
Abstract
Background
Hypoplastic right heart syndrome with pulmonary atresia is a rare cyanotic heart disease with poor prognosis requiring urgent intervention to establish the pulmonary blood flow. Pulmonary blood flow is achieved by BT shunt or percutaneous techniques like PDA stenting or pulmonary valve perforation. Various series have shown that early surgical intervention causes high mortality in these patients. Pulmonary valve perforation is a suitable, physiological alternative to surgical techniques in selected patients.
Case presentation
We report a case of hypoplastic right heart syndrome with pulmonary atresia and restrictive VSD presenting with cyanosis from birth and underwent pulmonary valve perforation successfully.
Conclusion
Duct-dependent pulmonary circulation is a pediatric emergency, palliative procedure for establishing adequate pulmonary blood flow is essential early in the life. In the management of duct-dependent pulmonary circulation, RVOT perforation is an effective and safe option in suitable high-risk subgroups. The induced pulmonary regurgitation along with established physiological antegrade flow would be beneficial in the remodeling of tripartite/hypertrophied small RV.
Similar content being viewed by others


Background
Hypoplastic right heart syndrome with pulmonary atresia is a rare form of cyanotic congenital heart disease with poor prognosis, high morbidity, and mortality. Early palliation for establishing adequate pulmonary flow is necessary for the relief of cyanosis and duct dependence. These patients are usually treated by aorto pulmonary shunt (BT shunt) followed by univentricular or 1.5 ventricular repair or biventricular repair at a later stage depending on the right ventricular growth if at all it grows. With the advent of use of radio frequency or laser-assisted perforation of pulmonary valve and balloon dilatation percutaneous techniques has garnered importance in achieving pulmonary flow. A series percutaneous techniques offered better outcome compared to surgical repair [1].
Case presentation
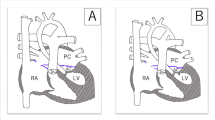
A Neonate presented to us on day 1 of life with cyanosis, mild respiratory distress, and with a saturation of 70% in all 4 limbs. He was not evaluated for cardiac abnormalities during fetal life. He was born at 32 weeks of gestation and was weighing 2.1 kg. All the peripheral pulses are equally felt. There was a grade 3/6 pansystolic murmur at base of the heart. His electrocardiogram showed RV predominance with normal sinus rhythm and chest skiagram showed pulmonary oligemia with increased cardiothoracic ratio. Echocardiogram showed situs solitus, levocardia, atrioventricular, and ventriculoarterial concordance. Tricuspid valve leaflets had restricted movements with z score of annulus − 1.94. There was a severe TR, with measured RVSP of 110 mmHg (+mean RAP). There was a restrictive muscular ventricular septal defect, hypertrophied, hypoplastic tripartite RV, and valvular pulmonary atresia. The branch pulmonary arteries were confluent measuring 2.7/2.5 mm each. The duct arising from the under surface of the left aortic arch, supplying pulmonary circulation (as shown in Fig. 1).

Echocardiogram showing hypoplastic and hypertrophied right ventricle with TV Z score-1.95. b Color doppler showing tricuspid regurgitation jet (RVSP 110 + mean RAP). b, c PDA seen filling the MPA. d CW showing the continuous flow across MPA due to its filling from PDA
He was immediately started on prostaglandin E1 infusion to maintain the patency of ductus arteriosus. After discussion in the combined meeting, neonate was considered to be ‘high-risk’ for BT shunt and option of PDA stenting was thought to be a safer option as stage I to ensure adequate pulmonary blood flow. Although, future single ventricular palliation was offered by the majority, some of us thought pulmonary valve perforation with induced pulmonary regurgitation would assist growth of this tripartite RV for future biventricular repair. Right femoral venous (6 Fr) and arterial lines (5 Fr) were established by digital palpation. A JR guiding catheter was crossed across the tricuspid valve and placed at the dimple of atretic pulmonary valve. The position was confirmed in orthogonal views with a hand injection in a diagnostic JR catheter placed in aortic arch. The course of the PDA showed the retrograde filling of MPA up to the atretic part reassuring the position of the guide. A GAIA (ASAHITM INTECC) wire was used to perforate the atretic pulmonary valve with serial dilatation using coronary balloons (4 × 12, 4.5 × 14, and 5 × 12) to obtain the desired diameter (as assessed preoperatively by echocardiogram). The final injection showed unobstructed contrast filling both branch pulmonary arteries with good bilateral arborization (as shown in Fig. 2a–f). The saturation increased from 70% to 94% with echo showing moderate pulmonary regurgitation, PDA still flowing (Fig. 3). The baby was managed with diuretics, extubated by 8 h. At 4 weeks follow-up, baby was doing fine with saturation of 87% although the right ventricle had not grown much. The duct had closed and RVSP by TR jet was 70+mean RA pressure.

