding site of this sulfate ion could be the starting point to predict inhibitor-PEX9 interactions using a structure-based computational design.
Before PEX9 had attracted any attention as the next target in the MMP9 inhibition, PEX1 was the first crystallised C-hemopexin domain in full length along with its catalytic domain (PDB 1FBL). These 2 domains are connected by 17 amino acids which might be flexible and free of secondary structure. As is with PEX9, PEX1 also contains 4 units of 4 antiparallel β-sheets which are stabilised by calcium on its fourfold axis, in which the blades are scarcely twisted [71]. Two PEX2 monomers were crystallised in the C-hemopexin domain only, which showed a homologous structure with the earlier PEX1. They showed an additional sodium, calcium and chloride ion bound to the protein by forming a channel-like opening (PDB 1GEN) [72]. In another PEX2 crystal (PDB 1RTG), 2 calcium and 1 chloride ions were found around the pseudo-fourfold axis [73]. When PEX13 was crystallised, it also revealed a similar pseudo-fourfold symmetry assigning 2 calcium, 2 chloride and additional sulfate ions in the central tunnel tube [74]. The dimer structure of PEX9 and the superposition of PEX1, PEX2 and PEX13 are illustrated in Fig. 4.

The 3D structure of hemopexin domains. a The dimer PEX9 showing hydrophobic interactions at blade 4 interface with sulfate ions bound at the blade 1 and blade 3. b The superposition of PEX1 (cyan ribbon), PEX2 (red ribbon) and PEX13 (magenta ribbon)
PEX14 is the latest hemopexin domain crystallised from the full protein MT1-MMP (also named as MMP14). This domain also shows homodimerisation in which blade 2 and blade 3 of chain A interact hydrophobically with blade 3 and 2 of chain B. The interruption of this homodimerisation such as amino acid mutagenesis affects the functionality of MT1-MMP in pro-MMP2 activation as well as collagen degradation. In contrast, the non-dependent dimer functions including gelatin film degradation and cell migration are not affected [75].
MMP9 inhibitors
Because it is known that MMPs are involved in many pathological processes, it was a matter of time before the first MMP inhibitors were developed. One of the members of the first generation of MMP inhibitors was batimastat (3) reported not to lead the expression of MMP9 and MMP2 at a dose of 30 mg/kg in lung tumour cells of mice [76], but the third phase of its clinical study revealed poor solubility as well as oral bioavailability, leading to its failure to be an anticancer agent. Marimastat (4) is an analogue of batimastat able to stop the expression of MMP2 and MMP9 in cerebral cortex cell of mice at a dose of 2 μM [77], developed for better oral bioavailability. However, this compound was found to be toxic during clinical phase [78]. The molecular structures of batimastat and marimastat (see Fig. 2) resemble peptides (peptidomimetic) and contain a hydroxamic acid group, which interacts directly with the zinc ion in the catalytic site of the MMP. Thus, they act as reversible competitive inhibitors towards the MMPs [5, 79, 80].
The next generation of MMP inhibitors was still designed to target the metalloenzyme at the catalytic site [81, 82]; therefore, the coming issue would be either drug resistance due to the non-selective binding mode of inhibitors towards the catalytic site or the adverse side effects such as musculoskeletal defects. The hemopexin domain in MMP9 has a different structure compared to the other MMPs, which means that this domain could be the next interesting target in the discovery of MMP9-selective inhibitors to treat cancer diseases [83]. Unfortunately, the only MMP9 inhibitor so far that has been approved for use by the US Food and Drug Administration is doxycycline hyclate (Periostat® = 5). Moreover, this inhibitor strongly reduces the expression of MMP1, MMP9 and MMP12 at 416 μM, which is only suitable for collagenases (not MMP9 since it is a gelatinase) [84], and the drug itself is indicated to treat periodontitis and not as anticancer [79]. AZD1236 is a possible next-generation MMP9 inhibitor. It underwent a clinical phase 2 study in the treatment of chronic obstructive pulmonary disease (COPD). This inhibitor was selective for MMP9 and MMP12 with a good safety index. However, more studies are necessary to be carried out for further developments [85]. The structures of some MMP inhibitors are presented in Fig. 2.
Kalva et al. utilised computational drug modelling to identify the pharmacophore model, which could be used as a query search for drugs that specifically target MMP9. The crystal structure being used was 1GKC, a complex of MMP9 with an inhibitor bearing a hydroxamate functional group. The structure-based pharmacophore modelling was incorporated with molecular docking studies, which revealed two aromatic rings and three hydrogen bond acceptors as the features of an MMP9 inhibitor, although this is for the catalytic domain and not for binding to PEX9. One of the natural product compounds being fit into the pharmacophore model was hinokiflavone which was followed by an in vitro study (IC50 = 43.08 μM) leading to the idea of using it as a lead compound needing optimisation for the development of a novel MMP9 inhibitor [86]. The pharmacophore model generated by Kalva was re-drawn in Fig. 5.

The pharmacophore model of hinokiflavone is composed of two aromatic rings (brown) and hydrogen bond acceptors (cyan) at the measured distances (in angstroms)
Focusing again on the PEX9 domain, a phage display peptide library with pro-MMP9 or a recombinant version was utilised to construct the potential PEX9 inhibitor. Uniquely, the peptide generated could not compete with TIMP-1 as the natural inhibitor; therefore, the peptide does not directly affect the proteolytic activity of MMP9. Furthermore, the prevention of pro-MMP9 activation followed by blocking of cell migration as well as in vivo tumour cell growth indicates the selectivity of PEX9 to be used as the target [87].
