
Abstract
Background
The 1,3,4-thiadiazoles are among the structural moieties that were found to be of utmost importance in the fields of pharmacy and agrochemicals because of their widespread biological activity that includes anti-tumor, antibacterial, anti-inflammatory, antihypertensive, anti-tuberculosis, anticonvulsant, and antimicrobial, among others.
Results
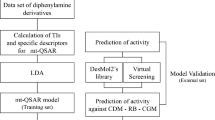
QSAR and molecular docking studies were carried out on thirty-two (32) derivatives of 2,5-disubstituted-1,3,4-thiadiazoles for their antifungal activities toward Phytophthora infestans. Using the “graphical user interface” of Spartan14 software, the structure of the compounds of the dataset is drawn and then optimized at DFT/B3LYP/6-31G* quantum mechanical method of the software. Molecular descriptors of the optimized compounds were calculated and later on divided into the training set and test sets (at a ratio of 3:1). The training set was used for model generation and the test set was for external validation of the generated model. Four models were generated by the employment of genetic function approximation (GFA) in which the optimal model (4) turned out to have the following statistical parameters: R2 = 0.798318, R2adj = 0.750864, cross-validation R2(Q2cv) = 0.662654, and external validation R2pred = 0.624008. On the molecular docking study of thiadiazole compounds with the target protein of Phytophthora infestans effector site (PDB ID: 2NAR ), compound 13 shows the highest binding affinity with − 9.3 kcal/mol docking score and composes hydrophobic as well as H-bond interactions with the target protein (2NAR).
Conclusion
The result of the QSAR study signifies the stability and robustness of the built model by considering the validation parameters and this gave an idea of template/ligand-based design while the molecular docking study revealed the binding interaction between the ligand and the protein site which gave an insight toward an “optimization method” of the structure-based design for the discovery of more potent compounds with better activity against Phytophthora infestans using the approach of computer-aided drug design (CADD) in plant pathology.
Similar content being viewed by others

1 Background
Phytophthora infestans (also called potato blight) may be the most destructive of all plant pathogens that excessively damage potato/Irish potato leading to famines and immigration in the nineteenth century [13, 14]. Some of the signs and symptoms of this disease can be seen as a white color in potato. P. infestans generate some sporanges on the steam and leaves of potato [15]. The sporanges always displayed at the lower superficies of the leaves. However, as in the case of tuber blight, the white hypha usually appears at the superficies of the tuber [11]. In normal circumstances, P. infestans perfect its life on potato or tomato leaves at approximately 5 days [22]. The sporanges formed at the surface of foliage thereby dispersing through plants at over 10 °C (50 °F) temperature and humidity of above 75–80% in two or more days. Sometimes the spores are washed away by the rain which gets into the soil and infect the early-stage tubers; and also, these spores can make long distances into the air which can easily get into another host. The premature levels of the blight may have disappeared. Some of the symptoms involve dark blotches displayed at the extreme end of the leaf and on the plant’s stem. A grey/dark patch developed on the affected tuber which covered the skin and rapidly decomposed it to an unpleasant odor. And apparently, healthy tubers may later become rotten while stored. According to the FOA report, the most thrusting/attacking problem in the third world apart from poverty must be food shortages. Farmers in Africa are encountering distinct limitations in food production as well as cash crops. Some of those limitations include damages from diseases and pests like fungi. In the search for food and the fight for human survival, the Irish potato has a significant role to play in food supply and, therefore, has been an instrument in addressing the issues of food insecurity, due to its performance in a given area and in a given time. This potato blight cause excessive economic loose, the annual economic loose caused by P. infestans in the developing countries begins to approach a $3-billion mark [5]. Due to its rapid adaptation to the various management skills (such as genetic resistivity), control of this plant pathogen is really challenging [10]. And this makes the synthesis of novel compounds that will inhibit the dangerous P. infestans to be among the most considerable in the field of agrochemicals. Some of these researches include computational studies.
