
Abstract
Objective
DEPDC5 together with NPRL2 and NPRL3 forms the GATOR1 which plays an important role in the the mechanistic target of rapamycin (mTOR) pathway. Deregulation of mTOR signalling has been associated to various neurological conditions, including epilepsy. Variants in the gene encoding GATOR1 complex, especially in DEPDC5, have been implicated in the pathogensis of several focal epilepsies. While there was little report on the electroencephalogram (EEG) feature of DEPDC5 related epilepsy, we decided to investigate the specific EEG pattern and the prognosis of DEPDC5 related epilepsy.
Methods
The records of 546 epilepsy patients with unknown causes who were admitted in Xijing Hospital and underwent whole exome sequencing (WES) from 2015 to 2019 were retrospectively reviewed. Finally, the clinical data of these 7 patients with DEPDC5 variants were collected in this study. We analyzed their clinical manifestations, EEG and magnetic resonance imaging (MRI).
Results
Seven DEPDC5 variants, including six novel mutations, were identified in seven individuals with focal epilepsy. Among these patients, one had family history. Four showed specific interictal EEG patterns, periodic-like sharp waves or spike waves, were found in four patients. Five out of seven patients (71.4%) were well-controlled by anti-epilepsy drugs while two patients with sleep-related hypermotor epilepsy had either drug resistance or relapse of epilepsy.
Conclusion
DEPDC5 variants were related to focal epilepsy in patients with or without family history. The EEG abnormalities of DEPDC5 related epilepsy were heterogeneous among different patients, while periodic-like sharp waves or spike waves might be the most characteristic interictal EEG pattern for DEPDC5 related epilepsies. In this study the prognosis of DEPDC5 related epilepsy was similar to other epilepsies. DEPDC5 variants may not predict the prognosis so far.
Similar content being viewed by others


Introduction
Epilepsy, as a chronic neurological disease caused by transient brain dysfunction, has become the second most common neurological disease. According to a recently research on global burden of epilepsy, there were 45.9 million patients with all-active epilepsy in 2016 [1]. In 2017, the International League Against Epilepsy (ILAE) updated the framework for classifying seizure types and epilepsy, and suggested that genetic factors may be the most important independent epileptic factors [2]. In recent years, with the concept of precision medicine and the development of next-generation sequencing technology, many epilepsy related genes have been localized and cloned.
DEPDC5 (Dishevelled, egl-10, and pleckstrin domain–containing protein 5), NPRL2 (Nitrogen permease regulator-like-2) and NPRL3 (Nitrogen permease regulator-like-3) together form the GATOR1 (GAP activity towards rags complex 1) complex. GATOR1 complex constitutes the amino acid-sensitive part of the mechanistic target of rapamycin (mTOR) pathway [3]. When cells stay in an environment of amino acids deprivation, this complex acts as a repressor of the mechanistic target of rapamycin complex 1 (mTORC1) pathway through interfering the localization of mTORC1 to the lysosome [3]. Mutations in DEPDC5, NPRL2 or NPRL3 may resulted in dysfunction of these proteins, which will cause over-activation of mTORC1. The over-activation of mTOR pathway in brain tissue will lead to altered neuronal morphology, enhanced cell size, and abnormal cortical lamination, resulting in focal cortical dysplasia (FCD) [4].
The relationship between genes encoding components of GATOR1 complex and epilepsy was firstly reported in familial focal epilepsies in 2013 [5]. From then on, heterozygous germline variants in the GATOR1-encoding genes have continuously been found to be associated with focal epilepsy syndromes, such as sleep-related hypermotor epilepsy (SHE) [6, 7], familial focal epilepsy with variable foci (FFEVF) [5, 8] and familial temporal lobe epilepsy (FTLE) [8]. Mutations in NPRL2 and NPRL3 causing focal epilepsy were first reported in 2015 [9, 10], which is later than DEPDC5 [5]. Recently, Baldassari etal reported 73 patients with GATOR1 variants including 63 patients carried mutations in DEPEC5, which is the largest cohort study ever reported [11]. However, in all of these studies, there was little report on the EEG feature of DEPDC5-related epilepsy. Hence, in this study, we investigate the clinical phenotype, genetic characteristics, the prognosis as well as the EEG features of epilepsy related to DEPDC5 variants, which may deepen the understanding of GATOR1-related epilepsy syndrome and lay the foundation of further researches.
