
Abstract
Background
Desmoplastic small-round-cell tumor (DSRCT) is an extremely rare and highly aggressive malignancy. It is of yet unclear origin, but it is assumed to be of a mesothelial origin based on its tendency for widespread metastasis in serosal linings.
Case presentation
In this report, we describe a young female who presented with bilateral ovarian masses that mimicked the classic clinical picture of ovarian cancer. The patient had a cytoreductive surgery done in the form of total abdominal hysterectomy, bilateral salpingo-oophorectomy, omentectomy, pelvic peritonectomy, low para-aortic and bilateral iliac lymphadenectomy. Postoperative course was smooth with no adverse events. The final pathology report revealed desmoplastic small-round-cell tumor. Afterwards, the patient was referred to medical oncologist to receive her adjuvant therapy.
Conclusions
DSRCT is still an unknown disease to us given the limited number of cases and poor survival. Given the lack of clear guidelines, treatment is offered based on the best available evidence and the collaborative effort of a multi-disciplinary team.
Similar content being viewed by others


Background
Desmoplastic small-round-cell tumor (DSRCT) is an extremely rare and highly aggressive malignancy that was first described in 1987 by Sesterhenn et al. [1]. Then, it was more elaborately described by Gerald and Rosai in 1989 [2]. It tends to have a predilection for adolescent males with an annual incidence of about 0.1 cases per million. It usually presents as intra-abdominal masses that might mimic gastrointestinal stromal tumors (GIST) or retroperitoneal sarcoma [3]. It is of yet unclear origin, but it is assumed to be of a mesothelial origin based on its tendency for widespread metastasis in serosal linings. An epithelial, neurogenic and blastomeric origin had also been reported [4]. This malignant entity with its atypical clinical, pathological, radiological features, aggressive course, and extensive intra-abdominal dissemination may confer a diagnostic dilemma when it is first encountered. Here, we describe a young female who was presented by bilateral ovarian masses that was later revealed to be DSRCT.
Case presentation
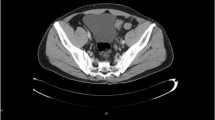
A 22-year-old female presented with abdominal pain. She had no significant medical and surgical history and had only been married for 4 months. Abdominal ultrasonography showed bilateral ovarian masses. CT scan and diffusion MRI revealed bilateral malignant-looking ovarian masses with associated peritoneal deposits and retroperitoneal lymphadenopathy. Also, an anomaly of horseshoe kidney was noted. Tumor markers cancer antigen 125 (CA-125) and CA19–9, carcinoembryonic antigen (CEA), lactate dehydrogenase (LDH), alpha-fetoprotein (AFP), and beta subunit of human chorionic gonadotropin (B-HCG) were all within the normal range. Upper and lower gastrointestinal endoscopies were done and excluded the possibility of gastrointestinal primary. After informed consent, surgical exploration aiming at complete cytoreduction was performed. Intraoperative exploration showed both ovaries to be totally replaced by malignant masses, with ascites and pelvic peritoneal deposits. The diaphragmatic surfaces and para-colic gutters were free from disease. One of the ovaries was excised and frozen section examination was requested which suggested the possibility of dysgerminoma. Cytoreductive surgery was done in the form total abdominal hysterectomy, bilateral salpingo-oophorectomy, omentectomy, pelvic peritonectomy, low para-aortic and bilateral iliac lymphadenectomy (high para-aortic lymphadenectomy could not be done due to the presence of horseshoe kidney). Apart from the high para-aortic lymph nodes, no gross residue was left at all.