Computational drug modelling targeting hemopexin of MMP9
Drug modelling aided by computers has been commonly used to rationalise the constructed model before a drug is chemically synthesised and biologically tested [88]. The advances of computer technology, complemented with robust data from bioinformatics and chemo-informatics, are extremely helpful in doing tasks involving drug design and discovery in respect to reducing the cost and increasing the speed of the investigation [89]. There are several software available that facilitate structure-based drug design (when the crystal structure of the protein is available) [90] and ligand-based drug design (when only the ligand with its biological activity is available) [91]. The software could be free, e.g. AutoDock [92], PLANTS [93] and ArgusLab [94], or bought under a purchased licence such as GOLD [95], Glide [96] and Flexx [97]. The bioinformatics data such as the protein crystal structure, either in apo-form or holo-form, is freely downloadable from protein databases such as the Protein Data Bank, commonly abbreviated as PDB (accessible at www.rscb.org). The ligands could also be freely downloaded from chemical databases such as ZINC [98], Natural Product Discovery (NADI) [99] and National Cancer Institute (NCI) [100]. Alternatively, if the ligand is a new chemical entity, its molecular structure could be directly drawn based on their chemical structures shown in published articles. The utilisation of a computer-aided drug modelling is able to shorten the time of investigations because the software can screen millions of ligands to be shortlisted as hit compounds by in silico testing. The hits could be further optimised either in silico or in vitro as the lead compound; therefore, the potential candidate of a drug is selected and/or optimised without needing to test all available compounds in a database.
The only structure of PEX9 available in PDB is 1ITV, a dimer of PEX9. The dimer is connected by the hydrophobic area between each blade 4 along the β-propeller forming the structure as an assymmetric plane [70]. Two studies utilised this crystal structure as the model in a virtual screening of the ZINC database in the effort to discover MMP9 inhibitors which can selectively bind to the PEX domain, as explained below.
Dufour et al. [101] have utilised one of the software available to screen ligands from the ZINC database by docking those ligands into the MMP9 PEX domain in an effort to discover a new MMP9 inhibitor that selectively binds at the PEX9. A hundred ligands had been selected from ZINC 2007 and five top in silico hits (6–10) which can be seen in Fig. 6 were selected for further in vitro testing. This was used to investigate their capabilities in inhibiting the activity of the PEX9 domain. The binding of five compounds towards PEX9 was observed upon fluorescence assay, revealing the dissociation constant (Kd) for each compound associated with their binding affinities. The compounds were then checked for their cytotoxicities against COS-1 cells to measure the LD50, and it was followed by cell proliferation determination upon treatment with the compounds. Finally, the in vivo experiment was carried out to evaluate the MMP9 expression interference in mice using a tumour xenograft model, i.e. human MDA-MB-435 cancer cells. The result showed five compounds (6, 7, 8, 9 and 10) that exhibited COS-1 migration inhibition; however, only compounds 8 and 10 demonstrated the control cells’ (GFP-transfected) migration inhibition. An early conclusion was taken that only compounds 6 and 7 did not cause a notable cytotoxicity in COS-1 cells; therefore, both compounds blocking the MMP9-induced cell migration were not because of their toxicities but in respect to their selectivity towards cancer cells. Further investigation showed that compound 7 did not inhibit MMP2 and MT1-MMP cell migration, proving that the selectivity of the compound was only towards cell migration caused by MMP9. The binding of compound 7 with PEX9 resulted in a Kd of 2.1 μM, confirming its potential PEX9 inhibitor activity in conjunction with its binding mode to PEX9 domain prediction upon docking. Structurally, compound 7 has a six-member heterocycle ring which provides a planar conformation that fits into the cavity of PEX9 domain blades, whereas the arylamide moiety facilitates a more flexible conformation, which bound to the surface near the cavity. Therefore, this experiment highly correlates with the in vitro study that compound 7 successfully disrupts the MMP9-induced cell migration via binding with the PEX domain as an allosteric inhibitor.

The five hit compounds active against PEX9, selected from ZINC database by Dufour et al. [101]
The work of Dufour in searching for PEX9 inhibitors was continued by Li [102] by using the National Cancer Institute (NCI) database to virtually screen the next hit ligands as PEX9 inhibitors. The molecular docking was carried out with a protocol similar to the one that was used by Dufour. There were two binding sites used by Li wherein the sulfate ion was bound to the interface of four blades (site 1) and at the interface of blade 3 and blade 4 (site 2). Most of the amino acid residues of PEX9 involved in the atomic interactions with the hit ligands were Ile12, Glu14, Val58, Glu60, Lys65, Ala104, Arg106, Val152 and Gln154 via hydrogen bonding interactions. The binding assay study revealed the Kd of four hit compounds (11, 12, 13 and 14) in low micromolar inhibitions (0.6 μM, 0.8 μM, 0.9 μM and 0.8 μM, respectively). The structure of those four compounds (11–14) are presented in Fig. 7. From the results, it is most likely that the steroid backbone is still dominant to construct the ligand-protein binding.

The latest finding on small molecule inhibitors targeting the PEX9 domain of latent MMP9 was carried out by [103], demonstrating a prevention of focal adhesion junction formation associating with specific protein-ligand binding. Computational docking is used to design the compound series which tightly dock to the PEX9 binding site. One of the interesting compounds (15) generated from 7 bears a flexible n-propyl chain within quinazolinone as the heterocyclic planar ring and p-fluoroarylamide ring. This compound demonstrated Kd 0.32 μM upon pro-MMP9 tryptophan fluorescence assay defining the compound affinity for the PEX9 binding domain due to the disruption of MMP9 homodimerisation to prevent the association of CD44 and α4β1 integrin yielding the dissociation of EFGR. The phosphorylation of SrC and its downstream is then decreased leading to the blocking of tumour cell growth, migration and invasion. According to the docking results, the fluorine atom was found deeply occupying within the binding pocket which is different from 7. This 7 shows solvent exposure surrounds difluoromethoxyphenol which decreases the free energy of binding. In addition, the planar quinazolinone was said to improve the ligand-binding efficiency as well as the longer flexible propyl chain. In cell migration assay, 15 demonstrates a very low average migrated cells per 10× fields compared to MMP2 (homologue) and MMP14 (non-homologue). These may contribute to the higher affinity of 15 than others. The structure of 15 is illustrated in Fig. 7.