The 1,3,4-thiadiazoles derivatives are among the structural moieties that were found to be of utmost importance in the fields of pharmacy and agrochemicals for their widespread biological activity such as anti-tumor [28], antibacterial [25], anti-inflammatory [19], antihypertensive [30], antituberculosis [23], anticonvulsant [18], and antimicrobial [2], among others. Furthermore, reports identify that compounds containing furan are intensively bioactive. Several researches on the derivatives of furan such as “pyrazole and triazole [6], diacyl-hydrazine derivatives [7]” containing 5-phenyl-2-furan moiety were carried out in which there appeared to have extensive biological activities including fungicidal and insecticidal activities, among others.
The Quantitative structure–activity relationship (QSAR) study aims to develop correlation models considering the activity of compounds and other chemical information in a statistical approach [16, 27] which will lead us to the design of new compounds. While molecular docking study is “a way of predicting the favorable orientation of one molecule to another when reacted to produce a stable complex”, it will also lead us to the design of more potent compounds.
Our aim in this research work is to predict highly active compounds by the employment of Genetic function approximation (GFA) and perform a molecular docking study between the 1,3,4-thiadiazole compounds and the 2NAR protein of P. infestans to predict their stable molecular orientation.
2 Methods
2.1 Dataset
Thirty-two derivatives of 2,5-disubstituted-1,3,4-thiadiazole derivatives containing 5-phenyl-2-furan used in this work were taken from the literature [8]. The activity of the compounds were reported in EC50 (g/L) values, which were converted to pEC50 (pEC50 = − log1/EC50). Presented in Fig. 1 and Table 1 are the molecular structures and their corresponding activities found in the dataset.

Parent structure of the dataset compounds
2.2 Molecular structure optimization
The structures of the compounds were optimized at the “Density function theory (DFT)” level, “Becke’s three-parameter Lee-Yang-Parr hybrid functional (B3LYP)” version together with the “6-31G*” basis set of Spartan14 [4]. In this process, all the molecular structures were drawn in the graphical user interface of Spartan14 software. The energies of the drawn molecules were minimized using Molecular Mechanics Force Field (MMFF) calculation [3].
2.3 Molecular descriptor calculations
Molecular descriptors are the properties of the molecule in numerical/mathematical values. PaDEL descriptor software was used to further calculate additional energy of those low-energy conformers, where a total of 1875 descriptors were calculated.
2.4 Dataset splitting
Using Kennard–Stone algorithm technics, the dataset of 32 compounds was split into two: the training set and the test set (70% of the training and 30% to the test set) which is found in DatasetDivision GUI 1.2 software. In this technic, the training and the test set were used for model development and its validation (externally) [12].
2.5 Model building
The training set in the dataset was used for model generation through the employment of the GFA method available in the material studio. The regression analysis occurs by considering the inhibition concentration (pEC50) as a dependent variable while the chosen descriptors served as independent variables.
2.6 Internal validation
Internal validation of 22 compounds of the training set took place in the software (Material studio) used for building the model. The validation parameters are as follows:
2.6.1 Cross-validation
This parameter was used to determine the ability of the QSAR model in predicting the activities of newly designed compounds. This indicates the stability of the built model.
where Yexp is the “observed/experimental activity”, Ypred is the “predicted activity”, and \( \overline{Y} \) is the “mean value of the observed activity”.
2.6.2 Friedman’s lack of fit (LOF)
The parameter describes the measure of the fitness of the model and it is given by equation. ii below:
where SEE is the standard error,
C is the “number of terms in the model”, d is the “user-defined smoothing parameter”, P is “the total number of descriptors in the model”, and M is “the number of molecules in the training set”.
The regression model is given by the straight line graphs’ equation, “(Y = mx + c)”,
where Y is the predicted activity (pEC50), D is the corresponding coefficients, x is the independent variable, and c is the regression constant [17].