Postoperative pathology examination revealed tumor tissue formed of malignant cells with intervening desmoplasia. The tumor cells were small with a marked degree of atypia and pleomorphism and high mitotic activity (Fig. 1). Three out of ten iliac lymph nodes and seven out of seven para-aortic lymph nodes were infiltrated by tumor tissue. Pelvic peritoneal nodules, uterine wall, and cervix were free from tumor infiltration. Immunohistochemical markers (IHC) were performed using monoclonal antibodies against pan-cytokeratin, cytokeratin 7, cytokeratin 20, placental alkaline phosphatase (PLAP), desmin, leukocyte common antigen (LCA), caudal type homeobox 2 (CDX-2), inhibin and GATA binding protein 3 (GATA-3), and Wilms’ tumor 1 (WT-1) (truncated human WT1 protein corresponding to N-terminal amino acids 1–181, FLEX, Monoclonal Mouse, Anti-Human Wilms’ Tumor 1 (WT1) Protein Clone 6F-H2 Ready-to-use (Dako Corporation)). Tumor tissue was positive for desmin and cytokeratin (Fig. 2) and negative for each of cytokeratin 7, cytokeratin 20, PLAP, WT-1, LCA, CDX-2, inhibin, and GATA-3 (Fig. 3) confirming the diagnosis of desmoplastic small-round-cell tumor.

The tumor cells were small with marked degree of atypia and pleomorphism and high mitotic activity (Hx&E)

a–c Tumor tissue was positive for IHC desmin, cytokeratin, and WT-1

Tumor tissue was negative for IHC PLAP
The patient had an insignificant postoperative course. Afterwards, the patient was referred to a medical oncologist to receive the appropriate adjuvant treatment. Adjuvant chemotherapy (VACA-IE) protocol: vincristine (Oncovin), doxorubicin (Adriamycin), cyclophosphamide (Cytoxan), doxorubicin (Adriamycin), ifosfamide (Ifex), and etoposide (Vepesid) respectively were ensued soon after full recuperation. Patient is still receiving the proposed chemotherapy protocol, and she is planned to receive adjuvant external beam radiation therapy.
Discussion
DSRCT is a rare and highly aggressive tumor that commonly affects the abdomen and or pelvis of young males. In their cases series, Al-Ibraheemi et al. reported 16 cases of extra-abdominal DSRCT. These reported sites included the head, neck, intracranial, thigh, axilla/shoulder, inguinal, paratesticular, intraosseous, and uterine body [5]. Moreover, Lae et al. reported DSRCTs originating from the ethmoidal sinus and soft tissue of the scalp [6]. To the best of our knowledge, DSRCT was reported to be originating from the ovary in 18 cases [7]. Interestingly, two of them presented during pregnancy and labor. Most of the reported cases presented with bilateral ovarian involvement with widespread peritoneal nodular infiltration [8, 9].
Its early presentation is usually non-specific, with symptoms varying between vague abdominal pain, distension, and altered bowel habits. Such a tendency to late presentation accounts for its difficult management, as it often manifests in advanced disease stage. Radiologic findings usually describe abdominal masses of variable sizes in association with peritoneal deposits, omental cakes, and ascites, findings that are usually found in the advanced colon, ovarian, and gastric cancer, hence the encountered diagnostic difficulty [10]. Differential diagnosis of small-round-cell tumor includes lymphoma, Ewing sarcoma, medulloblastoma, Wilms’ tumor, synovial sarcoma, neuroblastoma, and embryonal rhabdomyosarcoma [11]. Diagnosis is often confirmed after ultrasonographic- (U\S) or computerized tomography (CT)-guided biopsy from these lesions. Tumor markers’ elevation is non-specific. Only immunohistochemistry (IHC) and cytogenetic study are the tools that can confirm the diagnosis [12, 13]. The typical histopathological appearance of DSRCTs includes large necrotic area, sharply demarcated nests of different sizes containing small round cells with hyperchromatic nuclei and scanty faint eosinophilic cytoplasm, or spindle cells embedded in a desmoplastic stroma. These cells mostly exhibit epithelial, mesenchymal, and neural markers like cytokeratin, desmin, and smooth muscle actin (SMA) [14]. Desmoplastic small-round-cell tumor arises from mesenchymal cells of the abdominopelvic peritoneum. The gene fusion between Ewing sarcoma (EWS) and WT1 genes resulting in the characteristic translocation t(11;22)(p13;q12), is the unique molecular hallmark and no other genetic factor has been linked to this aggressive tumor [15]. Horseshoe