
Abstract
Background
Spinal muscular atrophy (SMA) is mostly caused by homozygous deletions in the survival motor neuron 1 (SMN1) gene. SMN2, its paralogous gene, is a genetic modifier of the disease phenotype, and its copy number is correlated with SMA severity. The purpose of the study was to investigate the number of copies of the SMN1 and SMN2 genes in a Venezuelan population control sample and in patients with a presumptive diagnosis of SMA, besides estimating the frequency of mutation carriers in the population.
Results
SMN1 and SMN2 gene copies were assessed in 49 Venezuelan dweller unrelated normal individuals and in 94 subjects from 29 families with a SMA presumptive diagnosis, using the quantitative PCR method. A SMN1 deletion carrier frequency of 0.01 and 0.163 of homozygous absence of the SMN2 gene were found in the Venezuelan control sample. Deletion of SMN1 exon 7 was confirmed in 15 families; the remaining 14 index cases had two SMN1 copies and a heterogeneous phenotype not attributable to SMN deletions. Based on clinical features of the index cases and the SMN2 copy number, a positive phenotype-genotype correlation was demonstrated. No disease geographical aggregation was found in the country.
Conclusion
The frequency of carriers of the deletion of exon 7 in SMN1 in the Venezuelan control population was similar to that observed in populations worldwide, while the frequency of 0 copies of the SMN2 gene (16.3 %) seems to be relatively high. All these findings have pertinent implications for the diagnosis and genetic counseling on SMA in Venezuela.
Similar content being viewed by others

Background
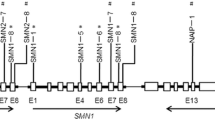
The SMN locus (Survival of motor neuron, OMIM #600354, #601627) is located within a large inverted duplication on chromosome 5q13. Normal individuals have two genes encoding SMN protein that are arranged in tandem on each chromosome: SMN1 (telomeric copy) and SMN2 (centromeric copy). Both genes share more than 99% nucleotide identity, differing only by five nucleotides. The protein encoded by the SMN genes is a RNA-binding protein required for the assembly of small nuclear ribonucleoprotein (snRNP) complexes involved in pre-mRNA splicing, with high expression levels in the motor neurons of the spinal cord.
Although both genes produce equal transcript amounts, a large proportion of SMN2 transcripts lack exon 7, due to the c.840 C>T nucleotide transition (rs1164325688) in exon 7 which causes its skipping, resulting in a truncated, less stable, and poorly functional protein.
Homozygous deletion of the entire SMN1 gene or loss of its exon 7 causes spinal muscular atrophy (SMA, OMIM #253300), a recessive inherited neuromuscular disease characterized by muscle weakness and atrophy due to the progressive degeneration and loss of the motor neurons of the anterior horn in the spinal cord (lower motor neurons), producing a system-wide atrophy of skeletal muscles. Patients with SMA are classified into four subtypes, based on the disease severity and age of onset: type I, the most severe infantile SMA, or Werdnig-Hoffmann disease; type II or infantile chronic SMA; type III, juvenile SMA, or Wohlfart-Kugelberg-Welander disease; and type IV or adult-onset Kufs SMA disease.
Absence of the SMN1 gene is partially compensated by SMN2; however, its lesser functional protein cannot fully replace the SMN protein function. Loss of SMN1 exon 7 can occur by gene conversion from SMN1 to SMN2, and this conversion event causes a variable number of copies of SMN2 in individuals, ranging from 0 to 5 or more. There is an inverse correlation between disease severity and number of copies of SMN2 [1, 2]; a SMN2 higher copy number results in a less severe phenotype [3].
The disease-prone mutation prevalence is higher among European Caucasoids, with a carrier frequency estimate of 1/47, followed by, Asian Indians 1/52, Asians 1/59, “Hispanics” 1/68, and African Americans 1/72 [4]. In Venezuela, a carrier frequency of 1/72 has been reported among gamete donors from a fertility clinic in two sisters whose ancestors came from Spain and from Italy [5]. To date, copy number variation of both genes in the admixed Venezuelan populations is still unknown.
