
Abstract
Background
Carcinoids at atypical sites are rare and mostly asymptomatic. In the majority, they show normal levels of serotonin and its metabolites in plasma and urine, resulting in a relatively late diagnosis and hence guarded prognosis. The relatively large size and repeated local recurrences are features of carcinoids at atypical locations and may direct the attention to its possible existence.
Patients and methods
A report of six cases of carcinoid tumors in the breast, abdominal paravertebral region, duodenum, ovary, and stomach is given. Blood serotonin, plasma, and urine 5-hydroxyindole acetic acid were determined for cases suspected to have carcinoid syndrome. Those presented with a mass were radiologically studied by CT or MRI, and if a biopsy was taken (or the mass removed), keratin, CEA, and chromogranin for argentaffin reaction were studied. Follow-up was for 15 months, and a second surgery was done for the two cases (1 and 5).
Results
Except for case 4, all patients were females. The symptoms and clinical findings in the six cases were vague and did not raise the possibility of a carcinoid. Repeated local recurrence was the clue for the diagnosis. The enrolled six cases had no common features that may point or suggest the presence of neither carcinoid tumor nor syndrome. Also, there was no relation between the site and size of the tumor and the detection of the manifestations of carcinoid syndrome. Additionally, in one case (case 1), the originally non-functioning lesion turned active when recurrence happened.
Conclusion
Carcinoid tumors at atypical sites are low-grade malignant neoplasms with a good long-term prognosis if correctly managed. Routine aggressive resection or even debulking is the treatment of choice whenever possible, even in recurrent cases. The survival of patients depends basically on the tumor size and/or the presence of distant metastases. Treatment by somatostatin analogy is usually mandatory if carcinoid syndrome is present.
Similar content being viewed by others

Introduction
Rare (0.15%) (Robertson et al. 2006), slowly growing with a high potential for local recurrences and metastasis, carcinoid tumors are of neuroendocrine origin (NETs). They are derived from primitive stem cells deep in the mucosa of the gut wall, lungs, mediastinum, thymus or liver, and even ovaries, kidneys, and Michel’s diverticulum (Broaddus et al. 2003). These cells are essential for the development of organs and the complex regulation of tissues (Guadagno et al. 2016). This may explain the diversity of symptoms and the different types of clinical presentations. Some of them present with distant metastases (22%), half of which have unknown primaries and produce a significant amount of beta-catenin, which enables tumor cell adhesion (Greenblatt et al. 2007). Also, they may be associated with carcinoid syndrome particularly if situated in the distal midgut in adults (Hlatky et al. 2004; Volpe et al. 2000). This occurs if the tumor cells secrete various vasoactive peptide hormones like 5-hydroxytryptamine (5-HT), kinins, substance P (SP), prostaglandins, and others. They may be argentaffin negative or positive. The etiology of carcinoid tumors is not known, but genetic abnormalities are suspected. Reported chromosomal abnormalities include changes in chromosomes, such as loss of heterogeneity, and numerical imbalances (Dreijerink et al. 2000; Hemminki and Li 2001). Tumor appearance takes an age-specific incidence with two peaks at 15–25 and 65–75 years. The overall 5-year survival rate for all carcinoid tumors—regardless of site or stage—ranges from 70 to 80%. Survival has improved over the last decades due to the introduction of treatment by octreotide (Sandostatin) particularly in metastatic disease (Modlin et al. 2003). Histologically, they have five patterns: solid, trabecular, glandular, tubular, poorly differentiated, or mixed. Immunohistochemically, these tumors have a strong positive reaction to keratin and other neuroendocrine markers. These include chromogranin, and synaptophysin (Rindi et al. 2006). A common finding is elastosis and fibrosis that surround the nests of the tumor cells and result in matting of the involved tissues and lymph nodes. They also have at least five somatostatin receptors frequently used for diagnosing the disease by octreotide-receptor scintigraphy and also in its treatment. Tumors have occurred in association with other familial or genetic disorders, such as multiple endocrine neoplasia type1 (MEN1) and Peutz-Jeghers syndrome (Igaz 2009). The sites most often affected by carcinoid tumors are the gastrointestinal tract (65%) followed by the bronchopulmonary tract (25%), but in about 10% of cases, the primary tumor site remains unknown. A substantial number of carcinoid tumors originate in less common anatomical sites and can range from indolent, unrecognized entities to highly active, metastatic secretory tumors. Their presentation within unfamiliar locations often results in clinical confusion. Examples are those of the esophagus, pancreas, liver, biliary tract, gallbladder, and Meckel’s diverticulum, as well as within the pelvic and otolaryngeal organs and in the breast. Organ distribution of carcinoid cases exhibited the most frequent site to be the respiratory system (19.8%), followed by the rectum (15.0%), jejunoileum (12.0%), stomach (11.4%), appendix (9.6%), and duodenum (8.3%). An extremely small number of cases (less than 0.7%) were found in the middle ear, testicle, kidney, and several other organs. The highest rate of metastases was noted in the ileocecum (75.3%), followed by the jejunoileum (65.2%), pancreas (64.2%), and finally the larynx (61.4%) (Soga 2003).