2.6.3 The correlation coefficient (R2)
This is another parameter used to assess the model. The closer the value of R2 to 1.0, the better the model generated. R2 is expressed as:
The value of R2 changes instantly with an increase in descriptors; therefore, the reliability of R2 in measuring the stability of a given model is very minimal. Thus, R2 has to be adjusted in order to have a fit and strong model. The following equation define adjusted R2 as [1]:
where P is the number of independent variables possessed by the model and n is the number of training sets’ compounds [21].
2.7 External validation
The model generated was further validated with the test set of the dataset in order to measure its level of competence in predicting the activity of new compounds. This was done by evaluating the values of the square of the calculated R2 of the test set values. The closer the R2 is to 1.0, the better the robustness, fitness, and the prediction capacity of the model as well. Though sometimes R2 value does not matter if the model fails other statistical analyses such as variance inflation factor (VIF) and mean effect, among others. The coefficient of determination R2pred is given by the following equation:
where \( {Y}_{{\mathrm{pred}}_{\mathrm{test}}} \) and\( {Y}_{\exp_{\mathrm{test}}} \) are the values of predicted and experimental activities for the test set and \( \overline{Y} \)train is the average activity for the training sets’ values [3].
2.8 Statistical analysis of the descriptors
2.8.1 Variance inflation factor (VIF)
VIF is defined as the measure of multicollinearity amongst the independent variables (i.e., descriptors). It quantifies the extent of correlation between one predictor and the other predictors in a model.
where R2 gives multiple correlation coefficient between the variables within the model. If the VIF is equal to 1, it means there is no intercorrelation in each variable, and if it ranges from 1 to 5, then it is said to be suitable and acceptable. But if the VIF turns out to be greater than 10, this indicates the instability of the model and need to be reexamined ([20, 26].
2.8.2 Mean effect (ME)
The average effect (mean effect) correlates the effect or influence of given molecular descriptors to the activities of the compounds that made up the model. The descriptor signs show the direction of their deviation toward the activity of the compounds. That is to say, an increase or decrease in the value of the descriptors will improve the activity of the compounds. The mean effect is defined by the following:
where Bj and Dj are the j-descriptor coefficient in the model and the values of each descriptor in training set, while m and n stand for the number of molecular descriptors as well as the number of molecules in a training set. To evaluate the significance of the model, the mean effect of each descriptor was calculated [9].
2.8.3 Applicability domain
To confirm the reliability of the model and to examine the outliers as well as the influential compounds, it is very important to evaluate the applicability domain of the built model. Its aim at predicting the uncertainty of a compound depends on its similarities to the compounds used in building the model and also the distance between the training and test set of the compounds. This can be achieved by employing William’s plot which was plotted using standardized residuals versus the leverages. The leverages for a particular chemical compound is given as follows:
where hi is the leverage for a particular compound and Zi is the matrix i of training set. Z is the nxk descriptor-matrix for a training set compound. ZT is the transpose of the Z matrix. The warning leverage (h*) that is the boundary for normal values of Z outliers is given by;
Where n is the number of molecules in the training set whereas p gives the amount of descriptors presence in the built model [17].
2.9 Molecular docking studies
With the aid of Autodock Vina of Pyrex software and Discovery Studio, a molecular docking study was performed between 2,5-disubstituted-1,3,4-thiadiazole derivatives and P. infestans effector target site to examine the interaction between the binding pocket of the effector and the compounds (i.e., the ligands). A highly resolute crystal structure of P. infestans was downloaded successfully from the protein databank (PDB Code: 2NAR). The downloaded substrate was carefully prepared using Discovery Studio which was later transported to the Pyrex for the docking calculation. With the aid of Spartan14 version 1.1.4, the optimized compounds of 2,5-disubstituted-1,3,4-thiadiazole derivatives (the ligands) were converted to PDB files [24]. The prepared structure of P. infestans effector site and prepared ligands were docked using Autodock Vina 4.2 [29]. Discovery Studio Visualizer was also used to visualize the docking results (Fig. 2).

X-ray structure of the downloaded protein and the prepared ligand
3 Results
3.1 Model building and validation
Below is the equation of the best-chosen model (4).
The validation parameters are shown in Tables 2 and 3 below.
