
Abstract
Background
Quantitative structure-activity relationships (QSAR) is a technique that is used to produce a model that connects biological activities of compounds to their chemical structures, and molecular docking is a technique that reveals the binding mode and interactions between a drug and its target enzyme. These techniques have been successfully applied in the design and development of many drug candidates and herein were employed to build a model that could help in the development of more potent antimalaria drugs.
Results
Descriptors of the compounds were calculated using the PaDEL-Descriptor software, and Genetic Function Algorithm (GFA) was used to select descriptors and build the model. A robust and reliable model was generated and validated to have internal and external squared correlation coefficient (R2) of 0.9622 and 0.8191, respectively, adjusted squared correlation coefficient (Radj) of 0.9471, and leave-one-out (LOO) cross-validation coefficient (Q2cv) of 0.9223. The model revealed that the antiplasmodial activities of 1,2,4,5-tetraoxane-8-aminoquinoline hybrids depend on MATS3m, GATS8p, GATS8i, and RDF50s descriptors. MATS3m, GATS8i, and RDF50s influenced the antiplasmodial activities of the compounds positively while GATS8p negatively with the greatest influence. The docking result shows strong interactions between 1,2,4,5-tetraoxane-8-aminoquinoline hybrids and Plasmodium falciparum lactate dehydrogenase (pfLDH) with binding affinities ranging from − 6.3 to − 10.9 kcal/mol which were better than that of chloroquine (− 6.1 kcal/mol), suggesting that these compounds could be better inhibitors of pfLDH than chloroquine.
Conclusion
The results of this study could serve as a model for designing new potent 1,2,4,5-tetraoxane-8-aminoquinolines with better antiplasmodial activities for the development of highly active antimalaria drugs.
Similar content being viewed by others

1 Background
Malaria accounts for most of the deaths in the world especially in children and pregnant women. This infection affected about 219 million persons in 2017 where 92% of this number was from WHO African countries [1]. And it claimed about 435,000 deaths worldwide in 2017 where 61% (266 000) was from children below the age of 5 years [1]. Five species of Plasmodium parasites cause malaria in humans, and these species are P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi [2]. Plasmodium falciparum is the most prevalent and the most deadly of the five species in the WHO regions of Africa, South-East Asia, Eastern Mediterranean, and the Western Pacific, which is responsible for 99.7%, 62.8%, 69%, and 71.9%, respectively, of the malaria cases in 2017, while P. vivax is the most prevalent in the WHO region of the America which is responsible for 74.1% [1, 2]. Medicinal chemists tested numerous compounds against Plasmodium parasites to find their most efficient inhibitors. Aminoquinolines such as chloroquine has been used for several decades as the first-line antimalarial drug [3]. Though its efficacy has been diminished due to Plasmodium falciparum resistance [4, 5], it remained efficacious in some Caribbean countries and Central America [6]. Since 2005, the World Health Organization recommended the use of artemisinin-based combination therapies (ACTs) for the treatment of P. falciparum malaria [7, 8]. These chemotherapies show excellent efficacy especially in the African region. But, the development and spread of parasites resistance to any antimalarial drug are very likely as experience with other antimalarial drugs [9]. Furthermore, resistance to ACTs has been observed and reported to be of an increase in Southeast Asia and its spread to other regions is seriously challenging [10,11,12], hence, the need for a promising antimalarial drugs.
Combinational therapies like ACTs are costly and have more toxic side effects than single drugs due to drug-drug interaction [13]. The alternatives to ACTs are hybrid compounds and molecules containing two or more active pharmacophores that can act simultaneously on two or more molecular targets [14,15,16,17]. Such molecules are active against erythrocytic and live stages of malaria infection; therefore, they can help in fighting resistance and meeting the agenda of eradicating malaria [18]. Nowadays, the search for antimalarials focuses on hybrid compounds containing quinoline which is one of the important pharmacophore acting against malaria [19,20,21].