Herein we report the results of qPCR quantification of the SMN1 and SMN2 genes in geographically heterogeneous Venezuelan population sample, as well as in SMA random families.
Methods
Sample
A quantitative analysis of SMN1 and SMN2 genes was performed in 94 individuals from 29 previously unselected non-related families, during the period of years 2002–2018; with a clinical diagnosis of presumptive spinal muscular atrophy, which were referred to the Laboratory of Human Genetics (LHG) at the Venezuelan Institute for Scientific Research (IVIC) for molecular diagnosis and genetic counseling; and in 49 unrelated control individuals, 47 of which had ancestors born in Venezuela, from different country states representing the main geographical regions: Andean, Occidental, North-Central, Eastern, and the Central Plains, as follows: Táchira (n = 3), Trujillo (n = 1), Zulia (n = 1), Falcón (n = 5), Lara (n = 3), Anzoátegui (n = 2), Bolívar (n = 2), Yaracuy (n = 1), Carabobo (n = 9), Aragua (n = 3), Capital District (Caracas) (n = 10), Barinas (n = 2), Guárico (n = 5), and two who had ancestors born in Colombia (Bucaramanga and Cúcuta).
The precise place of birth of remote ancestors was established for each index case, and a family history was recorded for phenotype and age of onset of symptoms.
Patients sample size was self-determined by the number of referred unrelated presumptive SMA subjects. Control individuals were extracted from the general population, which were pair-matched with the former, according to their similar (state) geographic place of birth.
The study was conducted according to the ethical standards of the Helsinki Declaration. Verbal consent was obtained from the parents of the patients to perform the diagnostic genetic testing. DNA samples of control individuals were included in the study, according to the institutional Bioethics Committee approval protocol; reference number GH-2261/27-11-2011.
DNA isolation and quantification
After voluntary informed consent from the research subjects, a 5 mL blood sample was withdrawn, EDTA-Na4 anti-coagulated, and the DNA was extracted by a saline method [6]. In some instances, prenatal diagnosis was performed in DNA extracted from amniotic fluid, using a Proteinase K protocol. A control sample with matched number of pregnancy weeks was always included.
DNA concentration was measured using a fluorescence photometer (Qubit ®, Invitrogen); each sample was diluted to a final DNA concentration of 3 ng/μL.
Quantitative real time PCR of SMN1 and SMN2
For the estimation of SMN1 and SMN2 copy numbers, a quantitative real-time PCR protocol was used based on primers that specifically amplify SMN1 and SMN2 genes. Primers designed by Feldkötter et al. [1] were selected to distinguish between both genes, based on the nucleotide differences in exon 7 at position 6 and intron 7 at position +214 [1]. As a DNA canonical sequence reference, exon 12 of the human serum albumin locus (ALB) was chosen [7]. Reactions were performed using a SYBR® Green-based qPCR.
The homozygous absence of SMN1 exon 7 was subsequently confirmed with the Dra I restriction enzyme, according to van der Steege et al. [8], since there is a strict cis allelic disequilibrium of an allele A in both mutation c.*239G>A, responsible for the SMN1 exon 7 deletion and the Dra I enzyme digestion of SNP rs1361078560 (c.*216G>A).
Gene dosage analysis
The analysis was performed by the comparative CT method [9] using the CT values obtained with the Eco software (Illumina). Patients, carriers, and unrelated unaffected individual samples were analyzed together with a calibrator sample in every assay. The relative gene copy numbers were estimated by the 2−ΔΔCT expression (Livak’s method) [9].
Segregation analysis
Microsatellite markers D5S610, D5S435, and D5S1556 [10, 11] were selected to assess the deletion segregation in one family with three consecutive generations of affected individuals. Genotyping was performed through conventional PCR, followed by polyacrylamide gel electrophoresis, and the fragments were revealed with silver nitrate.