kidney is the most common fusion anomaly of the kidneys accounting for 0.25% of the population [16]. One of the major concerns that affected the treatment decision in the present case is the encountered technical difficulty of retroperitoneal lymph node dissection. Also, the radiation oncologist should have a good radiotherapy field planning to avoid radiation nephritis as most of the renal parenchyma overlies the para-aortic lymph nodes. In the abovementioned case, the patient was presented by bilateral ovarian masses, a finding that usually first directs the oncologist’s attention to the possibility of either Krukenberg tumor or primary ovarian cancer [17]. Indeed, this case was handled as such and it was only the final pathology that revealed the diagnosis of this rare tumor. In this case, a complete surgical staging—as if it was a case of primary ovarian cancer—was done given the presenting intraoperative situation and the result of frozen section analysis. Young age in and of itself might raise the suspicion of such malignancy, yet the rarity of the disease should keep us in the lane of common differential diagnoses for such age in an algorithmic fashion. It should be noted that until now there is still no consensus regarding the optimal management of this disease. The treating physician should follow the previously reported multimodality treatment to achieve the best disease response [7]. Based on the available data from the literature, a multimodality therapy of neoadjuvant multiagent chemotherapy followed by cytoreductive surgery and external beam radiotherapy is now considered the standard of care for those patients without extra-abdominal spread [18]. Honoré et al. reported improved overall and progression-free survival in patients treated with multimodality treatment including whole abdominopelvic radiotherapy (WAP RT) of 30 Gy. Fewer side effects were encountered with intensity modulated radiotherapy (IMRT) than two and three dimensional radiotherapy [19]. Despite relapse which was reported in most of the patients during long-term follow-up, prolonged progression-free survival was best reported after multimodality treatment [20]. Prognosis is still dismal with 5-year survival barely exceeding 30%.
Conclusions
DSRCT is still an unknown disease to us given the limited number of cases and poor survival. Efforts are being maximized as much as possible to better find the optimal management. Given the lack of clear guidelines, treatment should be offered based on the best available evidence and the collaborative effort of a multi-disciplinary team.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- AFP:
-
Alpha-fetoprotein
- B-HCG:
-
Beta subunit of human chorionic gonadotropin
- CA 125:
-
Cancer antigen 125
- CA 19-9:
-
Cancer antigen 19-9
- CDX-2:
-
Caudal type homeobox-2
- CEA:
-
Carcinoembryonic antigen
- CT:
-
Computed tomography
- DSRCT:
-
Desmoplastic small-round-cell tumor
- EWS:
-
Ewing sarcoma
- GIST:
-
Gastrointestinal stromal tumor
- Gy:
-
Gray
- IHC:
-
Immune histochemical stains
- IMRT:
-
Intensity modulated radiotherapy
- LCA:
-
Leukocyte common antigen
- LDH:
-
Lactate dehydrogenase
- MRI:
-
Magnetic resonance imaging
- PLAP:
-
Placental alkaline phosphatase
- SMA:
-
Smooth muscle actin
- WAP-RT:
-
Whole abdominopelvic radiotherapy
- WT-1:
-
Wilms’ tumor-1
References
Sesterhenn I, Davis CJ, Mostofi FK. Undifferentiated malignant epithelial tumors involving serosal surfaces of escrotum and abdomen in young males. J Urol. 1987;137:214A.
Gerald WL, Rosai J. Case 2 desmoplastic small cell tumor with divergent differentiation. Pediatr Pathol. 1989;9(2):177–83.
Lettieri CK, Garcia-Filion P, Hingorani P. Incidence and outcomes of desmoplastic small round cell tumor: results from the surveillance, epidemiology, and end results database. J Cancer Epidemiol. 2014;2014:680126.
Layfield LJ, Lenarsky C. Desmoplastic small cell tumors of the peritoneum coexpressing mesenchymal and epithelial markers. Am J Clin Pathol. 1991;96(4):536–43.
Al-Ibraheemi A, Broehm C, Tanas MR, Horvai AE, Rubin BP, Cheah AL, et al. Desmoplastic small round cell tumors with atypical presentations: a report of 34 cases. Int J Surg Pathol. 2019;27(3):236–43.
Lae ME, Roche PC, Jin L, Lloyd RV, Nascimento AG. Desmoplastic small round cell tumor: a clinicopathologic, immunohistochemical, and molecular study of 32 tumors. Am J Surg Pathol. 2002;26(7):823–35.
