
Abstract
Background
Short stature is defined as a body height below the third percentile, based on chronological age, or 2 standard deviations (SD) below the national height standard. The prevalence of short stature is around 2% of children worldwide. Several gene deficiencies have been associated with the etiology of short stature. The SHOX is an important candidate gene for short stature, as its haploinsufficiency underlies syndromic and non-syndromic short stature. Partial and complete duplications of SHOX have been reported in patients with short stature. Proper genetic diagnosis of these children allows for appropriate therapeutic approaches to be administered. Since copy number variation (CNV) is a possible mechanism of interhuman variability and pathogenic disease, the multiplex ligation-dependent probe amplification technique (MLPA) can be used as an initial screening technique. Cartilage tissue expresses specific microRNAs (miRNAs), which play an essential role in the regulation of chondrocyte proliferation and differentiation during growth plate development. We aimed to assess the SHOX/PAR1 region using CNV profiling for non-syndromic short stature in Egyptian children with and without growth hormone deficiency using the MLPA technique and expression profiling of miR-1, miR-15a, and miR-140 using quantitative real-time polymerase chain reaction (qRT-PCR) in a group of Egyptian children with non-syndromic short stature.
Results
Of the fifty cases included in this study, different CNVs were detected in ten children (20%), in/outside the SHOX region. Moreover, in children with short stature, the expression level of miRNA-140 was significantly different from that of healthy controls.
Conclusions
This is one of the first studies that have assessed CNVs in the SHOX/PAR1 region in a group of Egyptian children with short stature. MLPA analysis of SHOX/PAR1 identified different CNVs in children with non-syndromic short stature, suggesting that the MLPA should be used as an initial screening technique in short children, as proper genetic diagnosis of these children leads to implementation of the appropriate therapeutic approach. Alterations in the levels of miRNA-140 in children with short stature suggest that changes in the expression levels of this miRNA are associated with the pathogenesis of short stature.
Similar content being viewed by others


Background
Short stature (SS) is defined as a body height below the third percentile, based on chronological age, or 2 standard deviations (SD) below the national height standard. Short stature affects 2% of children worldwide [1], and deficiencies in several genes have been associated with the etiology of SS [1]. Short-stature-homeobox-containing gene (SHOX, MIM 312865) is an important candidate gene for SS, as its haploinsufficiency underlies both syndromic and non-syndromic SS. The SHOX is located within the main pseudoautosomal region (PAR1) on the p arm of the X and Y chromosomes. SHOX is made up of five exons with a sixth exon that can be alternatively spliced to form exon 6a or b. SHOX escapes X-inactivation similar to other genes in PAR1. Therefore, 2 active forms of SHOX are expressed in both males and females. The SHOX protein acts as an important transcription factor during limb development [2, 3]. SHOX is the causative gene for short stature in Turner syndrome. SHOX functions in a dose-dependent manner, as haploinsufficiency with loss-of-function mutations in one allele of the SHOX leads to skeletal growth failure and SS. The most frequent SHOX defects include deletions of various sizes within SHOX or its regulatory enhancer region that is situated 50–250 kb downstream of the coding region. These deletions account for around 70–80% of the defects in SHOX. Heterozygous deletions or mutations are responsible for up to 60% of Lėri-Weill dyschondrosteosis (LWD) cases and about 1.5–15% cases of idiopathic SS (ISS). Homozygous or compound heterozygous deletions or mutations are responsible for a rare disorder, Langer mesomelic dysplasia (LMD). Missense and nonsense mutations account for 20–25% of the defects in SHOX. Partial or complete SHOX duplications, ranging from 13 to 346 kb, have been reported in patients with SS. Such defects are thought to result in decreased SHOX expression due to disruption of the interaction between its enhancer regions and promoter [2, 4,5,6,7].