3.1.1 Model 1
In the first model, pEC50 = 0.244535617 × BCUTp-1l − 22.874691031 × SCH-6 + 0.213428935 × WA.mass − 0.025525444 × Wgamma3.volume + 10.325883792, R2teat = 0.395084, R2train = 0.824826, R2adj = 0.783609, R2cv = 0.56979, Ntest = 10, Ntrain = 22, LOF = 0.12303, and m in experimental error for non-significant LOF (95%) = 0.12303.
3.1.2 Model 2
In the second model, pEC50 = 0.297814107 × nCl + 0.168441873 × nBondsS3 + 0.001197233 × PPSA-1 − 0.024107696 × Wgamma3.volume + 0.268877261, R2teat = 0.206664, R2train = 0.807874, R2adj = 0.762668, R2cv = 0.558932, Ntest = 10, Ntrain = 22, LOF = 0.12885, and min experimental error for non-significant LOF (95%) = 0.128845.
3.1.3 Model 3
In the third model, pEC50 = 0.139831691 × nHeavyAtom + 0.314911162 × nCl + 0.001443139 × PPSA-1 − 0.024455939 × Wgamma3.volume − 2.325579534, R2teat = 0.3681753, R2train = 0.800593, R2adj = 0.753674, R2cv = 0.516043, Ntest = 10, Ntrain = 22, LOF = 0.13126, and min experimental error for non-significant LOF (95%) = 0.131264.
Table 4 and Table 5 presented the external validation and calculation of predicted R2 of the chosen model.
3.2 Statistical analyses of the descriptors
The following are the different analyses: Pearson’s correlation, standard regression coefficients, standardized predicted activity against experimental activity, standardized residual against experimental activity (pEC50), and William’s plot.
3.3 The results of the docking study
The results can be seen in the receptor–ligand interaction, H-bond interactions, and hydrophobic and electrostatic interactions.
4 Discussion
4.1 QSAR model
The best QSAR model was generated using the GFA method. Four descriptors were used in building the model where four different models were generated and model 4 found to be the best following the statistical parameters. All the values obtained matches the minimum value for evaluating the QSAR model. These values signify that there is a high correlation between the predicted and experimental activity (pEC50, Fig. 3). Internal and external validations, as well as the other statistical analysis, made the model 4 to stand fit, reliable, and highly predictive.

Plot of predicted activity against experimental activity (pEC50)
From Tables 2 and 4, the R2 values of 0.79831800 (internal) and 0.624008 (external) indicate a strong relationship between the experimental and predicted activities. Additionally, the inhibition activities of the compounds increase by the addition of all the descriptors in the best chosen model.
4.2 Interpretation of descriptors
The 2D molecular descriptors AMR and SCH-7 defined as “Molar refractivity” and “Simple chain, order 7” are the first and second highest contributors toward the generation of the selected model with a positive mean effect of 0.52115 and 0.4413. Thus, the addition of those descriptors will significantly enhance the antifungal activity of the compound. 3D descriptors WG.unity and Wnu2.eneg with the mean effect of 0.01001 and 0.02754 have a low effect on the model therefore their increase will have no much significant on the activity of the compound. They are defined as “Non-directional WHIM, weighted by unit weights” and “Directional WHIM, weighted by Mulliken atomic electronegativities”.
Model 4 was examined as the optimal model considering the descriptors from test set compounds of the dataset.
The experimental activity, predictive activity, and residual values of the compounds are given in Table 6. The residual value is defined as the difference between the experimental and predicted activities. The lower residual values between the experimental and predicted activities indicate the high predictive power of the model.
4.3 Statistical analysis of descriptors
Pearson's correlation (Table 7) was performed between the descriptors of the chosen model in order to evaluate the relationship between each of the descriptors. The result of the correlation showed no intercorrelation among the descriptors with a correlation coefficient of less than 0.5, which signified that the descriptors used in the model were good enough. The VIF values are within the range of 1 to 5 which indicated that the descriptors and model are suitable and acceptable.