In addition to the current discovery of MMP inhibitors focusing on the PEX domain, MT1-MMP (MMP14) has also been studied alongside with PEX9. A total of 19 compounds screened from the NCI database via computational molecular docking against the PEX14 domain (PDB 3C7X) have been tested in vitro using fluorescence assay demonstrating remarkable IC50 values ranging from 1.41 to > 100 μM [104]. In this study, the ligands’ structures were designed to bind the PEX14 domain at the interface of four blades of beta-propellers, and the amino acid residues involved in this binding were Met328, Arg330, Asp376, Met422 and Ser470. It was suggested that the ligand intervention at this centre point could defect the flexibility of homodimerisation, thus affecting the MMP14 activity. However, the hit compounds dominantly bear the sulfonamide moiety which is quite similar with most of the active ligands working on the catalytic site. Therefore, further study in the selectivity of compounds, which bind to either the catalytic or hemopexin site, should be carefully managed.
Discussion and current research
Research in novel drug discovery for breast cancer by targeting the hemopexin domain of MMPs in general, and MMP9 specifically, is still less conducted. To the best of our knowledge, there are only three publications reporting compounds inhibiting PEX9 including pyrimidine-arylamide [101, 103] and steroid [102] scaffolds. This is in contrast with studies targeting MMP9 with the catalytic domain as the protein target covering diverse scaffolds such as pyridinone, pyridithiol, biaryl ether sulfonamide hydroxamate, aryl sulfone, pyrimidine, aromatic carboxylic acid and flavonoid which have been reviewed in our previous published article [13]. The distinction of structure amongst the hemopexin domains is promising a better target for selective inhibition rather than the catalytic domains, which share highly conserved amino acid residues in their binding sites. A potential compound from the ZINC database bearing a pyrimidinone scaffold extended by an arylamide moiety connected with a flexible S-alkyl chain was used to identify the pharmacophore which meets the requirements to be active as a PEX9 inhibitor. The pyrimidine ring has a planar character, which fits into the deep and stable hydrophobic pocket whereas the arylamide was said to be more flexible in the surface near to the cavity. Due to the unavailability of PEX9 in complex with its inhibitor, it is still not confirmed whether the binding site was correctly predicted by computational docking. In the case where the protein crystal structure is not in complex with its inhibitor, homology modelling could be another strategy to conduct a structure-based drug design. This could be done if only there is another protein crystal structure in complex with inhibitor having high similarity in amino acid sequence with the desired protein structure being available. This homologues crystal could be used as the template to model the desired 3D protein structure with the known binding site.
Intriguingly, two functional groups present in the compound that was identified by Dufour as being active against PEX9 (compound 7) had not been addressed. A propyl group attached to the pyrimidine ring and the OCF2 group at the para position of the arylamide were not assigned pharmacophores, which can be pointed out for further design. There exists another method in designing a novel drug called ligand-based drug design, wherein a series of a ligand’s structure along with the indicators of biological activity such as its IC50 or Ki can be studied for its quantitative-structure activity relationships (QSAR). For example, by using compound 7 by Dufour, we can maintain the pivotal functional groups (the planar ring and flexible arylamide) followed by modifying the smaller functional groups to test bioisosterism. The propyl group could be modified by extending the normal chain or cyclic chain, whereas the OCF2 could be modified by substituting F with another halogen. The series of those modifications with their respective bioactivities could be processed as a QSAR model explaining the correlation whether it gives the compound a higher or lower bioactivity and what type of pharmacophore contributes to that correlation. The pharmacophores that are marked as contributing to a negative value should be reduced or removed, whereas the positive correlations should be maintained or even added with the extra ones. The substitution and/or addition of functional groups should also consider the 3D circumstances such as stereochemistry. Therefore, the modified functional group can keep being stable and have the correct conformation associated with their binding. Finally, we can conclude that from the data available, PEX9 is the selective target for MMP9 inhibition, which appears to have a significant advantage compared to other targets, leading to a potentially successful discovery in novel anti-breast cancer agents.
Our in-progress research is also generating a series of compounds bearing the pharmacophore as identified by Dufour et al. [101] as well as Alford et al. [103] with modifications as illustrated in Fig. 7. Here, we utilised purine ring to have heterocyclic planar character instead of pyrimidine has been used in the lead compounds. One of the compounds in the series is designed by attaching a strong electron withdrawing group such as NO2 at the para-position of the arylamide ring (16). The docking study of this compound into the PEX9 binding pocket shows a low free energy of binding − 8.43 kcal/mol by interacting with important amino acid residues surrounding the binding site such as Glu14, Glu60, Arg106 and Gln154 (16a and 16b). This binding mode agrees with the study by Li [102] has been discussed previously. Interestingly, the in vitro inhibition assay of this compound towards MMP9 using fluorescence spectroscopy demonstrates IC50 3.20 μM indicating its potential compound to be developed as furthermore selective MMP9 inhibitor. The structures of 16, 16a and 16b are illustrated in Fig. 7.
Conclusions
Hemopexin MMP9 is an interesting domain of MMP9 to be targeted in searching for more selective drug for breast cancer. The current studies show that small molecule inhibitor selectively inhibits the activity of MMP9 compared to other homologous MMP2 as well as non-homologous MMP14. The utilisation of computational drug design is able to develop more effective MMP9 inhibitor with a lower cost which could be applied for further lead optimisation using either structure-based drug design or ligand-based drug design.