Methods
Patients
The records of 546 epilepsy patients with unknown causes who underwent whole exome sequencing (WES) in the Department of Neurology, Xijing Hospital of Fourth Military Medical School from 2015 to 2019 were retrospectively reviewed. The present study is a monocenter observational study, developed in accordance with the STROBE [12] guidelines for observational studies and those for epidemiological studies on epilepsy [13]. The study was approved by the local Ethical Committees. Due to the retrospective and noninterventional nature of the study, informed consent was not required.
Whole exome sequencing
Blood of the 546 epilepsy patients with unknown cause were collected and genomic DNA was extracted using the Blood Genome Column Medium Extraction Kit (Kangweishiji, China) according to the kit instructions. Liquid hybridization of the genomic DNA was performed using Roche Nimble Gen Seq EZ Exome Enrichment Kit V2.0 and Seq EZ Exome Enrichment Kit V2.0 capture probes (Roche, USA), and the target DNA fragments were enriched to construct exome library covering 19 119 genes with whole exons and partial introns. Each enriched region shared 40 Mb of targeted sequences. High-throughput sequencing was performed by Illumina NovaSeq 6 000 series sequencer (PE150), and no less than 99% of target sequences were sequenced. The sequencing process was performed by the Zhiyin Oriental Translational Medicine Research Center. The processes of bioinformatics analysis are as follows: (1) Quality control: Raw data were cleaned after adapters being removed; low-quality reads filtering and other quality control protocols. (2) Variants calling: The clean data were aligned to the NCBI human reference genome (hg18) using BWA and variants were called using GATK. Samtools and Pindel were used to call single nucleotide polymorphisms (SNPs) and indels, respectively. The clean data were than filtered, according to the quality of the sequencing, for further protocol. (3) Variants annotation and prediction: Nonsynonymous substitutions and SNPs with minor allele frequency (MAF) lower than 1% were filtered using SIFT. The function of mutated genes and their pathogenicity were then analyzed referencing to dbSNP, 1 000 Genomes Project, ExAC, ESP, OMIM, Swiss-var, HGMD, ClinVar and other disease databases. (4) The single base variants of unknown pathogenicity were analyzed by Provean, SIFT, Polyphen2-HVAR, Polyphen2-HDIV, Mutationtster and other protein structure prediction software. MaxEntScan was used to screen potential splice sites. All of these variants underwent Sanger sequencing to confirm.
Long term video electroencephalogram (VEEG)
EEG signals were recorded with 21 scalp electrodes placed according to the 10–20 system. Data from several recording systems (Nihon Kohden, biologic, Nicolet) were reviewed in an average reference montage. In some patients, one or two bipolar electromyographic signals were recorded in parallel from major muscles activated during motor seizures. Patient behavior was captured with a simultaneous video recording. EEG signals were recorded with pairs of 9-mm diameter silver/silver chloride electrodes placed 2-cm apart over muscle bellies. We recorded deltoid and quadriceps muscles according to the site of the most common movement disorders. EEG signals were band-pass filtered at 53–120 Hz. The patients took the 24 h VEEG annually. Twelve patients took at least twice 24 h VEEG. In the nine patients left, eight patients took one 24 h VEEG while one patient took one time of 15 h VEEG.
Classification of the prognosis
The response to treatment was classified into four categories: seizure freedom (defined as no seizures for at least 1 year), effectiveness (seizure reduction 50–99%), no beneficial effect, or increase of seizure frequency. Treatment success was defined as achieving ≥ 50% reduction in seizure frequency or seizure freedom after receiving treatment. Relapse was defined as the reappearance of epilepsy after stopping taking drug for at least three years. Refractory epilepsy is defined as epilepsy in which seizures persist and seizure freedom is very unlikely to be attained with further manipulation of anti-epileptic drugs (AEDs) therapy [14].