The majority of children with non-syndromic SS are categorized as having ‘idiopathic short stature’ (ISS), “small stature for the gestational age with failure of catch-up growth” (SGA) or those with systemic diseases or growth hormone deficiency. However, differentiation between these categories can be difficult. Children with systemic diseases or growth hormone deficiencies could be diagnosed if their complete medical history, a physical examination, and additional investigations were carried out sufficiently. Proper genetic diagnosis of children with non-syndromic SS allows implementation of the appropriate therapeutic approach. Since SHOX haploinsufficiency underlies syndromic and non-syndromic SS and growth hormone treatment could be useful for cases with SHOX haploinsufficiency, SHOX analysis should be one approach commonly used for diagnosis. As SHOX haploinsufficiency is more commonly due to submicroscopic copy number variations (CNVs), rather than point mutations, multiplex ligation-dependent probe amplification (MLPA) should be used as an initial screening technique to assess abnormalities in SHOX [2, 6, 8].
miRNAs are significant modulators of cellular pathways, including growth and proliferation [9]. Certain miRNAs are specifically expressed in cartilage tissue and play an essential role in the regulation of chondrocyte proliferation and differentiation during growth plate development [10]. miR-140 is expressed specifically in the cartilage and is upregulated during mesenchymal stem cell differentiation into chondrocytes. Therefore, miR-140 regulates cartilage development and homeostasis [11]. miR-1 acts by directly inhibiting histone deacetylase 4 at the post-transcriptional level, inducing expression of key regulators of endochondral bone formation [10]. miR-15a-5p recently emerged as a potential biomarker for osteoarthritis. miR-15a-5p promotes the degeneration of chondrocytes by targeting parathyroid hormone-related protein to alleviate the degeneration of chondrocytes via promotion of chondrocyte proliferation and inhibition of terminal differentiation [12].
The aim of our study was to carry out a CNV screening of the SHOX/PAR1 region using the MLPA technique, as well as to perform expression profiling of miR-1, miR-15a, and miR-140 using quantitative real-time polymerase chain reaction (qRT-PCR) in a group of Egyptian children with non-syndromic short stature with and without growth hormone deficiency.
Methods
This study was conducted at the author’s institution during a period of 3 years from October 2016 to June 2019, and was approved by the Medical Ethical Committee. Informed written consent was obtained from parents of the included cases. A total of 266 children with short stature attended our outpatient clinic over this period. LWD/LMD syndromes and skeletal dysplasia were excluded clinically. Conventional karyotyping was carried out to exclude Turner syndrome and any other chromosomal abnormalities. A total of 119 short children were diagnosed with non-syndromic short stature, with and without growth hormone deficiency. Only 50 cases consented to be included in this study and all of the 50 cases that were included had normal karyotypes. Thirty age and sex-matched healthy children were included as controls.
A full history was obtained from the patients. There was particular emphasis on collecting patients’ family history in order to construct a pedigree, including three consecutive generations, consanguinity status, and family history of similar conditions. Heights of the parents were documented. Complete medical history, physical examination, and nutritional status were assessed to exclude systemic diseases, dysmorphism, and clinically suspected syndromic cases. Birth weight, growth hormone profile, and bone age and height in the SD score were documented. Disproportionate SS were excluded from this study.
MLPA assay
DNA extraction was performed on 3 ml of peripheral blood lymphocytes from the 50 cases, their parents, and the reference samples. One reference sample was included for every 7 patient samples, with a minimum of 3 references per test. DNA extraction was carried out using the QIAamp DNA Mini kit, according to the manufacturer’s instructions. The quality and quantity of the DNA samples were determined using a NanoDrop spectrophotometer.
CNV evaluation in the SHOX/PAR1 region was carried out using the SALSA MLPA probemix P018-G1, SHOX, according to the manufacturer’s instructions (MRC-Holland). This probe mix contains 48 MLPA probes, with 26 probes located in the SHOX and Xp22 regions (including SHOX and its regulatory regions) and 13 probes located elsewhere on the X chromosome. One probe is present in each exon of human SHOX. In addition, one probe is included that detects the area just before the SHOX promoter region. Several probes are also located in SHOX regulatory regions, upstream and downstream of SHOX. Several probes for the X chromosome that can be used to characterize larger deletions and to distinguish SHOX deletions from a Turner syndrome karyotype are included in this probemix.