Availability of data and materials
This is a review article; therefore, data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Abbreviations
- CLL:
-
Chronic lymphocytic leukaemia
- ECM:
-
Extracellular matrix
- ERK:
-
Extracellular signal-regulated kinase
- Fi:
-
Fibronectin
- Her-2:
-
Human epidermal growth factor receptor 2
- HMECs:
-
Human mammary epithelial cells
- IGF-1:
-
Insulin-like growth factor 1
- IGFBP-1:
-
Insulin-like growth factor-binding protein 1
- LRP:
-
Lipoprotein receptor-related protein
- MMP:
-
Matrix metalloproteinase
- MT-MMP:
-
Membrane-type matrix metalloproteinase
- NADI:
-
Natural product discovery
- NCI:
-
National Cancer Institute
- OG:
-
O-glycosylated
- PARP:
-
Poly (ADP-ribose) polymerase
- PDB:
-
Protein Data Bank
- PEX:
-
Hemopexin
- TGF-β:
-
Transforming growth factor β
- TIMPS:
-
Tissue inhibitors of metalloproteinases
- TNF:
-
Tumour necrosis factor
- TSP-1:
-
Thrombospondin 1
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A (2015) Global cancer statistics, 2012. CA: Cancer J Clin 65:87–108. https://doi.org/10.3322/caac.21262
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136:E359–E386. https://doi.org/10.1002/ijc.29210
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA: Cancer J. Clin 65:5–29. https://doi.org/10.3322/caac.21254
Ministry of Health Republic of Indonesia: Situation of cancer disease. http://www.depkes.go.id/article/view/15021800011/situasi-penyakit-kanker.html: 2015 Accessed 28 February 2017.
Khalid A, Javaid MA (2016) Matrix metalloproteinases: new targets in cancer therapy. J Cancer Sci Ther 8:143–153. https://doi.org/10.4172/1948-5956.1000406
Krüger A, Kates RE, Edwards DR (2010) Avoiding spam in the proteolytic internet: future strategies for anti-metastatic MMP inhibition. BBA – Mol Cell Res 1803:95–102. https://doi.org/10.1016/j.bbamcr.2009.09.016
Singh P, Grewal AS, Pandita D, Lather V (2018) Synthesis and evaluation of a series of caffeic acid derivatives as anticancer agents. FJPS; available online. https://doi.org/10.1016/j.fjps.2017.11.002
Bauvois B (2012) New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: outside-in signaling and relationship to tumor progression. BBA – Rev Cancer 1825:29–36. https://doi.org/10.1016/j.bbcan.2011.10.001
Fanjul-Fernández M, Folgueras AR, Cabrera S, López-Otín C (2010) Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. BBA – Mol Cell Res 1803:3–19. https://doi.org/10.1016/j.bbamcr.2009.07.004
Gingras D, Béliveau R (2010) Emerging concepts in the regulation of membrane-type 1 matrix metalloproteinase activity. BBA – Mol Cell Res 1803:142–150. https://doi.org/10.1016/j.bbamcr.2009.04.011
Yang W, Li G (2012) The matrix metalloproteinases and cerebral ischemia. In: Balestrino M (ed) Advances in the preclinical study of ischemic stroke. Intech, London, pp 145–154
Aiken A, Khokha R (2010) Unraveling metalloproteinase function in skeletal biology and disease using genetically altered mice. BBA – Mol Cell Res 1803:121–132. https://doi.org/10.1016/j.bbamcr.2009.07.002
Hariono M, Yuliani SH, Istyastono EP, Riswanto FDO, Adhipandito CF (2018) Matrix metalloproteinase 9 (MMP9) in wound healing of diabetic foot ulcer: molecular target and structure-based drug design. Wound Med 22:1–13. https://doi.org/10.1016/j.wndm.2018.05.003
Nagase H, Visse R, Murphy G (2006) Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res 69:562–573. https://doi.org/10.1016/j.cardiores.2005.12.002
Tallant C, Marrero A, Gomis-Rüth FX (2010) Matrix metalloproteinases: fold and function of their catalytic domains. BBA – Mol Cell Res 1803:20–28. https://doi.org/10.1016/j.bbamcr.2009.04.003
Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES (2014) Tumor cell-produced matrix metalloproteinase 9 (MMP-9) drives malignant progression and metastasis of basal-like triple negative breast cancer. Oncotarget 5:2736–2749. https://doi.org/10.18632/oncotarget.1932
Roeb E, Behrmann I, Grötzinger J, Breuer B, Matern S (2000) An MMP-9 mutant without gelatinolytic activity as a novel TIMP-1-antagonist. FASEB J 14:1671–1673. https://doi.org/10.1096/fj.99-0947fje
Fields GB (2015) New strategies for targeting matrix metalloproteinases. Matrix Biol 44–46:239–246. https://doi.org/10.1016/j.matbio.2015.01.002
Brew K, Nagase H (2010) The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. BBA – Mol Cell Res 1803:55–71. https://doi.org/10.1016/j.bbamcr.2010.01.003
Yabluchanskiy A, Iyer RP, Hall ME, Lindsey ML (2013) Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiology 38:391–403. https://doi.org/10.1152/physiol.00029.2013
Strongin AY (2010) Proteolytic and non-proteolytic roles of membrane type-1 matrix metalloproteinase in malignancy. BBA – Mol Cell Res 1803:133–141. https://doi.org/10.1016/j.bbamcr.2009.04.009