Results
Genetic findings
In total, there were seven different DEPDC5 variants, which were considered pathogenic or likely pathogenic according to the American College of Medical Genetics and Genomics guideline [15], detected in seven patients, including four truncation variants, two splicing site variants and one frame-shift variant (Table 1). All these variants were validated by Sanger sequencing. In all these seven variants, six of them were novel variants and none of them can be located in the databases we chose. Splicing site variants, frame-shift variants and truncation variants were predicted by MutationTaster, MaxEntScan (data not shown). In the patients conducting WES-trios, there are no de novo mutations. All of these 7 variants were heterozygous variants. In addition, these variants spread across DEPDC5 protein and four of them located at the structural axis for binding arrangement (SABA) domain. The other three variants located at steric hindrance for enhancement of nucleotidase activity (SHEN) domain, C-terminal domain (CTD) and the Lasso B part between SHEN domain and Dishevelled, Egl-10 and Pleckstrin (DEP) domain (Fig. 1) [16].

Seven variants we found in the study. The variants in the red cycle indicates that it has been reported
Clinical findings
In this study, 7 patients with DEPDC5 pathogenic or likely pathogenic variants (disease duration, median 10 years, range 1–19 years) were selected from 546 patients who had underwent WES. The clinical data, including seizure type, EEG, neuroimaging characteristics, treatment and prognosis are listed in detail in Table 2 and Table 3. In these patients, only one patient was adult onset. The seizure onset age of the other six patients ranged from 1 to 12 (median 6.5) years, two thirds of them in childhood (4/6, 66.7%).
Seizure type
In these seven patients, all of them were diagnosed as focal epilepsy (Table 2). Among the seven patients, three of them only had this kind of seizure while the other four patients had different attack models. In patient 1 and 5, sleep-related focal hypermotor seizures were the most common type. Mostly, the epilepsy in patient 2 and 4 were presented as behavior arrest with impaired awareness. All of these patients mentioned above had focal seizures evolving into bilateral tonic-clonic seizures. In patient 3, only focal to bilateral tonic-clonic seizures were observed. In total, focal to bilateral tonic-clonic seizures were reported in six patients (6/7, 85.7%). Besides, focal clonic seizures without impaired awareness were observed in patient 6.
Neuroimaging and video electroencephalogram
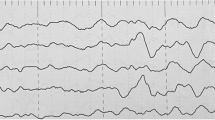
Brain magnetic resonance imaging (MRI) were available in all of these seven patients. Among them, four patients had normal MRI while the other three had different abnormalities. In patient 3, left hippocampus sclerosis was discovered. Bilateral paraventricular white matter demyelination was spotted. And in patient 6, FCD was found in the left paraventricular area. As for patient 1, besides MRI, she underwent positron emission tomography-computed tomography (PET-CT). On her PET-CT, a locus with lower metabolism was located in gyrus temporalis medius of left temporal lobe. All of these seven patients underwent long-term VEEG annually. At the last check, seizures were captured in two patients (patient 4 and 5). In patient 4, three seizures were recorded in his VEEG and concentrate in light sleep period. All of these seizures rise from the left temporal leads and spread to all leads and the patient started to develop tonic seizure lasting for 30 s with tachycardia. In patient 5, ten seizures were recorded in his VEEG. The moment paroxysmal rapid waves appeared in his right frontal leads, he started to developed hypermotor seizures which presented as pedaling, screaming and unconsciousness. As for the interictal EEG, four patients, including patient 1, 3, 4 and 7, had the same EEG pattern. Their interictal EEG were presented as periodic spike waves and sharp waves in their frontal leads. Three of them were diagnosed as frontal lobe epilepsy (FLE) while the other was diagnosed as temporal lobe epilepsy (TLE). As for patient 6, she underwent several EEGs. However, all of them showed normal EEG. Considering her clinical symptoms and the FCD on her MRI, she was diagnosed as unspecified focal epilepsy. All of the meaningful EEG pictures were presented in Fig. 3, while all of their EEG can be seen in the supplementary materials.