DNA denaturation and overnight hybridization of the MLPA probe mix was performed, followed by probe ligation and amplification the following day. Separation of amplified products was performed using the Genetic Analyzer ABI 3500 (USA). Coffalyser Net software (MRC-Holland) was used to interpret the results. MLPA ratios less than 0.75 were considered a deletion, MLPA ratios between 0.75 and 1.30 were considered normal, and MLPA ratios more than 1.30 were considered duplications. MLPA analysis of the parents of the positive cases was carried out in order to identify the origin of the CNVs that were detected.
miRNAs expression assay
miRNA was isolated from the sera of the 50 cases using the miRNeasy Mini kit (QIAgen), according to the manufacturer’s instructions. Reverse transcription (RT) and qPCR kits made specifically for accurate miRNA analysis (Applied Biosystems) were used to evaluate the expression of miRNA-1 (miR-1-3p), miRNA-15a (miR-15a-5), and miRNA-140 (miR-140-5p) from serum samples. RT reactions were performed using the TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems).
Real-time PCR was performed using a QuantStudio 12 k Flex real-time system (Applied Biosystems). Relative expression of target miRNAs was normalized to miRNA-16. The fold changes of candidate miRNA expression were calculated by the equation 2 −ΔΔCt.
Statistical methods
All data was converted and manipulated using the SPSS software program version 18.0. Data are presented as the mean and standard deviation for quantitative data. The quantitative variables were compared using a Student’s t test for parametric data. The Mann–Whitney U test was applied to non-parametric data. A p value less than 0.05 was considered statistically significant and a p value of 0.01 was considered highly statistically significant.
Results
Our study included 50 children with short stature, with and without growth hormone deficiency, with a total of 5 males and 45 females. Their age ranged between 2 and 16 years and their height ranged between −2.0 and − 5.5 SD. Thirty age and sex-matched healthy children were included as controls for miRNA profiling.
MLPA assay results
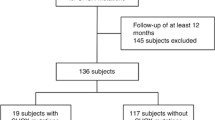
The SHOX CNVs were studied in 50 children with short stature. De novo CNVs of different clinical significance were detected in ten children (20% of the children studied). Five CNVs were pathogenic and related to SHOX. The remaining CNVs were variants of uncertain significance (VOUS) and outside the SHOX area (Fig. 1).

Schematic diagram and pie chart of the MLPA results of the children studied
Four children (8%) had CNVs in the SHOX area. The SHOX area is defined as the region where the SHOX and its regulatory elements are located (hg19; chrX: 398,000–980,000). Heterozygous complete deletion of the SHOX area was detected in one child, except for one probe (L05096) that showed homozygous deletion (Fig. 2a). This patient’s height was −2.4 SD. She had normal birth weight, low growth hormone levels, and delayed bone age. Three children had heterozygous duplication of exon one of SHOX, two children at exon 5 (Fig. 2b) and one child at exon 6 (Fig. 2c). The heights of these children were –3.5 SD, −4.2 SD, –2.8 SD, respectively. All of these children had normal birth weights and growth hormone levels. One child (2%) had a duplication in the SHOX area at hg19; chrX:1,029,698 (probe L24245) (Fig. 2d). His height was –2.5 SD and he had normal birth weight and growth hormone levels.

Ratio charts of MLPA results for children with SS using SALSA MLPA probemix P018-G1 SHOX. The spots represent MLPA probes. The upper blue line indicates a peak ratio of 1.3 and any probes (blue spot) above this line represent a duplication. The lower red line indicates a peak ratio of 0.75 and any probes (red spot) below this line represent a deletion. The probes between the two lines represent the normal two copies. a Represents complete SHOX deletion. b Represents SHOX ex5 duplication. c Represents SHOX ex6 duplication. d Represents SHOX area down duplication. e Represents ASMT, ex8 duplication. f Represents CRLF2, ex2/CSF2RA, ex4 duplication
The remaining five children (10%) had heterozygous duplication CNVs outside the SHOX area. Four of these children had heterozygous duplications in the ASMT, exon 8 (probe L00712) (Fig. 2e), and the fifth child had heterozygous duplications in CRLF2, exon 2 hg19; chrX:1327689 (probe L19678) and CSF2RA, exon 4 hg19; chrX:1401596 (probe L24246) (Fig. 2f). The heights of these children ranged between −2.2 and − 2.6 SD. All of these children had normal birth weights and growth hormone levels.
miRNA expression results
The expression levels of miRNA-140 were statistically significantly greater in the cases as compared to controls (p < 0.05). However, the levels of miRNA-1 and miRNA-15a were not statistically significantly different between the groups studied (p > 0.05) (Table 1). Figure 3 shows the distribution of miR-1, miR-15a, and miR-140 across the patients and controls.