Visse R, Nagase H (2003) Matrix metalloproteinases and tissue inhibitors of metalloproteinases. Struct Funct Biochem 92:827–839. https://doi.org/10.1161/01.RES.0000070112.80711.3D
Ugarte-Berzal E, Bailón E, Amigo-Jiménez I, Albar JP, García-Marco JA, García-Pardo A (2014) A novel CD44-binding peptide from the pro-matrix metalloproteinase-9 hemopexin domain impairs adhesion and migration of chronic lymphocytic leukemia (CLL) cells. J Biol Chem 289:15340–15349. https://doi.org/10.1074/jbc.M114.559187
Fassina G, Ferrari N, Brigati C, Benelli R, Santi L, Noonan D, Albini A (2000) Tissue inhibitors of metalloproteases: regulation and biological activities. Clin Exp Metastasis 18:111–120. https://doi.org/10.1023/A:1006797522521
Schwab RB, Koehler M, Ali SM, Murray BW (2016) Genomic profiling and treatment of HER2+, ER+, PgR+ “triple positive” breast cancer: a case report and literature review. Cancer Treat Res Commun 9:27–31. https://doi.org/10.1016/j.ctarc.2016.06.008
Ma X, Liu Y, Wang Q, Chen Y, Liu M, Li X, Xiang R, Wei Y, Duan Y, Han J (2015) Tamoxifen induces the development of hernia in mice by activating MMP-2 and MMP-13 expression. BBA - Mol Basis Dis 1852:1038–1048. https://doi.org/10.1016/j.bbadis.2015.02.006
Yousef EM, Tahir MR, St-Pierre Y, Gaboury LA (2014) MMP9 expression varies according to molecular subtypes of breast cancer. BMC Cancer 14:1–12. https://doi.org/10.1186/1471-2407-14-609
Benson CS, Babu SD, Radhakrishna S, Selvamurugan N, Sankar BR (2013) Expression of matrix metalloproteinases in human breast cancer tissues. Dis Markers 34:395–405. https://doi.org/10.3233/DMA-130986
Li HC, Cao DC, Liu Y, Hou YF, Wu J, Lu JS, Di GH, Liu G, Li FM, Ou ZL, Jie C, Shen ZZ, Shao ZM (2004) Prognostic value of matrix metalloproteinases (MMP-2 and MMP-9) in patients with lymph node-negative breast carcinoma. Breast Cancer Res Treat 88:75–85. https://doi.org/10.1007/s10549-004-1200-8
Mahmood NA (2015) Matrix metalloproteinases MMP2 and MMP9 expression in stages II-III breast cancer in Iraqi women. J Med Biol Sci Res 1:30–37
McGowan PM, Duffy MJ (2008) Matrix metalloproteinase expression and outcome in patients with breast cancer: analysis of a published database. Annu Oncol 19:1566–1572. https://doi.org/10.1093/annonc/mdn180
Stankovic S, Konjevic G, Gopcevic K, Jovic V, Inic M, Jurisic V (2010) Activity of MMP-2 and MMP-9 in sera of breast cancer patients. Pathol Res Pract 206:241–247. https://doi.org/10.1016/j.prp.2009.12.003
Hollborn M, Stathopoulos C, Steffen A, Wiedemann P, Kohen L, Bringmann A (2007) Positive feedback regulation between MMP-9 and VEGF in human RPE cells. Invest Ophthalmol Vis Sci 48:4360–4367. https://doi.org/10.1167/iovs.06-1234
Sa-nguanraksa D, O-charoenrat P (2012) The role of vascular endothelial growth factor A polymorphisms in breast cancer. Int J Mol Sci 13:14845–14864. https://doi.org/10.3390/ijms131114845
Castañeda-Gill JM, Vishwanatha JK (2016) Antiangiogenic mechanisms and factors in breast cancer treatment. J Carcinog 15:1. https://doi.org/10.4103/1477-3163.176223
Park JH, Rasch MG, Qiu J, Lund IK, Egeblad M (2015) Presence of insulin-like growth factor binding proteins correlates with tumor-promoting effects of matrix metalloproteinase 9 in breast cancer. Neoplasia 17:421–433. https://doi.org/10.1016/j.neo.2015.04.003
Mukherjee D, Zhao J (2013) The role of chemokine receptor CXCR4 in breast cancer metastasis. Am J Cancer Res 3:46–57
Deryugina EI, Quigley JP (2010) Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: contrasting, overlapping and compensatory functions. BBA – Mol Cell Res 1803:103–120. https://doi.org/10.1016/j.bbamcr.2009.09.017
Gialeli C, Theocharis AD, Karamanos NK (2011) Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J 278:16–27. https://doi.org/10.1111/j.1742-4658.2010.07919.x
Lawler J (2002) Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med 6:1–12. https://doi.org/10.1073/pnas.96.26.14888
Fleisch MC, Maxwell CA, Barcellos-Hoff MH (2006) The pleiotropic roles of transforming growth factor beta in homeostasis and carcinogenesis of endocrine organs. Endocr Relat Cancer 13:379–400. https://doi.org/10.1677/erc.1.01112
Kim ES, Kim MS, Moon A (2004) TGF-beta-induced upregulation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK signaling in MCF10A human breast epithelial cells. Int J Oncol 25:1375–1382. https://doi.org/10.3892/ijo.25.5.1375
Krstic J, Santibanez JF (2014) Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells. ScientificWorldJournal 2014:521754. https://doi.org/10.1155/2014/521754
Zarzynska JM (2014) Two faces of TGF-beta1 in breast cancer. Mediators Inflamm 2014: 141747. doi:org/https://doi.org/10.1155/2014/141747.