Familiy history
In these seven patients, one patient had certain family history. The genealogic tree for this patient was presented in Fig. 2. In Family a (proband: patient 3), the other affected individual (II-1) had the similar clinical characteristic with the proband and got seizure free after receiving AEDs. II-1 also carries the same variants found in the proband. Besides, the proband’s mother also carries the variant but doesn’t have any clinical symptoms.

a-d Genealogic tree of patient 3, 5, 6 and 7. The solid squares indicate patients with epilepsy while the hollow ones indicate healthy people. The letter “m” indicates the carrier of variants while “+” indicates wild type

EEG pictures for all patients. The first number represents the code of patient. While EEG of patient 6 was normal, it was not shown
Treatment and prognosis
In these seven patients, five patients responded well to AEDs, especially the therapeutic regimen based on levetiracetam. Three patients got seizure free after receiving AEDs (patient 2, 3 and 4). The seizure frequency of the other two patients also decreased for more than 90%. However, there was one refractory case (patient 1) who had already received five AEDs but still had seizure attacks for about 10 times per month. As for patient 5, he had been well controlled with sodium valproate and withdrawn all his medication at 2015. However, in 2018, his epilepsy relapsed. Until last follow-up, he still had seizures for about seven times per night, mainly as hypermotor seizures.
Discussion
The mTORC1 pathway is a central regulator of cell growth [17]. Nutrients, such as amino acids, signal to mTORC1 through the Rag GTPase heterodimer (RAGA or RAGB bound to RAGC or RAGD) [18]. DEPDC5 together with NPRL2 and NPRL3 forms GATOR1 complex regulating the activity of Rag GTPase heterodimer by acting as GTPase acticvating protein (GAP). In the situation of amino acid deprivation, GATOR1 complex will interfere the localization of mTOR complex 1 to the lysosomal surface, which prevents the activation of mTOR pathway [3]. According to the structure model presented by Shen et al. these three subunits, together with the Rag GTPase heterodimer, have complex interactions between each other and the intactness of GATOR1 is needed for its GAP function [16]. When there are mutations in any of these three subunits, the interactions are interfered and the intactness of GATOR1 is affected. The loss-of-function (LoF) of GATOR1 results in the over-activation of mTOR pathway. In brain, this over-activation may lead to FCD and epilepsy [4].
In our study, the age at epilepsy onset ranged from 1 to 20 years (median 10.5 years), mostly in their childhood (5/7, 71.4%), which was later than the group reported by Baldassari et al. recently [11]. We found seven different variants in DEPDC5. Four variants, including two truncation variants, one frame-shift variants and one splicing site variants, affects the SABA domain. This domain is in charge of the interaction between DEPDC5 and NPRL2 [16]. One truncation variant, c.2935G > T, affects SHEN domain. This domain plays an important role in the interaction between DEPDC5 and RAGA [16]. In the two variants left, one truncation variant (c.3214G > T) affects Lasso B part between SHEN and DEP domain while the other splicing site variant (c.4437–2(IVS41)_c.4437–1(IVS41)insG) affects CTD domain. These two variants may affect the intactness of DEPDC5, which also brings negative affect on the function of GATOR1. All in all, all these variants were thought to be related to patients’ symptoms.
In our study, there was one patient with family history. In this family, there was a patient (II-1) with the same variant and similar symptoms compared with the proband. However, mother of the proband who carried the same variant didn’t have any symptoms, which indicates incomplete penetrance of DEPDC5 variant. According to Baldassari et al. variants inherited from asymptomatic parents in 64% of the cases [11]. So, there is still a great need for future basic research to definitively settle on their pathogenicity.