a Distribution of miR-1 across the patients and controls. b Distribution of miR-15a across the patients and controls. c Distribution of miR-140 across the patients and controls
The miRNAs that were studied had no association with age or height. The expression of these miRNAs was also not significantly different between males and females (Table 2).
Discussion
A proper genetic diagnosis leads to implementation of the appropriate treatment for SS. SHOX is one of the main genes involved in human growth and is an important candidate gene used to test SS in children. The majority of SHOX defects are due to complete deletions (70–80%) or partial deletion (2–6%) of SHOX and/or its regulatory enhancer region. A total of 20–25% of SHOX defects is due to mutations [6]. SHOX enhancers are conserved non-coding DNA elements (CNEs). There are three CNEs enhancers upstream of SHOX (CNE-5, CNE-3, and CNE-2), and there are eight CNE enhancers downstream of SHOX, four of which have transcriptional activity (CNE4, CNE5, ECR1 (CNE7), and ECS4 (CNE9)). Recently, ZED, the fifth enhancer, has been reported as a more likely candidate than CNE9 as a critical limb enhancer in the analyzed downstream region [13, 14].
SHOX haploinsufficiency results in syndromic and non-syndromic SS. SHOX haploinsufficiency is primarily due to submicroscopic CNVs in PAR1, encompassing SHOX exons and/or the cis-acting enhancers [2]. However, only one case out of the 50 studied (2%) showed a large de novo SHOX/PAR1 deletion, including complete SHOX and its regulatory regions. This deletion is associated with normal birth weight and low growth hormone levels.
In contrast, duplications inside or outside the SHOX area were detected in 18% of the children studied. All of these children had normal birth weights and growth hormone levels. SHOX duplications have been described in individuals with SS. Studying LWD and ISS patients identified that duplications in SHOX and its associated elements are less frequent than deletions of SHOX. SHOX-associated elements are conserved non-coding elements (CNE) adjacent to the 3′ end (CNE-3,4,5,7,8,9) and 5′ end (CNE-2,3,5) of the gene [4, 7]. Previous studies have investigated the frequency and distribution of duplications within SHOX and its associated elements between population samples and merged groups of ISS/LWD patients [7]. This group proposed that partial SHOX duplications and small duplications encompassing the CNE-9 enhancer could be highly penetrant alleles associated with ISS and LWD development.
While intragenic SHOX duplications lead to SS, whole gene duplications are associated with more heterogeneous phenotypes [4, 13]. Upstream duplication of CNE-5 and CNE-3 and downstream duplication of ECS4/CNE-9 have been described in ISS [4, 15, 16]. Here, we report three heterozygous duplications (6%) in the SHOX area. These include one duplication at exon one of SHOX, and two cases with duplications at exon 5, and one case with a duplication at exon 6, both of which interfere with the SHOXa transcript.
Moreover, we report a heterozygous duplication (2%) at hg19; chrX:1,029,698 (probe L24245) (SHOX down area). While studying a cohort of 1200 patients, previous studies have reported a recurrent 325 kb duplication in the SHOX area (SALSA MLPA probemix P018-G1, probes L20176-L24245) in seven families presenting with SS [13]. Four families presented with LWD in the affected family members, and three families presented with familial short stature. The findings in this study suggest that duplications of upstream and downstream long-range cis-regulatory DNA elements can result in a SHOX-related phenotype. Controversially, Hirschfeldova and Solc described this duplication area only as a polymorphism, as they detected it in their population sample (0.40%) and in only one case from their LWD/ISS samples (0.28%) [7].