García-Pardo A, Opdenakker G (2015) Nonproteolytic functions of matrix metalloproteinases in pathology and insights for the development of novel therapeutic inhibitors. Metalloproteinases Med 2:19. https://doi.org/10.2147/MNM.S63629
Mantuano E, Inoue G, Li X, Takahashi K, Gaultier A, Gonias SL, Campana WM (2008) The hemopexin domain of matrix metalloproteinase-9 activates cell signaling and promotes migration of Schwann cells by binding to low-density lipoprotein receptor-related protein. Neuroscience 28:11571–11582. https://doi.org/10.1523/JNEUROSCI.3053-08.2008
Heermann S, Schwab MH (2013) Molecular control of Schwann cell migration along peripheral axons: keep moving! Cell Adh Migr 7:18–22. https://doi.org/10.4161/cam.22123
Cain C (2011) A bid to revive MMP inhibitors. SciBX 4:701. https://doi.org/10.1038/scibx.2011.701
Redondo-Muñoz J, Ugarte-Berzal E, García-Marco JA, del Cerro MH, Van den Steen PE, Opdenakker G, Terol MJ, García-Pardo A (2008) α4β1 integrin and 190-kDa CD44v constitute a cell surface docking complex for gelatinase B/MMP-9 in chronic leukemic but not in normal B cells. Blood 112:169–178. https://doi.org/10.1182/blood-2007-08-109249
Redondo-Muñoz J, Ugarte-Berzal E, Terol MJ, Van den Steen PE, Hernández del Cerro M, Roderfeld Roeb ME, Opdennaker G, García-Marco JA, García-Pardo A (2010) Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia B cell survival through Its hemopexin domain. Cancer Cell 17:160–172. https://doi.org/10.1016/j.ccr.2009.12.044
Ugarte-Berzal E, Bailón E, Amigo-Jiménez I, Vituri CL, del Cerro MH, Terol MJ, Albar JP, Rivas G, García-Marco JA, García-Pardo A (2012) A 17-residue sequence from the matrix metalloproteinase-9 (MMP-9) hemopexin domain binds α4β1 integrin and inhibits MMP-9-induced functions in chronic lymphocytic leukemia B cells. J Biol Chem 287:27601–27613. https://doi.org/10.1074/jbc.M112.354670
Ugarte-Berzal E, Vandooren J, Bailón E, Opdenakker G, García-Pardo A (2016) Inhibition of MMP-9-dependent degradation of gelatin, but not other MMP-9 substrates, by the MMP-9 hemopexin domain blades 1 and 4. J Biol Chem 291:11751–11760. https://doi.org/10.1074/jbc.M115.708438
Dufour A, Zucker S, Sampson NS, Kuscu C, Cao J (2010) Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J Biol Chem 285:35944–35956. https://doi.org/10.1074/jbc.M109.091769
Roeb E, Schleinkofer K, Kernebeck T, Pötsch S, Jansen B, Behrmann I, Matern S, Grötzinger J (2002) The MMP-9 hemopexin domain is a novel gelatin binding domain and acts as an antagonist. J Biol Chem 277:50326–50332. https://doi.org/10.1074/jbc.M207446200
Ezhilarasan R, Jadhav U, Mohanam I, Rao SS, Gujrati M, Mohanam S (2009) The hemopexin domain of MMP-9 inhibits angiogenesis and retards the growth of intracranial glioblastoma xenograft in nude mice. Int J Cancer 124:306–315. https://doi.org/10.1002/ijc.23951
Liu Y, Zhao Y, Lu C, Fu M, Dou T, Tan X (2015) Signatures of positive selection at hemopexin (PEX) domain of matrix metalloproteinase-9 (MMP-9) gene. J Biosci 40:885–890
Overall CM, Kleifeld O (2006) Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer 94:941–946. https://doi.org/10.1038/sj.bjc.6603043
Sela-Passwell N, Rosenblum G, Shoham T, Sagi I (2010) Structural and functional bases for allosteric control of MMP activities: can it pave the path for selective inhibition? BBA – Mol Cell Res 1803:29–38. https://doi.org/10.1016/j.bbamcr.2009.04.010
Rosenblum G, Van den Steen PE, Cohen SR, Grossmann JG, Frenkel J, Sertchook R, Slack N, Strange NW, Opdennaker G, Sagi I (2007) Insights into the structure and domain flexibility of full-length pro-matrix metalloproteinase-9/gelatinase B. Structure 15:1227–1236. https://doi.org/10.1016/j.str.2007.07.019
Bertini I, Fragai M, Luchinat C, Melikian M, Mylonas E, Sarti N, Svergun DI (2009) Interdomain flexibility in full-length matrix metalloproteinase-1 (MMP-1). J Biol Chem 284:12821–12828. https://doi.org/10.1074/jbc.M809627200
Overall CM, Butler GS (2007) Protease yoga: extreme flexibility of a matrixmetalloproteinase. Structure 15:1159–1161. https://doi.org/10.1016/j.str.2007.10.001
Bannikov GA, Karelina TV, Collier IE, Marmer BL, Goldberg GI (2002) Substrate binding of gelatinase B induces its enzymatic activity in the presence of intact propeptide. J Biol Chem 277:16022–16027. https://doi.org/10.1074/jbc.M110931200
Geurts N, Martens E, Van Aelst I, Proost P, Opdenakker G, Van den Steen PE (2008) β-Hematin interaction with the hemopexin domain of gelatinase B/MMP-9 provokes autocatalytic processing of the propeptide, thereby priming activation by MMP-3. Biochemistry 47:2689–2699. https://doi.org/10.1021/bi702260q
Rosenblum G, Meroueh S, Toth M, Fisher J, Fridman R, Mobashery S, Sagi I (2007) Molecular structures and dynamics of the stepwise activation mechanism of a matrix metalloproteinase zymogen: challenging the cysteine switch dogma. J Am Chem Soc 129:13566–13574. https://doi.org/10.1021/ja073941l
Lehti K, Lohi J, Juntunen MM, Pei D, Keski-Oja J (2002) Oligomerization through hemopexin and cytoplasmic domains regulates the activity and turnover of membrane-type 1 matrix metalloproteinase. J Biol Chem 277:8440–8448. https://doi.org/10.1074/jbc.M109128200