In the most recent article on epilepsy-related GATOR1 variants, Baldassari et al. reported 63 patients with DEPDC5 variants in 73 patients. In their research, focal seizures were described in most of these patients (60/63, 95.2%), including SHE, FLE, TLE and unspecified focal seizure and the most common phenotype is SHE (22/63, 34.9%) [11]. Drug resistance in the series of Baldassari et al. was observed in half of the probands, and up to 65% in probands with SHE, supporting previous studies based on smaller cohorts of families with SHE (7/9 drug-resistant individuals) [7]. In our study, two patients were diagnosed as SHE (patient 1 and 5, 2/7, 28.6%) and one of them was diagnosed as refractory epilepsy while the other patient was experiencing relapse of his epilepsy. These findings are in concordance with the conclusion made by Baldassari et al. [11]. As for the other seizure types, things were different. All of these patients responded well to the AEDs (71.4%) and three of them had already been seizure free for at least a year. So, the drug resistance is much smaller than Baldassari et al. had reported (54% for GATOR1 variants, 52.4% for DEPDC5 variants) [11]. This difference in drug resistance may related to the rather small sample size but it can also suggest that GATOR1 related epilepsy may be not as refractory as it was reported [11]. In these five patients with positive respond, four of them were given therapy based on LEV, which may indicate LEV as a preferential choice for patients with DEPDC5 variants. Considering the fact that the loss-of-function variants in DEPDC5 will lead to over-activation of the mTOR pathway, the mTOR inhibitor, such as sirolimus or everolimus, may be a complementary treatment for DEDPC5 related epilepsy. However, this assumption needs further confirmation.
In the previous studies, there was little information on the EEG characteristics of epilepsy induced by DEPDC5 variants. In 2018, Hu et al. created an animal model with a focal region with somatic DEPDC5 deletion [19]. Via this model, the authors found that the EEG features were highly clinically relevant to FCD IIA, which included low-voltage fast activity (LVFA) and and bursts of rhythmic spikes [20, 21]. In our study, the periodic spike waves or sharp waves were found in four patients (Patient 1, 3, 4 and 7), which may indicate that bursts of rhythmic spikes might be a specific EEG pattern for patients with DEPDC5 related epilepsy. Interestingly, the MRI of these patients were all normal. While patient 6, who had an abnormal MRI with deep-in FCD, had a normal EEG. As all of these patients underwent scalp-EEG and 1.5 T MRI, this situation may be related to the limited accuracy of the instrument.
Conclusion
In summary, here we reported seven patients with pathogenic or likely pathogenic DEPDC5 variants. Among these patients, only one patient had family history, which may indicate incomplete penetrance. The drug resistant epilepsy is mainly concentrated in patients with SHE, but the rate of drug resistance of DEPDC5 related epilepsy may not as high as we expected. However, considering the rather small sample size and the lack of basic research on the pathogenic molecular mechanism of the variants we found, there are still much to do in order to get a further understanding of the pathogenesis of DEPDC5 related epilepsy.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CTD:
-
C-terminal domain
- DEPDC5:
-
Dishevelled, egl-10, and pleckstrin domain–containing protein 5
- FCD:
-
Focal cortical dysplasia
- FFEVF:
-
Familial focal epilepsy with variable foci
- FLE:
-
Frontal lobe epilepsy
- FTLE:
-
Familial temporal lobe epilepsy
- GATOR1:
-
GAP activity towards rags complex 1
- ILAE:
-
The International League Against Epilepsy
- MRI:
-
Magnetic resonance imaging
- mTOR:
-
The mechanistic target of rapamycin
- mTORC1:
-
The mechanistic target of rapamycin complex 1
- NPRL2:
-
Nitrogen permease regulator-like-2
- NPRL3:
-
Nitrogen permease regulator-like-3
- PET-CT:
-
Positron emission tomography-computed tomography
- SABA:
-
Structural axis for binding arrangement
- SHE:
-
Sleep-related hypermotor epilepsy
- TLE:
-
Temporal lobe epilepsy
- AEDs:
-
Anti-epileptic drugs
- VEEG:
-
Video electroencephalogram
- WES:
-
Whole exome sequencing
References
Beghi E, Giussani G, Nichols E, Abd-Allah F, Abdela J, Abdelalim A, et al. Global, regional, and national burden of epilepsy, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019;18(4):357–75.