Table 8 showed the standard regression coefficients “bj”, the values of mean effect (MF), and confidence interval (p values). These give vital information on the effect and contribution of the descriptors toward the built model. The individual capability and inducing power of the selected descriptors toward the activity of the compounds depend on their values, signs, and their mean effects as well. The p values of the four descriptors (at a confidence limit of 95%) that made up the model are all less than 0.05; this implies that there is a significant relationship among the descriptors (as contrary to the null hypothesis) and the inhibitory concentration of the compounds.
Figure 4 which presented a graph of observed activity versus standardized residual shows a random dispersion at the baseline where the standardized residual is zero. Therefore, no systematic error occurred in the built model.

Plot of standardized residual against experimental activity (pEC50)
The graph of standardized residuals versus leverages (for all the training set and test set) termed the William’s plot shown in Fig. 5. The domain of applicability is established within a box at ± 3.0 limit for the residuals and a leverage threshold h* (h* = 0.68). This William’s plot functions to figure out the outliers as well as the influencing compounds in the model. Our results revealed that two compounds of the test set (with pEC50 of 1.84011 and 2.05115) were outside the applicability domain which signified that the compound may be structurally different from other compounds in the dataset. Thus, the compound was outside the warning leverage h* which was found calculated as 0.68.

William’s plot
4.4 The docking study
Molecular docking was run between the protein of P. infestans effector target site (PDB ID: 2NAR; >95% purity) and the ligands to investigate/examine the mode of interaction of the ligands with the macromolecular target site of the protein. The interaction of all the 32 compounds with the receptor active site was carried out in which the receptor–ligand interactions with lower energy, i.e., those with better docking scores, were recorded in Table 9. The table consists of the ligands with their binding affinity, the H-bonds, H-bond distances, as well as their hydrophobic and electrostatic interactions. The binding affinity for all the compounds is between the range of − 8.2 to − 9.3 kcal/mol. Compound 13 possessed the highest binding score with − 9.3 kcal/mol and showed an interaction mode with H-bonds (GLU88 with H-bond distance of 2.78089Å and GLN67Å with H-bond distance of 2.91512Å), hydrophobic interaction mode of TYR87 (4.7572Å), TYR71, LEU52, TYR87 (4.88051Å), and ALA69 residues.
Figure 6 showed a receptor–ligand interaction while Fig. 7 is the 2D structure which shows that H-bond interaction exists between the receptor and the compound 13 which has a better binding affinity and showed a better interaction with the macromolecular target site of the residue when compared with other compounds as well.

Receptor–ligand interaction

2D structure showing H-bond interactions between receptor and compound 13
5 Conclusion
This research involves a QSAR and molecular docking studies on 32 compounds of 2,5-disubstituted-1,3,4-thiadiazole derivatives against P. infestans effector site. After using DFT to optimize the compounds, GFA was used to generate the built model. Among the four generated models, the fourth model was found to be the optimal, having appreciable statistical parameters with R2 = 0.798318, R2adj = 0.750864, cross-validation R2 (Q2cv) = 0.662654, and external validation R2pred = 0.624008. Descriptors AMR and SCH-7 were the first and second highest contributors toward the generation of the selected model, and thus, their increase will increase the activity of the compound while WG.unity and Wnu2.eneg have a low effect on the model, therefore, their increase will have no much significance on the activity of the compound against P. infestans.
According to the docking scores, almost all the ligands (compounds) showed high binding affinity/strong inhibition activity against P. infestans effector site. However, ligands 11, 13, 14, 15, 17, 24, 26, and 30 showed higher binding affinity ranging from − 8.9 to − 9.3 kcal/mol. With ligand 13 having the highest binding energy of − 9.3 kcal/mol. This compound [13] was able to strongly dock at the binding pocket of the P. infestans effector site (2NAR) producing an H-bond as well as hydrophobic interaction with the target site.
The generated QSAR model provides a worthy idea on ligand-based design whereas the molecular docking analysis suggested an approach toward the structure-based design of novel and more potent compounds against P. infestans.