Conventional drug discovery methods are expensive and time-consuming requiring the sacrifice of animals or compounds in their pure forms [22]. Effective and efficient techniques that can screen chemical databases of molecules with known activities against a particular infection are necessary [23]. Quantitative structure-activity relationship (QSAR) modeling and molecular docking studies have been successfully used in the development of drugs as cost- and time-effective techniques [24, 25]. QSAR is a significant modeling method for structural optimization and drug design [23, 26]. Herein, we conducted a QSAR study of tetraoxane-8-aminoquinoline hybrids as dual-stage antimalarial agents to produce a model that could be used to design new potent antimalaria therapy. We also carried out a molecular docking study of the hybrid compounds with Plasmodium falciparum lactate dehydrogenase (pfLDH) enzyme to investigate the interaction of the hybrids with potential target enzyme. Tetraoxane-8-aminoquinoline hybrids were reported to be metabolically stable and active against both erythrocytic and liver-stage malaria parasites [21].
2 Methods
2.1 Data collection
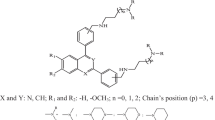
Twenty-two compounds of 1,2,4,5-tetraoxane-8-aminoquinoline hybrids and their in vitro antimalarial activities (EC50) against intraerythrocytic P. falciparum W2 strain were obtained from the paper published by Capela and coworkers [21] and used herein. The antiplasmodial activities of the compounds reported in EC50 (μM) were transformed to pEC50 (pEC50 = − logEC50) for the purpose of this research. Structures of the molecules and their activities were presented in Table 1.
2.2 Geometric optimization
The structures of the molecules shown in Table 1 were drawn and optimized using the ChemDraw version 12.0.2 software [27] and Spartan 14 Version 1.1.4 software with semi-empirical (PM3) quantum mechanics method [28], respectively.
2.3 Molecular descriptor calculation
A total of 1875 molecular descriptors of the optimized molecules of 1,2,4,5-tetraoxane-8-aminoquinoline hybrids were computed with PaDEL-Descriptor software version 2.20 [29].
2.4 Normalization and data pretreatment
Using Eq. (1), the obtained descriptors were normalized so that each variable will have equal opportunity in influencing the construction of a good model [30].
where X is the normalized descriptors, Xi is the descriptor’s value for each molecule, Xmin and Xmax are the minimum and maximum value for each descriptor. To eliminate redundancy in the normalized data, it was then pretreated using the Data Pretreatment software gotten from Drug Theoretical and Cheminformatics Laboratory (DTC Lab).
2.5 Data division
Kenard and Stone’s algorithm [31] was employed to divide the pretreated data into a training set (70%) for model generation and a test set (30%) for external validation of the model. This was achieved using the Data Division software gotten from Drug Theoretical and Cheminformatics Laboratory (DTC Lab).
2.6 Model generation
Using the genetic function approximation (GFA) technique in the Material Studio software, regression analysis was carried out to generate the model (using training set) with the activities in pEC50 as the dependent variable and the descriptors of independent variable.
2.7 Internal validation of the model generated
The model generated was assessed using Friedman formula [32] and defined as:
where LOF is the Friedman’s lack fit (a measure of fitness of a model), SEE is the standard error of estimation, p is the total number of descriptors in the model, d is the user-defined smoothing parameter, c is the number of terms in the model, and M is the number of compound in the training set.
SEE is defined as:
which is the same as the standard deviation of the model and if its value is low, a model is said to be good.
The correlation coefficient R2 of a built model is another parameter considered and the closer it is to 1.0, the better the model is built. R2 is expressed as:
where Yprd, Yexp, and Ymtrn are the predicted, experimental, and mean experimental activities in the training set, respectively.
The value of R2 is directly proportional to the number of descriptors; hence, the stability of the model is not reliable on it. Thus, to have a reliable and stable model, R2 is adjusted according to the expression:
where p is the number of descriptors in the model and n is the number of compounds used in the training set.
The cross-validation coefficient, Q2cv, is expressed as:
where Yprd, Yexp, and Ymtrn are the predicted, experimental, and average experimental activity in the training set, respectively.
2.8 External validation of the model generated
The generated model was assessed (using test set) for external validation by the value of R2test expressed as:
where Yprd and Yexp are respectively the predicted and experimental activities of the test set, and Ymtrn is the mean experimental activity of the training set. The closer the value is to 1.0, the better the model generated [33].