Van Den Steen PE, Van Aelst I, Hvidberg V, Piccard H, Fiten P, Jacobsen C, Moestrup SK, Fry S, Royle L, Wormald MR, Wallis R, Rudd PM, Dwek RA, Opdenakker G (2006) The hemopexin and O-glycosylated domains tune gelatinase B/MMP-9 bioavailability via inhibition and binding to cargo receptors. J Biol Chem 281:18626–18637. https://doi.org/10.1074/jbc.M512308200
Itoh Y, Takamura A, Ito N, Maru Y, Sato H, Suenaga N, Aoki T, Seiki M (2001) Homophilic complex formation of MT1-MMP facilitates proMMP-2 activation on the cell surface and promotes tumor cell invasion. EMBO J 20:4782–4793. https://doi.org/10.1093/emboj/20.17.4782
Olson MW, Bernardo MM, Pietila M, Gervasi DC, Toth M, Kotra LP, Massova I, Mobashery S, Fridman R (2000) Characterization of the monomeric and dimeric forms of latent and active matrix metalloproteinase-9 differential rates for activation by stromelysin 1. J Biol Chem 275:2661–2668. https://doi.org/10.1074/jbc.275.4.266
Vandooren J, Born B, Solomonov I, Zajac E, Saldova R, Senske M, Ugarte-Berzal E, Martens E, Van den Steen PE, Damme JV, García-Pardo A, Froeyen M, Deryugina EI, Quigley JP, Moestrup SK, Rudd PM, Saqi I, Opdenakker G (2015) Circular trimers of gelatinase B/matrix metalloproteinase-9 constitute a distinct population of functional enzyme molecules differentially regulated by tissue inhibitor of metalloproteinases-1. Biochem J 465:259–270. https://doi.org/10.1042/BJ20140418
Cha H, Kopetzki E, Huber R, Lanzendörfer M, Brandstetter H (2002) Structural basis of the adaptive molecular recognition by MMP9. J Mol Biol 320:1065–1079. https://doi.org/10.1016/S0022-2836(02)00558-2
Li J, Brick P, O’hare MC, Skarzynski T, Lloyd LF, Curry VA, Clark IM, Biqq HF, Hazleman BL, Cawston TE (1995) Structure of full-length porcine synovial collagenase reveals a C-terminal domain containing a calcium-linked, four-bladed β-propeller. Structure 3:541–549. https://doi.org/10.1016/S0969-2126(01)00188-5
Libson AM, Gittis AG, Collier IE, Marmer BL, Goldberg GI, Lattman EE (1995) Crystal structure of the haemopexin-like C-terminal domain of gelatinase A. Nat Struct Biol 2:938–942. https://doi.org/10.1038/nsb1195-938
Gohlke U, Gomis-Rüth F-X, Crabbe T, Murphy G, Docherty AJP, Bode W (1996) The C-terminal (haemopexin-like) domain structure of human gelatinase A (MMP2): structural implications for its function. FEBS Lett 378:126–130. https://doi.org/10.1016/0014-5793(95)01435-7
Gomis-Rüth FX, Gohlke U, Betz M, Knäuper V, Murphy G, López-Otín C, Bode W (1996) The helping hand of collagenase-3 (MMP-13): 2.7 Å crystal structure of its C-terminal haemopexin-like domain. J Mol Biol 264:556–566. https://doi.org/10.1006/jmbi.1996.0661
Tochowicz AM (2006) Structural analysis of the cancer promoting matrix metalloproteinase – 9 in complexes with novel pharmacological inhibitors, PhD Diss, Max-Plank-Institute fürBiochemie.
Krüger A, Soeltl R, Sopov I, Kopitz C, Arlt M, Magdolen V, Harbeck N, Gänsbacher B, Schmitt M (2001) Hydroxamate-type matrix metalloproteinase inhibitor batimastat promotes liver metastasis. Cancer Res 61:1272–1275
Sinno M, Biagioni S, Ajmone-Cat MA, Pafumi I, Caramanica P, Medda V, Tonti G, Minghetti L, Mannello F, Cacci E (2012) The matrix metalloproteinase inhibitor marimastat promotes neural progenitor cell differentiation into neurons by gelatinase-independent TIMP-2-dependent mechanisms. Stem Cells Dev 22:345–358. https://doi.org/10.1089/scd.2012.0299
Cathcart J, Pulkoski-Gross A, Cao J (2015) Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis 2:26–34. https://doi.org/10.1016/j.gendis.2014.12.002
Jacobsen JA, Jourden JLM, Miller MT, Cohen SM (2010) To bind zinc or not to bind zinc: an examination of innovative approaches to improved metalloproteinase inhibition. BBA – Mol Cell Res 1803:72–94. https://doi.org/10.1016/j.bbamcr.2009.08.006
Kumar A, Bhatnagar S, Kumar A (2010) Matrix metalloproteinase inhibitor batimastat alleviates pathology and improves skeletal muscle function in dystrophin-deficient mdx mice. Am J Pathol 177:248–260. https://doi.org/10.2353/ajpath.2010.091176
Gupta A, Kaur CD, Jangdey M, Saraf S (2014) Matrix metalloproteinase enzymes and their naturally derived inhibitors: novel targets in photocarcinoma therapy. Ageing Res Rev 13:65–74. https://doi.org/10.1016/j.arr.2013.12.001
Löffek S, Schilling O, Franzke CW (2011) Series “matrix metalloproteinases in lung health and disease” edited by J. Müller-Quernheim and O. Eickelberg number 1 in this series: Biological role of matrix metalloproteinases: a critical balance. Eur Resp J 38:191–208
Björklund M, Koivunen E (2005) Gelatinase-mediated migration and invasion of cancer cells. BBA – Rev Cancer 1755:37–69. https://doi.org/10.1016/j.bbcan.2005.03.001
Li H, Ezra DG, Burton MJ, Bailly M (2013) Doxycycline prevents matrix remodeling and contraction by trichiasis-derived conjunctival fibroblasts. IOVS 54:4675–4682. https://doi.org/10.1167/iovs.13-11787
Dahl R, Titlestad I, Lindqvist A, Wielders P, Wray H, Wang M, Samuelsson V, Mo J, Holt A (2012) Effects of an oral MMP-9 and-12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: a randomised controlled trial. Pulm Pharmacol Ther 25:169–177. https://doi.org/10.1016/j.pupt.2011.12.011