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21.
Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. A Tumor Suppressor Complex with GAP Activity for the Rag GTPases That Signal Amino Acid Sufficiency to mTORC1. Sci (80- ). 2013;340(6136):1100–6.
Iffland PH, Baybis M, Barnes AE, Leventer RJ, Lockhart PJ, Crino PB. DEPDC5 and NPRL3 modulate cell size, filopodial outgrowth, and localization of mTOR in neural progenitor cells and neurons. Neurobiol Dis. 2018;114:184–93.
Dibbens LM, de Vries B, Donatello S, Heron SE, Hodgson BL, Chintawar S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. 2013;45(5):546–51.
Tinuper P, Bisulli F, Cross JH, Hesdorffer D, Kahane P, Nobili L, et al. Definition and diagnostic criteria of sleep-related hypermotor epilepsy. Neurol. 2016;86(19):1834–42.
Picard F, Makrythanasis P, Navarro V, Ishida S, de Bellescize J, Ville D, et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurol. 2014;82(23):2101–6.
Ishida S, Picard F, Rudolf G, Noé E, Achaz G, Thomas P, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet 2013;45(5):552–5.
Sim JC, Scerri T, Fanjul-Fernández M, Riseley JR, Gillies G, Pope K, et al. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann Neurol. 2016;79(1):132–7.
Ricos MG, Hodgson BL, Pippucci T, Saidin A, Ong YS, Heron SE, et al. Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol. 2016;79(1):120–31.
Baldassari S, Picard F, Verbeek NE, van Kempen M, Brilstra EH, Lesca G, et al. The landscape of epilepsy-related GATOR1 variants. Genet Med. 2019;21(2):398–408.
Vandenbroucke JP, von Elm E, Altman DG, Gøtzsche PC, Mulrow CD, Pocock SJ, et al. Strengthening the reporting of observational studies in epidemiology (STROBE): explanation and elaboration. Int J Surg. 2014;12(12):1500–24.
Guidelines for epidemiologic studies on epilepsy. Commission on Epidemiology and Prognosis, International League Against Epilepsy. Epilepsia. 1993;34(4):592–6.
Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. 2010;51(6):1069–77.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–23.
Shen K, Huang RK, Brignole EJ, Condon KJ, Valenstein ML, Chantranupong L, et al. Architecture of the human GATOR1 and GATOR1–rag GTPases complexes. Nat. 2018;556(7699):64–9.
Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nat. 2015;517(7534):302–10.
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–501.
Hu S, Knowlton RC, Watson BO, Glanowska KM, Murphy GG, Parent JM, et al. Somatic Depdc5 deletion recapitulates electroclinical features of human focal cortical dysplasia type IIA. Ann Neurol. 2018;84(1):140–6.
Gambardella A, Palmini A, Andermann F, Dubeau F, Da Costa JC, Quesney LF, et al. Usefulness of focal rhythmic discharges on scalp EEG of patients with focal cortical dysplasia and intractable epilepsy. Electroencephalogr Clin Neurophysiol. 1996;98(4):243–9.
Chassoux F, Landré E, Mellerio C, Turak B, Mann MW, Daumas-Duport C, et al. Type II focal cortical dysplasia: electroclinical phenotype and surgical outcome related to imaging. Epilepsia. 2012;53(2):349–58.
Acknowledgements
Funding
This study was funded by National Key R&D Program of China, Precision medicine program——Cohort study on nervous system diseases under Grant2017YFC0907700 (2017–2021).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Due to the retrospective and noninterventional nature of the study, informed consent was not required. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this article is consistent with those guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wei, Z., Wang, L., Qiao, X. et al. The clinical phenotypes and genetic characteristics of seven epilepsy patients related to heterozygous DEPDC5 variants in China. Acta Epileptologica 2, 2 (2020). https://doi.org/10.1186/s42494-020-0011-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42494-020-0011-9