The precise mechanisms of the pathogenic effects of microduplications are not yet understood. These duplications most likely exert a negative effect on SHOX transcription, which leads to the reduction of its expression, ultimately resulting in SHOX haploinsufficiency [13]. Partial SHOX duplications appear to have a more deleterious effect than complete SHOX duplications, due to the disruption of the gene sequence [4, 13].
Small duplications at the CNE-9 enhancer are associated with SS. This duplication could decrease effective communication between the CNE-9 enhancer and the SHOX promoter, negatively effecting SHOX expression [7]. However, no duplication of the CNE-9 enhancer was detected in the group of children that we studied.
Five children out of the 50 children studied (10%) had copy number gain outside of the SHOX area. Four of these children had an exon 8 duplication in the ASMT. Acetyl serotonin methyltransferase (ASMT) is the last enzyme in the melatonin biosynthesis pathway and is located in the PAR1 region of the sex chromosomes outside the SHOX area. Melatonin is a synchronizer of many physiological processes and abnormal melatonin signaling is associated with a broad range of diseases. There is data that suggests that there is a relationship between melatonin and growth hormone (GH) secretion, with reciprocal negative feedback regulation [17, 18]. Hirschfeldova et al. reported a recurrent duplication of the ASMT probe (SALSA MLPA probemix P018-G1, probe L00712) with a frequency of 2.3% in their cohort of 352 LWD/ISS patients [3]. However, Hirschfeldova and Solc detected this duplication at a frequency of 2.0% in their population of 250 individuals [3]. These frequencies are comparable to frequencies obtained by Cai et al., where the duplication of ASMT was reported to be associated with autism spectrum disorders [19]. None of the individuals from the Hirschfeldova et al. and Hirschfeldova and Solc studies that carry the ASMT duplication suffer from autism spectrum disorders [3, 7]. Similarly, none of the four cases in the present study that carry the ASMT duplication suffer from autism spectrum disorders. All these findings suggest the possible involvement of the ASMT in SS. Further studies with a larger sample size are needed to develop a better understanding of the role of the ASMT in SS.
The fifth child with a copy number gain outside of the SHOX area had CRLF2, (exon 2 hg19; chrX:1327689) and CSF2RA (exon 4 hg19; chrX:1401596) duplications. Mitka et al. reported an ISS patient with a duplication encompassing the Xp22.33-PAR1 regulatory region and CRLF2 and CSF2RA, which comprises 1.8% of the cases in this study [20].
Our study demonstrated that there was significantly higher expression of serum miR-140 in children with SS as compared to age and sex-matched healthy subjects (p < 0.05). Miyaki et al. identified that miR-140 null mice exhibit long bone growth retardation, which suggests reduced chondrocyte proliferation, although the structure of the articular joint cartilage appeared grossly normal [11]. Loss of miR-140 in mice leads to growth defects of the endochondral bones, resulting in dwarfism and craniofacial deformities [21]. Integrated study of miRNA expression and mRNA expression during chondrogenesis highlights the importance of miR-140, particularly the −5p strand, and identified positive regulation of the Wnt signaling pathway by miR-140-5p. Wnt signaling has a complex role in chondrogenesis and skeletal development [22]. The significantly high serum miR-140 expression levels observed in children with SS in the present study suggest that serum miRNA is associated with the pathogenesis of SS.
Conclusions
This is one of the first studies assessing CNVs in SHOX/PAR1 that has been performed on a group of Egyptian children with short stature. However, a limitation of this study is the relatively small sample size. MLPA analysis of SHOX/PAR1 led to the identification of different CNVs in children with non-syndromic short stature, with and without GH deficiency. These findings suggest that MLPA should be used as an initial screening technique in short children. Proper genetic diagnosis of these children leads to implementation of appropriate therapeutic approaches. Future studies with larger sample sizes are needed to confirm these findings and assess other regions that are considered major regulators of longitudinal growth, such as GH and insulin-like growth factor 1 (IGF-1). miR-140 expression levels were significantly different in children with short stature, suggesting that alterations in its expression level are associated with the pathogenesis of short stature.
Availability of data and materials
Data and material are available upon request.