a JR guide at the atretic RVOT, with contrast filling of the RV and ending as bling pouch. b Injection in the arch of the aorta showing filling of the PA branches through PDA and also confirming guide position at the RVOT. c RVOT perforation with coronary guidewire. d Successful crossing of the guidewire across the atretic RVOT after perforation and wire parked in the DTA E Serial Balloon dilatation of the RVOT. f Final angiogram showing good antegrade pulmonary blood flow and arborization

Post-procedure echocardiogram showing good antegrade flow across the pulmonary valve
Discussion
Hypoplastic right heart syndrome (HRHS) is a rare cyanotic heart disease due to the underdevelopment of right heart structures (Tricuspid valve, RV, Pulmonary valve and pulmonary artery). The spectrum of disorders varies from simple hypoplastic RV with or without pulmonary stenosis to grossly underdeveloped RV with pulmonary and/or tricuspid atresia [1, 2]. Ventricular size can vary from grossly hypoplastic to normal-sized chambers [3]. HRHS is usually associated with ASD (atrial septal defect), VSD (ventricular septal defect), AVCD (atrioventricular cushion defect) and other complex defects. Clinical presentation depends on the extent of development of RV, pulmonary circulation, and usually present early in the life. The goals of early palliation include relief of cyanosis and duct dependence by establishing adequate pulmonary flow for development of right-sided structures. Treatment options include surgical systemic pulmonary shunt and percutaneous techniques like PDA stenting or pulmonary valve perforation. The difficulty in ductal stenting stems from the fact that ducts in these patients are tortuous and so incomplete stenting, stent migration can occur. Surgical correction at neonatal stage has higher mortality [4]. In the subset of patients with short-segment atresia management, options are well studied. Transcatheter pulmonary valve perforation offers a physiological approach for pulmonary flow and development of pulmonary arteries for future palliative surgeries. It also helps in the growth of the RV by the induced pulmonic regurgitation to a near normal size [3]. Also, the authors have experience of PV perforation and stenting of the RVOT in a sick infant (unpublished work); we felt that PV perforation offers a better palliative procedure short of surgery. The advantage with pulmonary valve perforation is that the pulmonary flow is more physiological and the induced pulmonary regurgitation helps in the growth of the right ventricle for future biventricular repair. In a series of 13 patients who underwent RVOT perforation, there was significant growth of the TV annulus and reduction in TR during at a mean follow-up of 13.2 months [5]. On the contrary there have been studies where there was no significant growth of the right ventricle following perforation [6,7,8]. The surgical aorto pulmonary shunting is a well-established palliative surgery in HRHS, but carries an increased risk in under-weight neonates. Complication with surgical shunting include pulmonary over-circulation, stenosis at the anastomosis site, asymmetric development of pulmonary vasculature, distortion of the vessels, nerve injury, and chylothorax [9]. In these patient’s assessment of the atretic pulmonary valve for its thickness, diameter of the RVOT, MPA, and branch PA’s is essential for selecting suitable patients. Perforation using hydrophilic coronary wires or radiofrequency ablation has been described in the literature [10,11,12]. Surgical correction (pulmonary valvotomy) at neonatal prove to be detrimental in some series [13]. Humpl et al. in their series of 50 patients of PA with IVS, attempted PV perforation in 30 patients by either stiff end of the coronary guide wire or RF ablation with a procedural success in 27 patients. They could find that with PV perforation though RV did not increase according to body growth at early follow up, it is adequate to maintain a biventricular circulation [10]. In a series of 33 patients of PA with IVS by Alwi et al. where RF ablation with balloon dilatation was compared with surgical valvotomy and BT shunt, percutaneous procedure was more efficacious and safe [1]. Procedural success has been described as establishment of adequate pulmonary flow, weaning from prostaglandin, and without requiring reintervention [8]. Complications are rare including perforation with tamponade, arrhythmia, and vascular injury [10]. Surgical correction (pulmonary valvotomy) at early stage prove to be detrimental in some series [13]. In our case, we proceeded with percutaneous intervention, as less invasive procedure in a sick neonate is probably safer. However, the child succumbed to pneumonia-sepsis after 5 weeks of procedure.