Data analyses
General data analyses (average, standard deviation, Pearson’s correlation coefficient, Chi-square test) were performed using the statistical function tool of Microsoft® Excel.
A discriminant equation for quantitative values [12] was developed to calculate the cutoff value of 2−∆∆CT between 1 and 2 gene copy categories, using the mathematical expression L = [(XC) (SDNC) + (XNC) (SDC)]/SDNC + SDC, where XC is the arithmetic mean of the 2−ΔΔCT values of the obligatory carriers, SDNC is the standard deviation of the 2−ΔΔCT values of the healthy non-carrier individuals, XNC is the arithmetic mean of non-carrier 2−ΔΔCT values, and SDC is the standard deviation of the obligatory carrier 2−ΔΔCT values.
The equation compares the 2−ΔΔCT values of the obligatory deletion carriers (parents of patients with the homozygous deletion of SMN1) with the range of values of individuals with 2 copies of SMN1.
Results
The number of families included in the study represents the random prevalence figure of SMA along 16 years. All families had remote ancestors born in the country, except for 2 mentioned instances from different foreign geographical regions.
SMA clinical diagnosis
Fifteen index cases out of 29 studied families showed full, typical clinical signs of the disease: hypotonia, EMG findings and/or muscle biopsy compatible with SMA, poor fetal movements during pregnancy, weakness/loss of cephalic support, and symmetrical limb muscle weakness; additional findings varied according to the age of onset of the disease. All the remaining index cases (aged between 3 months and 14 years old, being 80% of them below 5 years of age) had severe hypotonia accompanied by diverse non-specific clinical signs of SMA.
Quantitative analysis
The results obtained according to the quantitative analyses showed that the ranges of 2−ΔΔCT values did not overlap with those corresponding to each category of gene copy numbers. Table 1 shows the range of values of 2−ΔΔCT of SMN1 and SMN2 genes in non-related control individuals. One out of those 49 analyzed subjects (98 chromosomes) was considered to be a carrier (heterozygous) for the SMN1 deletion, with a value of 2−ΔΔCT = 0.35; giving an estimated allelic frequency of 0.01 in the Venezuelan sample.
Thirty-seven individuals (75.5%) were unequivocally classified as carriers of two copies of the SMN1 gene, two individuals (4.1%) were classified as carriers of 3 copies of SMN1, two as carriers of 4–5 copies, and one with a value of 2−ΔΔCT eventually corresponding to a dose of 6 copies of that gene.
Results for the SMN2 gene showed that 27 subjects (55.1%) carried a single copy of the gene, 7 individuals (14.3%) possessed 2 copies, and 5 (10.2%) had 3 copies. A sample number considered high (16.3%, 8 subjects) had no copies of SMN2; in them, the values of 2−ΔΔCT were equal or very close to 0.00, indicating the homozygous absence of the gene.
In the control sample (Table 1), there were 6 borderline cases in which the classification into one or two gene copy category could not be a priori unequivocally established. In them, the average values of 2−∆∆CT (0.687) did not overlap however with that of the obligatory carrier parents figure (0.43) of the affected subjects. Nonetheless, the upper limits in the ranges were very close, so it was necessary to establish the cutoff value of each category calculating it through the use of a discriminant equation. A threshold value L = 0.66 was estimated, which permitted to classify those individuals within the most probable gene copy number category. That is, with values below 0.66, it was considered likelier to belong to the carrier range (1 gene copy), and with values above it, it was likelier to belong to the range of 2 copies. Therefore, according to this final classification, the estimated frequency of carriers of 2 SMN1 copies in the Venezuelan control sample was 87.8%.
The use of an additional technique as multiplex ligation-dependent probe amplification (MLPA) could validate this classification.
All fourteen index cases without typical clinical findings of SMA had two copies of the SMN1 gene. In them, the etiology of the hypotonia was diverse but not attributable to a lower motor neuron impairment; thus, further differential diagnoses were assessed.