Abbreviations
- B3LYP:
-
Becke’s three-parameter read Yang-Parr hybrid
- DFT:
-
Density function theory
- GFA:
-
Genetic function approximation
- PDB:
-
Protein data bank
- P. infestans :
-
Phytophthora infestans
- QSAR:
-
Quantitative structure–activity relationship
References
Adeniji SE, Uba S, Uzairu A (2018) QSAR modeling and molecular docking analysis of some active compounds against Mycobacterium tuberculosis receptor (Mtb CYP121). J Pathog 2018(1018694):24
Almajan GL, Barbuceanu SF, Saramet I, Draghici C (2010) New 6-amino-[1, 2, 4] triazolo [3, 4-b] [1, 3, 4] thiadiazines and [1, 2, 4] triazolo [3, 4-b] [1, 3, 4] thiadiazin-6-ones: synthesis, characterization and antibacterial activity evaluation. Eur J Med Chem 45(7):3191–3195
Arthur DE (2017) Toxicity modelling of some active compounds against k562 cancer cell line using genetic algorithm-multiple linear regressions. J Turkish Chem Soc, Section A: Chem 4(1):355–374
Benarous N, Cherouana A, Aubert E, Durand P, Dahaoui S (2016) Synthesis, characterization, crystal structure and DFT study of two new polymorphs of a Schiff base (E)-2-((2, 6-dichlorobenzylidene) amino) benzonitrile. J Mol Struct 1105:186–193
CABI International, (2018). Phytopthora infestans (Phytopthora blight). Retrieved from https://www.cabi.org/isc/datasheet/40970.
Cui ZN, Shi YX, Zhang L, Ling Y, Li BJ, Nishida Y, Yang XL (2012) Synthesis and fungicidal activity of novel 2, 5-disubstituted-1, 3, 4-oxadiazole derivatives. J Agric Food Chem 60(47):11649–11656
Cui Z, Li X, Tian F, Yan X (2014) Synthesis and bioactivity of 5-substituted-2-furoyl diacylhydazide derivatives with aliphatic chain. Int J Mol Sci 15(5):8941–8958
Cui ZN, Li YS, Hu DK, Tian H, Jiang JZ, Wang Y, Yan XJ (2016) Synthesis and fungicidal activity of novel 2, 5-disubstituted-1, 3, 4-thiadiazole derivatives containing 5-phenyl-2-furan. Sci Rep 6:20204
Edache EI, Hambali HU, Arthur DE, Oluwaseye A, Chinweuba OC (2016) In-silico discovery and simulated selection of multi-target Anti-HIV-1 inhibitors. Int Res J Pure Appl Chem 11(1):1–15
Fry W (2008) Phytophthora infestans: the plant (and R gene) destroyer. Mol Plant Pathol 9(3):385–402
Fry WE, Grünwald NJ (2010) Introduction to oomycetes. The Plant Health Instructor. https://doi.org/10.1094/PHI-I-2010-1207-01.