2.9 Y-randomization test
Random multi-linear regression models were generated (using training set) in the Y-randomization test whose average R2 and Q2 values have to be low for the QSAR model to be robust [33]. Coefficient of determination, cR2p, whose value has to be greater than 0.5 for passing this test is also calculated in the Y-randomization test and is expressed as:
where R is the correlation coefficient for Y-randomization and R2r is the average “R” of the random models.
2.10 Applicability domain of the generated model
Leverage (hi) method was used in describing the applicability domain of the QSAR models [34]; and for a chemical compound, it is expressed as:
where Xi is training compounds matrix of i. X is the n × k descriptor matrix of the training set compound, and XT is the transpose matrix of X used to generate the model. The warning leverage, h*, is the maximum value for X and is expressed as:
where n is the number of training compounds and p is the number of descriptors in the model.
2.11 Quality assurance of the generated model
Internal and external validation parameters presented in Table 2 give the minimum required values for a QSAR model to be predictable and reliable [34].
2.12 Docking study
To elucidate the interaction of 1,2,4,5-tetraoxane-8-aminoquinolines with a possible molecular target, the molecular docking study of the hybrid compounds was conducted with Plasmodium PfLDH which is a potential target enzyme for antimalarials because the parasite relies on glycolysis to produce energy [35]. The Discovery Studio software was used to prepare the crystal structure of the enzyme obtained from protein data bank (PDB ID: 1CET) as the receptor, and the compounds are prepared as the ligands. Autodock Vina in the Pyrx software was used to dock the receptor and the ligands [36]. The docking result was visualized and analyzed with the aid of Discovery Studio Visualizer.
3 Results
Genetic function algorithm (GFA) of the material studio software was used to build four QSAR models to study how the chemical structure of 1,2,4,5-tetraoxane-8-aminoquinoline hybrids relates with their biological activities as potent antimalaria. One of the built models was selected for its statistical significance and reported herein as follows:
pEC50 = 33.566456798 * MATS3m
− 18.570253404 * GATS8p
+ 16.287782272 * GATS8i
+ 0.044070689 * RDF50s
+ 6.676939310
Table 3 presents the validation parameters of the model which satisfied the minimum required values presented in Table 2.
4 Discussion
The model contained 2D autocorrelation descriptors (MATS3m, GATS8p, and GATS8i) and radial distribution function (RDF50s) descriptor. MATS3m was Moran autocorrelation of lag 3 weighted by atomic masses, GATS8p, and GATS8i were Geary autocorrelation of lag 8 weighted by atomic polarizabilities and first ionization potential, respectively. The 2D autocorrelation descriptors explained how the values of certain functions (topological distance) at intervals equal to the lag (atomic properties) were correlated. These descriptors of type GATSd and MATSd are slightly different but generally describe how the considered property was distributed along the topological structure [37, 38]. RDF50s was 3D radial distribution function at 5.0 inter-atomic distance weighted by relative I-state. RDF-type descriptors of a molecule indicate the probability distribution of finding an atom in a spherical volume of radius R [39]. RDF50s indicated the existence of a linear relationship between the antiplasmodial activities of 1,2,4,5-tetraoxane-8-aminoquinoline hybrids and the 3D molecular distribution of the relative inductive effect of atoms or group of atoms in the molecules calculated at the radius of 5.0 Å from the geometrical centers of each hybrids molecule.
Table 4 shows the experimental and predicted activities of 1,2,4,5-tetraoxane-8-aminoquinoline hybrids as potent multidrug-resistant Plasmodium falciparum W2 strain inhibitors with the residual values. The high predictability of the model was indicated by the low residual value between the experimental and predicted activity of the compounds.
Pearson’s correlation matrix, variance inflation (VIF) factor, and mean effect (ME) of the four descriptors in the model were presented in Table 5. The correlation matrix shows no significant inter-correlation among the descriptors used in building the model as corroborated by the VIF values which were less than 10 for all the descriptors. Hence, the descriptors used in building the model were good, and the model is stable without multi-co-linearity problem. The ME indicates the magnitudes and directions of influence of the descriptors on the antiplasmodial activities of the compounds. The descriptors MATS3m, GATS8i, and RDF50s with positive sign ME values vary directly with the activities of the molecules, while the descriptor GATS8p with negative sign ME values varies inversely with the activities of the molecules. The descriptor ME magnitudes indicated the extent of their respective influences where GATS8p had a greater influence on the antiplasmodial activities of the compounds. Y-randomization test result presented in Table 6 confirmed that the built QSAR model was reliable, robust, and stable for the low R2 and Q2 values for several trials. The result also shows that the model is good and not gotten by chance for the value of cR2p (> 0.5).