Three out of the fifteen families in whom a molecular confirmation of SMA was established were consultand parents whose first child had died in the first months of life, with severe hypotonia and typical signs of SMA, but in whom a DNA diagnosis of the disease had not been previously made. All of them resulted to carry a single copy of SMN1. In 11 families, SMA was confirmed in the index case by homozygous deletion of exon 7 of SMN1 (Table 2). In total, 16 individuals with this genotype were detected. Table 3 shows the clinical features of those patients. Only in one instance, a patient with typical clinical signs of the disease did not have a homozygous SMN1 exon 7 deletion; he had instead 2 SMN1 copies but no SMN2 copies; both the EMG and the muscle biopsy findings were compatible with a SMA diagnosis.
In one of the families, a reliable prenatal diagnosis of SMA could be established in an amniotic fluid sample of a second pregnancy of the carrier mother whose first affected child had no copies of SMN1 and a single SMN2 gene.
One of the studied families (referred to as family 7, Table 3) had 5 members with homozygous absence of the SMN1 gene. Of them, a brother (III-8) and a cousin (III-3) of the index case were severely affected, but conversely not so were their mothers (II-3 and II-5), who had homozygous absence of the SMN1 gene having each 4 or more copies of the SMN2 gene (Fig. 1; Table 2).

Pedigree of SMA family 7, showing the copy number of the SMN genes of each studied member and the in-phase haplotype constructed with the SMA multicopies flanking markers D5S610, D5S1556, and D5S435. It is shown that the index case grandmother’s in-phase haplotype (122, 110/114, 140) was transmitted to the two SMA asymptomatic daughters (II-3 and II-5). In the transmission to the index case and his brother (III-7 and III-8, respectively), events of double crossing over appeared independently twice. The grandfather I-1 was not available for SMN gene testing; I-2 needed a wheelchair after her 6th decade of age due to weakness of her lower limbs. II-3 and II-5 remained asymptomatic at ages 46 and 57, respectively. Their husbands II-4 and II-6 were not biologically related, the II-6 remote ancestors came from a different geographic region than that of I-1 and I-2; the geographic origin of the ancestors of II-4 was unknown. III-6 died at 4 months of age without a molecular diagnosis of SMA. III-3, III-7, and III-8 were still alive at ages 18, 30, and 34, respectively, when the study was performed
Genotype-phenotype correlation
Based on the genotypes established by the real-time PCR analysis and the clinical signs of the disease in each case (Table 3), the genotype-phenotype correlation could be assessed; a correlation coefficient of 0.416 indicated indeed a positive correlation.
Discussion
The identification of the genotype constitution (SMN1: SMN2 copy numbers) in patients diagnosed with SMA has important implications for the genetic counseling of families with or without a history of the disease. Approximately 94–95% of individuals with SMA are homozygous carriers of a SMN1 exon 7 deletion (OMIM #253300; ORPHA #70). On the other hand, SMN2 is a SMA modifier gene, and a strong inverse correlation between its copy number and disease severity has been demonstrated: with a larger number of copies, the better the prognosis and the later the age of onset [3, 13, 14].
In our study, the number of SMN2 copies and the carrier frequency of the SMN1 deletion in a random sample of the Venezuelan populations were assessed, as well as in families with a presumptive diagnosis of SMA.
Gene dosages
The frequency of carriers of 2 SMN1 copies in the Venezuelan control sample was 87.8%, very similar to the observed pan-ethnic frequencies (84.84%) [4].
It has been reported that about 2–5% of SMA carriers have two SMN1 copies in cis in one chromosome and none in the other (silent carriers 2/0).This could produce a lower detection rate [15, 16]. Neither MLPA nor qPCR techniques allow detection of the carriers of this condition, giving a false negative result. Thus, in partners of known SMA carriers, it is important to test their parents and additional family members to exclude the presence of a cryptic 2/0 genotype [17].