Xie YP, Shen YM. Ovarian involvement of a desmoplastic small round cell tumor of unknown primary origin with lymph node and lung metastases: a case report. Oncol Lett. 2016;11(2):1125–9.
Church DN, Bailey J, Hughes J, Williams CJ. Desmoplastic small round cell tumour: obstetric and gynecological presentations. Gynecol Oncol. 2006;102(3):583–6.
Neff RT, Kellert B, Isley M, Backes F. Management of a rapidly enlarging new adnexal mass: a rare case of desmoplastic small round cell tumor of the ovary arising in pregnancy. Gynecol Oncol Rep. 2016;17:23–5.
Zhang WD, Li CX, Liu QY, Hu YY, Cao Y, Huang JH. CT, MRI, and FDG-PET/CT imaging findings of abdominopelvic desmoplastic small round cell tumors: correlation with histopathologic findings. Eur J Radiol. 2011;80(2):269–73.
Reynolds CP, Smith RG, Frenkel EP. The diagnostic dilemma of the “small round cell neoplasm”: catecholamine fluorescence and tissue culture morphology as markers for neuroblastoma. Cancer. 1981;48(9):2088–94.
Arnold MA, Schoenfield L, Limketkai BN, Arnold CA. Diagnostic pitfalls of differentiating desmoplastic small round cell tumor (DSRCT) from Wilms tumor (WT): overlapping morphologic and immunohistochemical features. Am J Surg Pathol. 2014;38(9):1220–6.
Barnoud R, Sabourin J-C, Pasquier D, Ranchère D, Bailly C, Terrier-Lacombe MJ, et al. Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol. 2000;24(6):830–6.
Ferreira EN, Barros BDF, de Souza JE, Almeida RV, Torrezan GT, Garcia S, et al. A genomic case study of desmoplastic small round cell tumor: comprehensive analysis reveals insights into potential therapeutic targets and development of a monitoring tool for a rare and aggressive disease. Hum Genomics. 2016;10(1):36.
Wakahashi S, Sudo T, Ichida K, Sugita S, Hasegawa T, Nagao S, et al. Diagnosis of desmoplastic small-round-cell tumor by cytogenetic analysis: a case report. Clin Case Rep. 2016;4(5):520–3.
Kim JW, Park JH, Cho HJ, Kwon JH, Koh Y, Kim SJ, et al. A case of desmoplastic small round cell tumor diagnosed in a young female patient. Cancer Res Treat. 2009;41(4):233–6.
Schiappacasse G, Aguirre J, Soffia P, Silva C, Zilleruelo N. CT findings of the main pathological conditions associated with horseshoe kidneys. Br J Radiol. 2014;88(1045):20140456.
Thway K, Noujaim J, Zaidi S, Miah AB, Benson C, Messiou C, et al. Desmoplastic small round cell tumor: pathology, genetics, and potential therapeutic strategies. Int J Surg Pathol. 2016;24(8):672–84.
Honoré C, Amroun K, Vilcot L, Mir O, Domont J, Terrier P, et al. Abdominal desmoplastic small round cell tumor: multimodal treatment combining chemotherapy, surgery, and radiotherapy is the best option. Ann Surg Oncol. 2015;22(4):1073–9.
Kushner BH, LaQuaglia MP, Wollner N, Meyers PA, Lindsley KL, Ghavimi F, et al. Desmoplastic small round-cell tumor: prolonged progression-free survival with aggressive multimodality therapy. J Clin Oncol. 1996;14(5):1526–31.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
AA was responsible for writing the manuscript; KG was responsible for the revision and reformatting of the manuscript; MZ was responsible for writing, revising, and reformatting the manuscript and was an assistant in the operation; KA was an assistant in the operation and was responsible for the patient follow-up and manuscript reformatting; WE was responsible for the pathological interpretation; SA was responsible for writing the manuscript and the case presentation; BG was responsible for the revision of the manuscript; and BR was the principal operator and was responsible for the revision and final approval of manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
A written informed consent was taken from the patient included in this report.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Atef, A., Gaballa, K., Zuhdy, M. et al. Primary desmoplastic small-round-cell tumor of the ovary. J Egypt Natl Canc Inst 31, 4 (2019). https://doi.org/10.1186/s43046-019-0001-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43046-019-0001-4