Conclusion
Duct-dependent pulmonary circulation is a pediatric emergency, palliative procedure for establishing adequate pulmonary blood flow is essential early in the life. In the management of duct-dependent pulmonary circulation, RVOT perforation is an effective and safe option in suitable high-risk subgroups. The induced pulmonary regurgitation along with established physiological antegrade flow would be beneficial in the remodelling of tripartite/hypertrophied small RV.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- HRHS:
-
Hypoplastic right heart syndrome
- PV:
-
Pulmonary valve
- VSD:
-
Ventricular septal defect
- ASD:
-
Atrial septal defect
- AVCD:
-
Atrioventricular cushion defect
- PDA:
-
Patent ductus arteriosus
- RV:
-
Right ventricle
- RVOT:
-
Right ventricular outflow tract
- MPA:
-
Main pulmonary artery
- JR:
-
Judkin’s right
- Fr:
-
French units
References
Alwi M, Geetha K, Bilkis AA, Lim MK, Hasri S, Haifa AL et al (2000) Pulmonary atresia with intact ventricular septum percutaneous radiofrequency-assisted valvotomy and balloon dilation versus surgical valvotomy and blalock taussig shunt. J Am Coll Cardiol 35(2):468–476
Prasad K, Singh M, Radhakrishnan S (1992) Hypoplastic right ventricle with mild pulmonary stenosis in an adult. Int J Cardiol 37(2):260–262
Jacobstein MD, Fletcher BD, Goldstein S, Riemenschneider TA (1985) Magnetic resonance imaging in patients with hypoplastic right heart syndrome. Am Heart J 110(1 Pt 1):154–158
Leonard H, Derrick G, O'Sullivan J, Wren C (2000) Natural and unnatural history of pulmonary atresia. Heart 84(5):499–503
El Shedoudy S, El-Doklah E (2018) Transcatheter perforation of atretic pulmonary valve by the stiff end of a coronary wire in neonates with pulmonary atresia with intact ventricular septum: A solution in developing countries. J Saudi Heart Assoc 30(3):222–232
Alcíbar-Villa J, Rubio A, Peña N, Galdeano JM, Luis M, Arriola J et al (2007) Pulmonary atresia with intact ventricular septum. Perforation and pulmonary valvuloplasty using a modified mechanical technique. Medium-term follow-up. Rev Esp Cardiol 60(8):833–840
Chen RHS, K. T. Chau A, Chow PC, Yung TC, Cheung YF, Lun KS (2018) Achieving biventricular circulation in patients with moderate hypoplastic right ventricle in pulmonary atresia intact ventricular septum after transcatheter pulmonary valve perforation. Congenit Heart Dis 13(6):884–891
Drighil A, Aljufan M, Slimi A, Yamani S, Mathewson J, AlFadly F (2010) Echocardiographic determinants of successful balloon dilation in pulmonary atresia with intact ventricular septum. Eur J Echocardiogr 11(2):172–175
Petrucci O, O'Brien SM, Jacobs ML, Jacobs JP, Manning PB, Eghtesady P (2011) Risk factors for mortality and morbidity after the neonatal Blalock-Taussig shunt procedure. Ann Thorac Surg 92(2):642–651 discussion 51-2
Humpl T, Söderberg B, McCrindle BW, Nykanen DG, Freedom RM, Williams WG et al (2003) Percutaneous Balloon Valvotomy in Pulmonary Atresia With Intact Ventricular Septum. Circulation. 108(7):826–832
Rosenthal E, Qureshi SA, Chan KC, Martin RP, Skehan DJ, Jordan SC et al (1993) Radiofrequency-assisted balloon dilatation in patients with pulmonary valve atresia and an intact ventricular septum. Br Heart J 69(4):347–351
Hausdorf G, Schulze-Neick I, Lange PE (1993) Radiofrequency-assisted "reconstruction" of the right ventricular outflow tract in muscular pulmonary atresia with ventricular septal defect. Br Heart J 69(4):343–346
Weldon CS, Hartmann AF Jr, Clark RE, Ferguson TB (1970) Surgical management of the hypoplastic right heart syndrome. Ann Thorac Surg 10(6):489–502
Acknowledgements
None.
Funding
No funding has been received for the current study.
Author information
Authors and Affiliations
Contributions
KP was involved in collection of data, preparing the first draft, and revising the manuscript for important intellectual content. KP, PB, JV, and SN were involved in collection of data and preparing the first draft. PB and SN were involved in the conception of the study, revising the manuscript for important intellectual content, and approving the manuscript. All the authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was not required since it is an accepted procedure.
Consent for publication
Written consent has been obtained to publish the case report from the guardian. The consent copy is available with the authors and ready to be submitted if required.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barwad, P., Prasad, K., Vijay, J. et al. Is there a transcatheter solution for a sick neonate with hypoplastic right heart syndrome?: Pulmonary valve perforation in a neonate with hypoplastic right ventricle with pulmonary atresia, restrictive VSD—a case report. Egypt Heart J 72, 64 (2020). https://doi.org/10.1186/s43044-020-00097-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43044-020-00097-7