One of the 49 studied control subjects had 1 copy of the gene, being thus a carrier of the deletion. This corresponds to an allelic frequency of 0.01 in this sample (n = 98 chromosomes), for a carrier frequency of 2.04% (1/49) in the Venezuelan populations, being close to those values reported in diverse populations: Brazil 2.7% [18]; USA Caucasoids 2.7%; African Americans 1.1%; Ashkenazi Jews 2.2%; and 1.8% in Asians [19]. In sub-Saharan populations, the frequencies are lower: 1.6% in Nigerians, 0.83% in Kenyans, 0.48% in Malian [20]; in France, it is 2.9% in the general population [21]. Moreover, the frequency of homozygotes with deletion of SMN1 in the Venezuelan sample calculated by extrapolation of the Hardy-Weinberg equation is 1/9091, being similar to several studies, in which the estimated prevalence has been around 1/6000 to 1/10000 livebirths [22, 23].
Around 4% of the control individuals carried 3 copies of SMN1 as has been found in most populations with variable frequencies (5.3 to 41.3%) [24]. Three individuals had 4 or more copies of SMN1; they did not carry any copy of SMN2. This inverse correlation, caused by gene conversion events between both genes [2], suggests that the increase in the number of copies of SMN1 is associated with the number of copies of SMN2 in the general population. Furthermore, 16% of control individuals of the Venezuelan population sample lack SMN2 copies. According to the results obtained in other assessed populations [24], the average of SMN2 = 0 in the main ethnic groups are European = 6.8%, Asian = 3.9%, African = 23.1%, and American = 7.2% (the last category only includes Colombian and Peruvian subjects, total n = 100). The admixed populations of Venezuela have ancestral Amerindian, African, and Spanish genetic contributors, in varying proportions depending on the geographic regions. Our study is the first that assessed SMN copy number variation in a Venezuelan control sample. Although the sample is small (n = 98 chromosomes), it informs about the frequencies in our random admixed populations, which in the studied cohort seems to be close to the American, but higher, and African ones. A larger population sample should thus be studied to confirm these findings.
Genotype-phenotype correlation
SMA is a disease with a wide inter- and intra-family phenotypic heterogeneity. The motor dysfunction associated with the loss of motor neurons and skeletal muscle atrophy is a defining feature of SMA throughout the clinical spectrum of the disease. The symptoms of the studied cases were characterized by generalized muscular hypotonia, areflexia, weakness of the intercostal muscles, progressive proximal weakness, inability to remain seated or standing, scoliosis, and electromyography (EMG) results compatible with SMA. In 10 out of 14 cases, SMA was confirmed by homozygous deletion of SMN1. In a single instance, the index case did not have any SMN1 deletion despite having all the typical features of the disease; he probably is a carrier of a point mutation in the gene, a possibility which should be confirmed by a Sanger sequencing.
The analysis of the number of SMN2 copies in the studied individuals homozygous for the SMN1 deletion showed that, although the phenotype-genotype correlation is quite clear, it is not the only genetic factor influencing it. The correlation coefficient estimated from the SMN2 number of copies and the SMA subtype was 0.416, indicating a significance, but not complete correlation between the genotype and the clinical phenotype, attributable to the eventual presence of positive modifiers, such as the SMN2 variants c.859G>C (p.Gly287Arg) in exon 7 and g.69372304A > G in intron 6; both variants produce a moderate increase in exon 7 inclusion [25, 26]. Additionally, the PLP3 gene is known to influence the phenotype [27]. However, the number of SMN2 copies is the strongest known modifier of the SMA phenotype [3] and it is relevant for current clinical trials using nusinersen (SpinrazTM). This is the first approved drug for SMA treatment. Nusinersen is an antisense oligonucleotide that binds specifically a silencer sequence in intron 7 allowing the inclusion of exon 7 in the mature mRNA from SMN2 [17], increasing thus the amount of a functional full-length SMN protein. The available data show that the efficacy of the treatment is enhanced when it is applied before or soon after the onset of symptoms [28, 29]. Targeted treatment is recommended for all individuals who have two or three copies of SMN2; contrarily, for individuals with four or more copies, targeted treatment can be deferred until symptom onset [30].