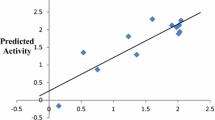
Figure 1 presents the plot of predicted activity against the experimental activity of both training and test set. The linearity of this plot indicated the high predictive power of the built model. The plot of standardized residual against experimental activity presented in Fig. 2 shows the dispersal of standardized residual values on both sides of zero; hence, there was no systematic error in the generated model [40]. Figure 3 shows the Williams plot of the standardized residuals versus the leverages. It was clear that all compounds were within the applicability domain and have no influential compounds. This implies that any of the compounds can be used in designing new 1,2,4,5-tetraoxane-8-aminoquinoline hybrids with highly potent antiplasmodial activities.

Plot of predicted activity against experimental activity of both training and test set

Plot of standardized residual activity against experimental activity

Plot of the standardized residuals against the leverages (Williams plot).
Table 7 presents the result of the molecular docking study carried out between PfLDH (receptor) and 1,2,4,5-tetraoxane-8-aminoquinoline hybrid compounds (ligands). The result shows strong interactions of the ligands with the active sites of the receptor with binding affinities ranging from − 6.3 to − 10.9 kcal/mol having important hydrogen bonding and hydrophobic interaction with the amino acids of the protein. The binding affinities of all the hybrids are better than that of chloroquine. Figure 4 shows the 2D and 3D interaction of ligand 22 with the receptor. This interaction had the best binding affinity of − 10.9 kcal/mol containing two conventional hydrogen bonds, one of which was between one of the oxygen atom of the tetraoxane moiety as the H-acceptor and ARG109 residue as the H-donor, and the other was between the NH of the quinoline moiety as the H-donor and the residue PRO246 as the H-acceptor. The interaction also contains a carbon-hydrogen bond between the methoxide carbon atom of the quinoline moiety as the H-donor and ASN140 residue as the H-acceptor. The ligand also formed two hydrophobic interactions of alkyl-alkyl type with the ALA236 amino acid of the receptor and halogen-type interaction between ASP53 and bromine atom of the ligand. The hydrogen bond and the hydrophobic interactions of ligand 22 with the receptor are depicted in Figs. 5 and 6, respectively.

3D and 2D 22-pfLDH interactions

Hydrogen bond 22-pfLDH interactions

Hydrophobic 22-pfLDH interactions
5 Conclusion
QSAR and molecular docking studies were conducted on 1,2,4,5-tetraoxane-8-aminoquinoline hybrids as potent antimalaria. A stable, reliable, and robust model was generated and found to be influenced by MATS3m, GATS8p, GATS8i, and RDF50s descriptors. MATS3m, GATS8i, and RDF50s were found to influence the antiplasmodial activities of the compounds positively while GATS8p negatively with the greatest influence. The molecular docking study revealed the mode of interaction of the hybrid compounds with Plasmodium falciparum lactate dehydrogenase as the potential target. The result shows strong interaction of the compounds with the receptors. The QSAR model couple with the docking result can be employed in designing new 1,2,4,5-tetraoxane-8-aminoquinoline hybrids with highly potent activities against Plasmodium falciparum.
Availability of data and materials
All the data generated during the current study are included in this published article, [and its supplementary file] and the datasets used during the study are available from the article: Capela R, Magalhaes J, Miranda D, Machado M, Sanches-Vaz M, Albuquerque IS, Sharma M, Gut J, Rosenthal PJ, Frade R, Perry MR, Moreira R, Prudencio M, and Lopes F (2018) endoperoxide-8-aminoquinoline hybrids as dual-stage antimalarial agents with enhanced metabolic stability. European Journal of Medicinal Chemistry 149:69-78. https://doi.org/10.1016/j.ejmech. 02.048
Abbreviations
- QSAR:
-
Quantitative structure activity relationships
- GFA:
-
Genetic function algorithm
- pfLDH:
-
Plasmodium falciparum lactate dehydrogenase
- EC50 :
-
Half maximal effective concentration
- ME:
-
Mean effect
- VIF:
-
Variance inflation factor
References
World malaria report 2018; https://www.who.int/malaria/media/world-malaria-report-2018 Accessed 3 September 2019.