Abbreviations
- ASMT:
-
Acetyl serotonin methyltransferase
- CNE:
-
Conserved non-coding elements
- CNV:
-
Copy number variation
- GH:
-
Growth hormone
- IGF-1:
-
Insulin-like growth factor 1
- ISS:
-
Idiopathic SS
- LMD:
-
Langer mesomelic dysplasia
- LWD:
-
Lėri-Weill dyschondrosteosis
- miRNA, miR:
-
Micro Ribonucleic acid
- mRNA:
-
Messenger Ribonucleic acid
- MLPA:
-
Multiplex ligation-dependent probe amplification
- PAR1 :
-
Pseudoautosomal region
- qPCR:
-
quantitative real-time polymerase chain reaction
- RT:
-
Reverse transcription
- SGA:
-
Small for gestational age with failure of catch-up growth
- SD:
-
Standard deviations
- SHOX:
-
Short-stature HOmeoboX
- SS:
-
Short stature
References
Marchini A, Rappold G, Schneider KU (2007) SHOX at a glance: from gene to protein. Arch Physiol Biochem 113(3):116–123 Epub 2007/10/09
Fukami M, Seki A, Ogata T (2016) SHOX Haploinsufficiency as a cause of syndromic and nonsyndromic short stature. Mol Syndromol 7(1):3–11 Epub 2016/05/20
Hirschfeldova K, Florianova M, Kebrdlova V, Urbanova M, Stekrova J (2017) Detection of SHOX gene aberrations in routine diagnostic practice and evaluation of phenotype scoring form effectiveness. J Hum Genet 62(2):253–257 Epub 2016/10/07
Benito-Sanz S, Barroso E, Heine-Suner D, Hisado-Oliva A, Romanelli V, Rosell J et al (2011) Clinical and molecular evaluation of SHOX/PAR1 duplications in Leri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J Clin Endocrinol Metabol 96(2):E404–E412 Epub 2010/12/15
Binder G (2011) Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Hormone Res Paediatr 75(2):81–89 Epub 2011/02/18
Caliebe J, Broekman S, Boogaard M, Bosch CA, Ruivenkamp CA, Oostdijk W et al (2012) IGF1, IGF1R and SHOX mutation analysis in short children born small for gestational age and short children with normal birth size (idiopathic short stature). Hormone Res Paediatr 77(4):250–260 Epub 2012/05/11
Hirschfeldova K, Solc R (2017) Comparison of SHOX and associated elements duplications distribution between patients (Leri-Weill dyschondrosteosis/idiopathic short stature) and population sample. Gene. 627:164–168 Epub 2017/06/21
Wit JM (2011) Definition and subcategorization of idiopathic short stature: between consensus and controversy. Hormone Res Paediatr 76(Suppl 3):3–6 Epub 2011/09/23
Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF (2009) miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA 106(29):12103–12108 Epub 2009/07/04
Li P, Wei X, Guan Y, Chen Q, Zhao T, Sun C et al (2014) MicroRNA-1 regulates chondrocyte phenotype by repressing histone deacetylase 4 during growth plate development. FASEB J. 28(9):3930–3941 Epub 2014/05/23
Miyaki S, Sato T, Inoue A, Otsuki S, Ito Y, Yokoyama S et al (2010) MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev 24(11):1173–1185 Epub 2010/05/15
Duan ZX, Huang P, Tu C, Liu Q, Li SQ, Long ZL et al (2019) MicroRNA-15a-5p regulates the development of osteoarthritis by targeting PTHrP in chondrocytes. BioMed Res Int 2019:3904923 Epub 2019/04/06
Bunyan DJ, Baffico M, Capone L, Vannelli S, Iughetti L, Schmitt S et al (2016) Duplications upstream and downstream of SHOX identified as novel causes of Leri-Weill dyschondrosteosis or idiopathic short stature. Am J Med Genet A. 170A(4):949–957 Epub 2015/12/25
Skuplik I, Benito-Sanz S, Rosin JM, Bobick BE, Heath KE, Cobb J (2018) Identification of a limb enhancer that is removed by pathogenic deletions downstream of the SHOX gene. Sci Rep 8(1):14292 Epub 2018/09/27
Hirschfeldova K, Solc R, Baxova A, Zapletalova J, Kebrdlova V, Gaillyova R et al (2012) SHOX gene defects and selected dysmorphic signs in patients of idiopathic short stature and Leri-Weill dyschondrosteosis. Gene. 491(2):123–127 Epub 2011/10/25