Kalva S, Singam ERA, Rajapandian V, Saleena LM, Subramanian V (2014) Discovery of potent inhibitor for matrix metalloproteinase-9 by pharmacophore based modeling and dynamics simulation studies. J Mol Graph Model 49:25–37. https://doi.org/10.1016/j.jmgm.2013.12.008
Björklund M, Heikkilä P, Koivunen E (2004) Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J Biol Chem 279:29589–29597. https://doi.org/10.1074/jbc.M401601200
Liao C, Sitzmann M, Pugliese A, Nicklaus MC (2011) Software and resources for computational medicinal chemistry. Future Med Chem 3:1057–1085. https://doi.org/10.4155/fmc.11.63
Taboureau O, Baell JB, Fernández-Recio J, Villoutreix BO (2012) Established and emerging trends in computational drug discovery in the structural genomics era. Chem Biol 19:29–41. https://doi.org/10.1016/j.chembiol.2011.12.007
Anderson AC (2003) The process of structure-based drug design. Chem Biol 10:787–797. https://doi.org/10.1016/j.chembiol.2003.09.002
Cozza G (2017) The development of CK2i: from traditional pharmacology to in silico rational drug design. Pharmaceuticals 10:26. https://doi.org/10.3390/ph10010026
Morris G, Huey R (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comp Chem 30:2785–2791. https://doi.org/10.1002/jcc.21256
Exner TE, Korb O, Ten BT (2009) New and improved features of the docking software PLANTS. Chem Cent J 3:16. https://doi.org/10.1186/1752-153X-3-S1-P16
Chikhi A, Bensegueni A (2008) Docking efficiency comparison of Surflex, a commercial package and Arguslab, a licensable freeware. J Comput Sci Syst Biol 1:81–86. https://doi.org/10.4172/jcsb.1000007
Jones G, Willett P, Glen RC, Leach AR, Taylor R (1997) Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267:727–748. https://doi.org/10.1006/jmbi.1996.0897
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47:1739–1749. https://doi.org/10.1021/jm0306430
Bissantz C, Folkers G, Rognan D (2000) Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J Med Chem 43:4759–4767. https://doi.org/10.1021/jm001044l
Irwin JJ, Shoichet BK (2005) ZINC− a free database of commercially available compounds for virtual screening. J Chem Inform Model 45:177–182. https://doi.org/10.1021/ci049714+
Ikram NKK, Durrant JD, Muchtaridi M, Zalaludin AS, Purwitasari N, Mohamed N, Rahim ASA, Lam CK, Normi YM, Rahman NA, Amaro RE, Wahab HA (2015) A virtual screening approach for identifying plants with anti H5N1 neuraminidase activity. J Chem Inform Model 55:308–316. https://doi.org/10.1021/ci500405g
Rabow AA, Shoemaker RH, Sausville EA, Covell DG (2002) Mining the National Cancer Institute’s tumor-screening database: identification of compounds with similar cellular activities. J Med Chem 45:818–840. https://doi.org/10.1021/jm010385b
Dufour A, Sampson NS, Li J, Kuscu C, Rizzo RC, DeLeon JL, Zhi J, Jaber N, Liu E, Zucker S, Cao J (2011) Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res 71:4977–4988. https://doi.org/10.1158/0008-5472.CAN-10-4552
Li Z (2012) Identification of small molecules that bind to the hemopexin domain of matrix metalloproteinase-9. The Graduate School, Stony Brook University, Stony Brook, NY
Alford VM, Kamath A, Ren X, Kumar K, Gan Q, Awwa M, Tong M, Seeliger MA, Cao J, Ojima I, Sampson NS (2017) Targeting the hemopexin-like domain of latent matrix metalloproteinase-9 (proMMP-9) with a small molecule inhibitor prevents the formation of focal adhesion junctions. ACS Chem Biol 12:2788–2803. https://doi.org/10.1021/acschembio.7b00758
Remacle AG, Golubkov VS, Shiryaev SA, Dahl R, Stebbins JL, Chernov AV, Cheltsov AV, Pellecchia M, Strongin AY (2012) Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res 72:2339–2349. https://doi.org/10.1158/0008-5472.CAN-11-4149
Acknowledgements
We acknowledge the Language Centre of Sanata Dharma University for the help in proof reading.
Funding
This article is a part of a grant by the Center of Research and Community Service (LPPM) 2017, Sanata Dharma University, Indonesia, which provides financial support for the data collection and also for writing the manuscript. Indonesia Toray Science Foundation 2017 has roles in funding the study design, analysis and data interpretation.
Author information
Authors and Affiliations
Contributions
MH organised the project and the article writing. CFA, DPKSL and EA are students under MH supervision who prepared the manuscript. RIJ and BAN contributed to the writing style and proof reading. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The authors declare that the work did not involve humans or animals which required ethical approval or consent to participate.
Consent for publication
The authors declare that the work did not involve human’s data which required ethical consent for publication.
Competing interests
The authors declare they have no competing interest regarding this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Adhipandito, C.F., Ludji, D.P.K.S., Aprilianto, E. et al. Matrix metalloproteinase9 as the protein target in anti-breast cancer drug discovery: an approach by targeting hemopexin domain. Futur J Pharm Sci 5, 1 (2019). https://doi.org/10.1186/s43094-019-0001-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-019-0001-1