Cohen JM, Smith DL, Cotter C, Ward A, Yamey G, Sabot OJ, Moonen B (2012) Malaria resurgence: a systematic review and assessment of its causes. Malar J 11:122
Krafts K, Hempelmann E, Skórska-Stania A (2012) From methylene blue to chloroquine: a brief review of the development of an antimalarial therapy. Parasito Res 11:1–6
Kondaparlaa S, Sonib A, Manhasb A, Srivastavab K, Purib SK, Kattia SB (2016) Synthesis and antimalarial activity of new 4-aminoquinolines active against drug resistant strains. R soc chem 10(49):12 https://doi.org/10.1039/C6RA14016E
Uhlemann AC, KrishnaS (2005) Antimalarial multi-drug resistance in Asia: mechanisms and assessment. Curr Top Microbiol Immunol 295:39–53
Houa X, Chena X, Zhanga M, Yana A (2016) QSAR study on the antimalarial activity of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitors. SAR QSAR Environ Res 27(2):101–124
Rogerson SJ, Menendez C (2006) Treatment and prevention of malaria in pregnancy: opportunities and challenges. Expert Rev Anti Infect Ther 4:687–702
Martinelli A, Moreira R, Cravo PVL (2008) Malaria combination therapies: advantages and shortcomings. Mini Rev Med Chem 8:201–212
Ekland EH, Fidock DA (2008) In vitro evaluations of antimalarial drugs and their relevance to clinical outcomes. Int J Parasitol 38:743–747
Mbengue A, Bhattacharjee S, Pandharkar T, Liu HN, Estiu G, Stahelin RV, Rizk SS, Njimoh DL, Ryan Y, Chotivanich K, Nguon C, Ghorbal M, Lopez-Rubio JJ, Pfrender M, Emrich S, Mohandas N, Dondorp AM, Wiest O, Haldar K (2015) A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 520:683–687
`Fairhurst RM, Dondorp AM (2016) Artemisinin-resistant Plasmodium falciparum malaria. Microbiol Spectr 4(3). https://doi.org/10.1128/microbiolspec.El10-0013-2016.
Kar S, Kar S (2010) Control of malaria. Nat Rev Drug Discov 7:511–512
Krstulovic L, Stolic I, Jukic M, Opacak-Bernardi T, Starcevic K, Bajic M, Glavas-Obrovac L (2017) New quinoline-arylamidine hybrids: synthesis, DNA/RNA binding and antitumor activity. Eur J Med Chem 137:196–210. https://doi.org/10.1016/j.ejmech.2017.05.054
Meunier B (2008) Hybrid molecules with a dual mode of action: dream or reality? Accounts. Chem Res 41:69–77
Muregi FW, Ishih A (2009) “Next-generation antimalarial drugs: hybrid molecules as a new strategy in drug design.” Drug Dev Res 71:20-32
Kaur H, Machado M, de Kock C, Smith P, Chibale K, Prudencio M, Singh K (2015) Primaquine-pyrimidine hybrids: synthesis and dual-stage antiplasmodial activity. Eur J Med Chem 101:266–273
Ribeiro CJA, Espadinha M, Machado M, Gut J, Goncalves LM, Rosenthal PJ, Prudencio M, Moreira R, Santos MMM (2016) Novel squaramides with in vitro liver stage antiplasmodial activity. Bioorg Med Chem 24:1786–1792
Alonso PL, Djimde A, Kremsner P, Magill A, Milman J, Najera J, Plowe CV, Rabinovich R, Wells T, Yeung S, drugs MCG (2011) A research agenda for malaria eradication: drugs. PLoS Med 8(1):e1000406. https://doi.org/10.1371/journal.pmed.1000406
Mishra A, Batchu H, Srivastava K, Singh P, Shukla PK, Batra S (2014) Synthesis and evaluation of new diaryl ether and quinoline hybrids as potential antiplasmodial and antimicrobial agents. Bioorg Med Chem Lett 24(7):1719–1723
Rania A, Legacb J, Rosenthalb PJ, Kumara V (2019) Substituted 1,3-dioxoisoindoline-4-aminoquinolines coupled via amide linkers: synthesis, antiplasmodial and cytotoxic evaluation. Bioorg Chem 88:102–912
Capela R, Magalhaes J, Miranda D, Machado M, Sanches-Vaz M, Albuquerque IS, Sharma M, Gut J, Rosenthal PJ, Frade R, Perry MR, Moreira R, Prudencio M, Lopes F (2018) Endoperoxide-8-aminoquinoline hybrids as dual-stage antimalarial agents with enhanced metabolic stability. Eur J Med Chem 149:69–78. https://doi.org/10.1016/j.ejmech%2002.048