Fukami M, Naiki Y, Muroya K, Hamajima T, Soneda S, Horikawa R et al (2015) Rare pseudoautosomal copy-number variations involving SHOX and/or its flanking regions in individuals with and without short stature. J Hum Genet 60(9):553–556 Epub 2015/06/05
Munoz-Hoyos A, Hubber E, Escames G, Molina-Carballo A, Macias M, Valenzuela-Ruiz A et al (2001) Effect of propranolol plus exercise on melatonin and growth hormone levels in children with growth delay. J Pineal Res 30(2):75–81 Epub 2001/03/29
Botros HG, Legrand P, Pagan C, Bondet V, Weber P, Ben-Abdallah M et al (2013) Crystal structure and functional mapping of human ASMT, the last enzyme of the melatonin synthesis pathway. J Pineal Res 54(1):46–57 Epub 2012/07/11
Cai G, Edelmann L, Goldsmith JE, Cohen N, Nakamine A, Reichert JG et al (2008) Multiplex ligation-dependent probe amplification for genetic screening in autism spectrum disorders: efficient identification of known microduplications and identification of a novel microduplication in ASMT. BMC Med Genomics 1:50 Epub 2008/10/18
Mitka M, Bednarek M, Kaluzewski B (2016) Diagnostics of SHOX gene rearrangement in 46,XX women with idiopathic short stature. Endokrynologia Polska. 67(4):397–402 Epub 2016/07/09
Nakamura Y, Inloes JB, Katagiri T, Kobayashi T (2011) Chondrocyte-specific microRNA-140 regulates endochondral bone development and targets Dnpep to modulate bone morphogenetic protein signaling. Mol Cell Biol 31(14):3019–3028 Epub 2011/05/18
Barter MJ, Tselepi M, Gomez R, Woods S, Hui W, Smith GR et al (2015) Genome-wide microRNA and gene analysis of mesenchymal stem cell chondrogenesis identifies an essential role and multiple targets for miR-140-5p. Stem Cells. 33(11):3266–3280 Epub 2015/07/16
Acknowledgements
Not Applicable
Funding
This research was financially supported by 11th In-house Research Project grant 2016-2019, grant number: 11010161, National Research Centre, Egypt. We would also like to express our gratitude to the funding agent, for giving us the chance to accomplish this study with the help of its updated equipment and instrumentation.
Author information
Authors and Affiliations
Contributions
OE: providing the idea of this research, participated in preparing the design of the research, conducting the laboratory work, interpretation of the data, and preparing the paper for submission and final approval of the version to be published. ME: participated in performing the laboratory work, and preparing the paper for submission. AA: participated in preparing the design of the research, participated in conducting the laboratory work, statistical analysis, and preparing the paper for submission. RA: participated in conducting the laboratory work, and preparing the paper for submission. MF: participated in conducting the laboratory work and preparing the paper for submission. RM: participated in conducting the laboratory work and preparing the paper for submission. MM: participated in conducting the laboratory work and preparing the paper for submission. AK: participated in conducting the laboratory work and preparing the paper for submission. IM: participated in the clinical evaluation of the patients and preparing the paper for submission. HE: participated in the clinical evaluation of the patients and preparing the paper for submission. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the ethical committee of the National Research Centre, Egypt (16-274), which is in accordance with the ethical standards of the Declaration of Helsinki. All participants gave informed written consent before their inclusion in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eid, O.M., Eid, M.M., Abdelrahman, A.H. et al. Screening of the SHOX/PAR1 region using MLPA and miRNA expression profiling in a group of Egyptian children with non-syndromic short stature. Egypt J Med Hum Genet 21, 47 (2020). https://doi.org/10.1186/s43042-020-00090-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-020-00090-4