A clear SMN2 genotype-phenotype correlation was observed in family 7 (Table 3), which had four affected siblings (Fig. 1), with ages of onset between 6 and 11 months of age. Patient III-6 was born without motor activity and died at 4 months of age with respiratory insufficiency. It was surprising to find that the mothers (II-3 and II-5, Fig. 1) of the affected children despite being also homozygous carriers of the deletion had very few clinical manifestations of the disease: fine tongue tremor and decreased muscle strength in the hands. Quantification of SMN2 copy number revealed 4–5 in each case, clearly illustrating the phenotypic modulation of SMN2 on the disease severity.
On the other hand, the haplotype analysis in the family revealed that a double recombinant event had occurred in the transmission of the affected gene from one of the mothers (II-5, Fig. 1) to two of her affected children (III-7 and III-8). The presence of a large inverted repeat as well as multiple smaller repeats in the 5q13 region makes the SMN locus highly susceptible to unequal recombination, causing de novo deletions of SMN1 [2]. This mechanism could be acting in this family, which showed an unexpected number of independent deletion carriers. Husbands of the affected mothers (II-3 and II-5, Fig. 1) are not biologically related, carrying different in-phase haplotypes.
The findings in this family show the importance of performing genetic tests to different relatives at risk in specific families, even if they did not have clinical manifestations of the disease. Unfortunately, grandfather I-1 could not be studied but his spouse I-2 was informative enough for inferring genotype origins in their immediate descendants.
Geographic distribution
There was neither close nor remote kinship between the parents in any of the families. Regarding the possible geographic aggregation of SMN1 gene deletion, in only one family, it was observed an eventual “geographic focus” [31], since two grandparents from different ancestral lines of the index case were born in Maracay, State of Aragua. Ancestors of the control individual, carrier of the SMN1 deletion, were born in the central states of the country: Guárico (Calabozo) and Aragua (Villa de Cura), this latter city being very close to Maracay city, suggesting a higher mutation prevalence in that eventual geographic focus. The remaining families had quite diverse places of birth of their ancestors throughout the country, without any clear or even suspected aggregation.
Conclusions
The results of the present study showed that the frequency of deletion carriers of exon 7 in SMN1 seems to be relatively high in the Venezuelan populations, as is also the frequency of 0 copies of the SMN2 gene (16.3%). The range of values of 2−∆∆CT allowed a reliable estimation of the number of copies of each SMN gene, which is a valuable information for the proper genetic counseling of at risk couples and subjects, as well as for establishing a timely therapeutic intervention. The geographic distribution of the SMA family ancestors did not show any strong geographic aggregation, being them scattered throughout the whole country.
Limitations
In spite of the randomness, which thus rules out any bias in both cohort samples, the total size is barely almost 200 chromosomes. However, most results—with a single significant deviation—are close to the expected ones that have been observed in major populations worldwide. Conversely, other methods with high analytical power were not additionally tested due to lack of the timely financial support. Availability of such resource would have allowed an eventual confirmation of the results herein obtained by applying quite simple and economic although informative techniques. Such actions rendered the first disposable data on the geographic distribution and dosage effect of the SMN1-SMN2 genes for the Venezuelan populations under risk of SMA, which require more objective diagnostic and prognostic information from genetic counseling.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- SMA:
-
Spinal muscular atrophy
- SMN :
-
Survival of motor neuron
- SMN1 :
-
Survival motor neuron 1 gene
- SMN2 :
-
Survival motor neuron 2 gene
- EMG:
-
Electromyography
- qPCR:
-
Quantitative polymerase chain reaction
- RFLP-PCR:
-
Polymerase chain reaction–restriction fragment length polymorphism
- Ct:
-
Cycle threshold
References
Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B (2002) Quantitative analyses of SMN1 and SMN2 based on real-time light Cycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 70(2):358–368. https://doi.org/10.1086/338627
Ogino S, Gao S, Leonard DG, Paessler M, Wilson RB (2003) Inverse correlation between SMN1 and SMN2 copy numbers: evidence for gene conversion from SMN2 to SMN1. Eur J Hum Gen 11(3):275–277. https://doi.org/10.1038/sj.ejhg.5200957
Calucho M, Bernal S, Alías L, March F, Venceslá A, Rodríguez-Álvarez FJ, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2,834 reported cases, Neuromuscul Disord 2018;208-215. doi.org/10.1016/j.nmd.2018.01.003.
Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM et al (2012) A pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72 400 specimens. Eur J Hum Gen 20(1):27. https://doi.org/10.1038/ejhg.2011.134
Urbina MT, Benjamin I, Medina R, Jiménez J, Trías L, Lerner J (2017) Expanded carrier screening in gamete donors of Venezuela. JBRA Assisted Reproduction 21(4):356. https://doi.org/10.5935/1518-0557.20170062
Lahiri D, Nurnberger J (1991) A rapid and non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res 19:5444. https://doi.org/10.1093/nar/19.19.5444
Lee TM, Kim SW, Lee KS, Jin HS, Koo SK, Jo I, Kang S et al (2004) Quantitative analysis of SMN1 gene and estimation of SMN1 deletion carrier frequency in Korean population based on real-time PCR. J Korean Med Sci 19(6):870–873. https://doi.org/10.3346/jkms.2004.19.6.870
van der Steege G, Grootscholten PM, van der Vlies P, Draaijers TG, Osinga J, Cobben JM et al (1995) PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet 345:985–986. https://doi.org/10.1016/s0140-6736(95)90732-7
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
DiDonato CJ, Morgan K, Carpten JD, Fuerst P, Ingraham SE, Prescott G et al (1994) Association between Ag1-CA alleles and severity of autosomal recessive proximal spinal muscular atrophy. Am J Hum Genet 55(6):1218–1229
Wirth B, Hahnen E, Morgan K, DiDonato CJ, Dadze A, Rudnik-Schöneborn S et al (1995) Allelic association and deletions in autosomal recessive proximal spinal muscular atrophy: association of marker genotype with disease severity and candidate cDNAs. Hum Mol Genet 4(8):1273–1284. https://doi.org/10.1093/hmg/4.8.1273
Barrai I. 1984. Metodi di regressione e classificazione in biometria. Prima edizione. Edagricole. Italia. Pp 95-105.
Mailman MD, Heinz JW, Papp AC, Snyder PJ, Sedra MS, Wirth B et al (2002) Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med 4(1):20–26. https://doi.org/10.1097/00125817-200201000-00004
Wirth B, Brichta L, Schrank B, Lochmüller H, Blick S, Baasner A et al (2006) Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet 119(4):422–428. https://doi.org/10.1007/s00439-006-0156-7
Alías L, Barceló MJ, Bernal S, Martínez-Hernández R, Also-Rallo E, Vázquez C et al (2014) Improving detection and genetic counseling in carriers of spinal muscular atrophy with two copies of the SMN1 gene. Clin Genet 85(5):470–475. https://doi.org/10.1111/cge.12222
Verhaart I, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC et al (2017) Prevalence, incidence and carrier frequency of 5q–linked spinal muscular atrophy – a literature review. Orphanet J Rare Dis 12:124. https://doi.org/10.1186/s13023-017-0671-8
Serra-Juhe C, Tizzano EF (2019) Perspectives in genetic counseling for spinal muscular atrophy in the new therapeutic era: early pre-symptomatic intervention and test in minors. Eur J Hum Genet 27:1774–1782. https://doi.org/10.1038/s41431-019-0415-4
Bueno KC, Gouvea SP, Genari AB, Funayama CA, Zanette DL, Silva WA Jr et al (2011) Detection of spinal muscular atrophy carriers in a sample of the Brazilian population. Neuroepidemiology 36(2):105–108. https://doi.org/10.1159/000324156
Hendrickson BC, Donohoe C, Akmaev VR, Sugarman EA, Labrousse P, Boguslavskiy L et al (2009) Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Gen 46(9):641–644. https://doi.org/10.1136/jmg.2009.066969
Sangaré M, Hendrickson B. Sango HA, Chen K, Nofziger J, Amara A, et al. Genetics of low spinal muscular atrophy carrier frequency in sub-Saharan Africa. Ann Neurol 2014; 75(4):525-532. doi:https://doi.org/10.1002/ana.24114