Jitender V, Vijay MK, Evans CC (2010) 3D-QSAR in drug design - a review. Curr Top Med Chem 10:95–115
Tropsha A (2010) Best practices for QSAR model development, validation, and exploitation. Mol Inf 29:476–488
Talele TT, Santosh AK, Alan CR (2010) Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr Top Med Chem 10:127–141
Ibezim E, Duchowicz PR, Ortiz EV, Castro EA (2012) QSAR on arylpiperazine derivatives with activity on malaria. Chemomet Intell Lab Syst 110:81–88
Basak SC, Mills D, Garg R, Bhhatarai B (2010) A QSAR study of HIV protease inhibitors using theoretical descriptors. Curr Comput Aided Drug Des 6:269–282
Li Z, Wan H, Shi Y, Ouyang P (2004) Personal experience with four kinds of chemical structure drawing software: review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J Chem Inf Comput Sci 44:1886–1890
Shao Y, Molnar LF, Jung Y, Kussmann J, Ochsenfeld C, Brown ST, Gilbert AT, Slipchenko LV, Levchenko SV, O’Neill DP (2006) Advances in methods and algorithms in a modern quantum chemistry program package. Phys Chem Chem Phys 8:3172–3191
Yap CW (2011) PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J Comput Chem 32:1466–1474
Singh P (2013) Quantitative structure-activity relationship study of substituted-[1, 2, 4] oxadiazoles as S1P1 agonists. J Curr Chem Pharm Sci 3
Kenard RW, Stone LA (1969) Computer aided design of experiments. technometrics 11(1):137-148. https://doi.org/10.1080/00401706.1969.10490666
Friedman JH (1991) Multivariate adaptive regression splines. Ann Stat:1–67
Tropsha A, Gramatica P, Gombar VK (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. Mol Inform 22:69–77
Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK (2011) Validation of QSAR models-strategies and importance. Int. J. Drug Des. Discov. 3:511–519
Penna-Coutinho J, Cortopassi WA, Oliveira AA, Franc TCC, Krettli AU (2011) Antimalarial activity of potential inhibitors of plasmodium falciparum lactate dehydrogenase enzyme selected by docking studies. PLoS ONE 6(7):e21237 1010.1371/journal.pone.0021237
Trott O, Olson AJ (2010) Autodock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput chem 22:455–461
Moreau G, Broto P (1980) The autocorrelation of topological structure: a new molecular descriptor. Nouv J Chimie-New. J Chem 4:359–360
Broto P, Moreau G, Vandycke C (1984) Molecular structure: perception, autocorrelation descriptor and SAR studies. Eur J Med Chem 19:79–84
Hemmer MC, Steinhauer V, Gasteiger J (1999) Deriving the 3D structure of organic molecules from infrared spectra. Vib Spectrosc 19(350):151–164
Jalali-Heravi M, Kyani A (2004) Use of computer-assisted methods for the modeling of the retention time of a variety of volatile organic compounds: a PCA-MLR-ANN approach. J Chem Inf Comput Sci 44:1328–1335
Acknowledgements
The authors wish to acknowledge the support of Kaduna polytechnic, Nigeria.
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
AWM and AU conceived and designed the study. AWM and GAS drew and optimized the structures. AWM calculated descriptors and generated the model. AWM and GAS performed the statistical analysis. AWM and AU conducted the docking study. AWM and GAS wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mahmud, A.W., Shallangwa, G.A. & Uzairu, A. In silico modeling of tetraoxane-8-aminoquinoline hybrids active against Plasmodium falciparum. Beni-Suef Univ J Basic Appl Sci 9, 19 (2020). https://doi.org/10.1186/s43088-020-00044-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43088-020-00044-0