Gramatica P, Cassani S, Roy PP, Kovarich S, Yap CW, Papa E (2012) QSAR modeling is not “push a button and find a correlation”: a case study of toxicity of (benzo-) triazoles on algae. Mol Inform 31(11-12):817–835
Griffiths RG, Dancer J, O'Neill E, Harwood JL (2003) A mandelamide pesticide alters lipid metabolism in Phytophthora infestans. New Phytol 158(2):345–353
Haas BJ, Kamoun S, Zody MC, Jiang RH, Handsaker RE, Cano LM et al (2009) Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461(7262):393
Henfling JW (1987) Late blight of potato: Phytophthora infestans, Technical Information Bulletin 4. International Potato Center, Lima
Ibezim EC, Duchowicz PR, Ibezim NE, Mullen LMA, Onyishi IV, Brown SA, Castro EA (2009) Computer-aided linear modeling employing QSAR for drug discovery. Sci Res Essays 4(13):1559–1564
Ibrahim MT, Uzairu A, Shallangwa GA, Ibrahim A (2018) Computational studies of some biscoumarin and biscoumarin thiourea derivatives as α-glucosidase inhibitors. J Eng Exact Sci 4(2):0276–0285
Jatav V, Mishra P, Kashaw S, Stables JP (2008) CNS depressant and anticonvulsant activities of some novel 3-[5-substituted 1,3,4-thiadiazole-2-yl]-2-styryl quinazoline-4 (3H)-ones. Eur J Med Chem 43(9):1945–1954
Kadi AA, Al-Abdullah ES, Shehata IA, Habib EE, Ibrahim TM, El-Emam AA (2010) Synthesis, antimicrobial and anti-inflammatory activities of novel 5-(1-adamantyl)-1,3,4-thiadiazole derivatives. Eur J Med Chem 45(11):5006–5011
Karthikeyan C, Moorthy NHN, Trivedi P (2009) QSAR study of substituted 2-pyridinyl guanidines as selective urokinase-type plasminogen activator (uPA) inhibitors. J Enzyme Inhib Med Chem 24(1):6–13
Mustapha A, Shallangwa G, Ibrahim MT, Bello AU, Ebuka DA, Uzairu A, Mamza P (2018) QSAR studies on some C14-urea tetrandrine compounds as potent anti-cancer against leukemia cell line (K562). J Turkish Chem Soc, Section A: Chem 5(3):1387–1398
Nowicki M, Lichocka M, Nowakowska M, Kłosińska U, Kozik EU (2012) A simple dual stain for detailed investigations of plant-fungal pathogen interactions. Vegetable Crops Res Bull 77:61–74
Oruç EE, Rollas S, Kandemirli F, Shvets N, Dimoglo AS (2004) 1,3,4-thiadiazole derivatives. Synthesis, structure elucidation, and structure−antituberculosis activity relationship investigation. J Med Chem 47(27):6760–6767
Parvatham K, Veerakumari L, Shoba G (2015) Molecular docking studies of acetate-succinate CoA-transferase of Ascaris lumbricoides with a few phytochemicals and anthelmintics. J Comput Methods Mol Design 5(4):1–10
Plech T, Wujec M, Kosikowska U, Malm A, Kaproń B (2012) Studies on the synthesis and antibacterial activity of 3,6-disubstituted 1,2,4-triazolo [3, 4-b]1,3,4-thiadiazoles. Eur J Med Chem 47:580–584
Pourbasheer E, Aalizadeh R, Ganjali MR, Norouzi P (2014) QSAR study of IKKβ inhibitors by the genetic algorithm: multiple linear regressions. Med Chem Res 23(1):57–66
Roy K, Kar S, Das RN (2015) A primer on QSAR/QSPR modeling: fundamental concepts. Springer
Taher AT, Georgey HH, El-Subbagh HI (2012) Novel 1,3,4-heterodiazole analogues: synthesis and in-vitro antitumor activity. Eur J Med Chem 47:445–451
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461
Vio L, Mamolo MG, Laneve A (1989) Synthesis and antihypertensive activity of some 1,3,4-thiadiazole derivatives. Farmaco (Societa chimica italiana: 1989) 44(2):165–172
Acknowledgements
The authors acknowledge the technical effort of the Department of Chemistry, Ahmadu Bello University, Zaria, Nigeria, and Muhammad Tukur Ibrahim for his useful advice toward the successful completion of the research.
Availability of data and material
Not applicable
Funding
The authors declare that no funding has been received.
Author information
Authors and Affiliations
Contributions
YI contributed throughout the research work. AU gave directives and technical advices. SU partook in the technical activities. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Isyaku, Y., Uzairu, A. & Uba, S. QSAR and molecular docking studies of novel 2,5-distributed-1,3,4-thiadiazole derivatives containing 5-phenyl-2-furan as fungicides against Phythophthora infestans. Beni-Suef Univ J Basic Appl Sci 9, 11 (2020). https://doi.org/10.1186/s43088-020-0037-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43088-020-0037-5