Cusin V, Clermont O, Gerard B, Chantereau D, Elion J (2003) Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. J Med Genet 40(4):e39–e39. https://doi.org/10.1136/jmg.40.4.e39
Prior TW, Snyder PJ, Rink BD, Pearl DK, Pyatt RE, Mihal DC et al (2010) Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A. 152A:1608–1616. https://doi.org/10.1002/ajmg.a.33474
Verhaart I, Robertson A, Leary R, McMacken G, KÖnig K, Kirschner J et al (2017) A multi-source approach to determine SMA incidence and research ready population. J Neurol 264:1465–1473. https://doi.org/10.1007/s00415-017-8549-1
Vijzelaar R, Snetselaar R, Clausen M, Mason AG, Rinsma M, Zegers M et al (2019) The frequency of SMN gene variants lacking exon 7 and 8 is highly population dependent. PLoS ONE 14(7):e0220211. https://doi.org/10.1371/journal/pone.0220211
Bernal S, Alías L, Barceló MJ, Also-Rallo E, Martínez-Hernández R, Gámez J et al (2010) The c.859>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J Med Genet 47:640–642. https://doi.org/10.1136/jmg.2010.079004
Wu X, Wang SH, Sun J, Krainer AR, Hua Y, Prior TW (2017) 2017. A-44G transition in SMN2 intron 6 protects patients with spinal muscular atrophy. Hum Mol Genet 26:2768–2780. https://doi.org/10.1093/hmg/ddx166
Stratigopoulos G, Lanzano P, Deng L, Guo J, Kaufmann P, Darras B et al (2010) Association of plastin 3 expression with disease severity in spinal muscular atrophy only in postpubertal females. Arch Neurol 67(10):1252–1256. https://doi.org/10.1001/archneurol.2010.239
Dangouloff T, Servais L (2019) Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther Clin Risk Manag 15:1153–1161. https://doi.org/10.2147/TCRM.S172291
Müller-Felber, Villa K, Schwartzb O, Gläserc D, Uta Nennstield U, Wirth B et al. Infants diagnosed with spinal muscular atrophy and 4 SMN2 copies through newborn screening–opportunity or burden? J Neuromuscul Diseas Preprint 2020; 1-9. doi: 10.3233/JND-200475
Glascock J, Sampson J, Haidet-Phillips A, Connolly A, Darras B, Day J, et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis 2018; 5(2):145–158. doi: 0.3233/JND-180304
Arias S. 1994. Selective detection among the high risk populations, instead of mass screening proposed for countries with various homogeneous origins and a stratified distribution of abnormal genes. In Farriaux JP, Dhondt JL. (eds). New horizons in neonatal screening, Int. Cong. Ser. 1041:97-100. Excerpta Med. Amsterdam.
Acknowledgements
The authors thank all the participants and families for accepting to participate in this study.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
YY performed part of the laboratory work, the statistical analysis, and wrote a preliminary version of the manuscript. IP conceived the study, performed part of the laboratory work, and corrected the manuscript. SA diagnosed the patients, had important intellectual contribution to the study, and corrected the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted according to the ethical standards of the Helsinki Declaration. Verbal consent was obtained from the parents of the patients to perform the diagnostic genetic testing. DNA samples of control individuals were included in the study, according to the Bioethics Committee approval; reference number GH-2261/27-11-2011.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yépez, Y., Paradisi, I. & Arias, S. Spinal muscular atrophy in Venezuela: quantitative analysis of SMN1 and SMN2 genes. Egypt J Med Hum Genet 21, 26 (2020). https://doi.org/10.1186/s43042-020-00070-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-020-00070-8