Patients and methods
Six cases (five females and one male) were enrolled in the study from February 2016 to January 2017. The youngest was 33 and the oldest was 61 years. Routine laboratory investigations were done to the six patients; in addition, whole blood serotonin together with plasma and urine 5-hydroxyindole acetic acid was made for the cases suspected to have carcinoid syndrome (Kulke and Mayer 1999). Those presented with a mass were radiologically studied by CT or MRI, and if a biopsy was taken, keratin, CEA, and chromogranin for argentaffin reaction were studied. The same was applied to any removed masses, primary or revisional. Follow-up was for 15 months, and when recurrence took place, all investigations were repeated. Re-surgery was done for the two cases (1 and 5).
Results
-
a.
Availability of data and materials
Data supporting the present findings can be found in the surgical patients’ archives at the Cairo University Hospitals. Both authors have no limitations in sharing data with others.
-
b.
Case introduction
The six cases studied have no common features that may point to or suggest the presence or absence of a carcinoid tumor or its syndrome. Except case 4, all patients were females. Also, there was no relation between the site and size of the tumor and the detection of the manifestations of carcinoid syndrome. Additionally, in one case (case 1), the originally non-functioning lesion turned active when recurrence happens.
-
c.
Case details (Table 1)
-
1.
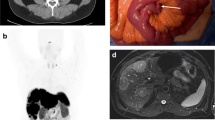
Case 1: A female, 35 years old, presented with a mass in the upper outer quadrant of her left breast (Fig. 1). The mass was excised and pathologically proved to be a carcinoma with endocrine differentiation. Breast conservation surgery was performed followed by both radio and chemotherapy. Two months later, unfortunately, the mass recurred at the same site, implicating the previous scar and necessitating aggressive local resection, and was proved to be a malignant carcinoid
-
2.
Case 2: A female, 66 years old, presented because of weight loss, attacks of fainting, and melena. Gastroscopy revealed a 2 × 1-cm malignant-looking ulcer on the lesser curve; a biopsy was taken and chromogranin staining proved its carcinoid nature. Open wedge excision was done. She received chemotherapy and did not develop carcinoid symptoms up to 11 months.
-
3.
Case 3: A female, 44 years old, presented with left sided abdominal pain and diarrhea. CT revealed a paravertebral mass close to the left suprarenal gland. At the operation, an 8 × 10-cm bosselated mass was found separate from the gland and related to the left paraaortic region. It was totally removed and histologically was paraganglionoma. Three months later, the patient developed recurrent severe diarrhea with face flushing and asthma-like attacks. X-ray showed a fracture involving the left sixth rib. Abdominal MRI revealed local recurrence at the site of the previous surgery. The involved segment of the rib was excised and proved to be a metastasis from a primary carcinoid tumor. Elevated 5-HIAA in urine (threefold the normal) confirmed the presence of carcinoid syndrome (Kulke and Mayer 1999). The recurrent mass was re-excised, and Sandostatin analog was given. Her symptoms disappeared.
-
4.
Case 4: A Sudanese female, 61 years old, presented with lower abdominal pain of 1-month duration referred to the left lower limb. MRI revealed an oblong mass 16 × 3 cm in the left sacral paravertebral region. Only tumor debulking was possible. Histologically, it was paraganglionoma and histochemically proved to be malignant trabecular carcinoid. She received a full course of radiotherapy. The patient left to her country and was not amenable for follow up.
-
5.
Case 5: A male, 45 years old, presented with postprandial epigastric and lumbar pains with moderate diarrhea and infrequent bouts of dark stools of 7 months’ duration. MRI revealed an oblong mass in the transverse colon close to the splenic flexure with coloduodenal fistula. Extended right hemicolectomy combined with duodenal repair was performed, and the tumor (12 cm) was histologically an adenocarcinoma. Symptoms improved for 1 month and then recurred again. Exploration detected an irresectable highly vascular ulcerating mass involving the third part of the duodenum surrounded by extensive local fibrosis. Through a duodenotomy, wedge biopsy was taken that confirmed its carcinoid nature. He received chemotherapy and died after 3 months.
-
6.
Case 6: A female, 33 years old, presented with a large (18 cm) partially cystic mass involving the left ovary necessitating ovariectomy. Histologically, it was a trabecular carcinoid with no malignant features. Follow-up for 6 months detected no recurrence.

Case 1, recurrent left breast carcinoid: a young lady with a mass in the upper outer quadrant of left breast pathologically diagnosed as carcinoma with endocrine differentiation and treated by surgery and adjuvant chemoradiotherapy. The mass recurred at the same site implicating the previous scar necessitating aggressive local resection. It proved to be a malignant carcinoid
Discussion
Though sometimes aggressive, carcinoids are relatively slow-growing tumors, and even in the presence of metastatic disease, patients can survive for several years. Unless the characteristic peptides are directly poured into the systemic circulation, they remain asymptomatic causing delayed diagnosis (Carling et al. 2002). In three cases of this report (cases 1, 3, and 4), the diagnosis was made only by chance when local recurrence developed combined with diarrhea and flushing. Repeated local recurrence is probably a feature of neuroendocrinal tumors including carcinoids. It is judicious to consider them as a possibility that should exist and thought of in most of recurrent benign or malignant tumors present at abnormal sites, particularly in the abdomen. Familiarity with such unusual sites—particularly if diarrhea is troublesome—will facilitate appropriate recognition allowing for timely intervention (King et al. 1985). High urinary 5-HIAA values—as a part of the systemic study in such unusual cases—was the clue to correct diagnosis in the present report and in the work of others (Kulke and Mayer 1999). This elevation may not be present from the start, or may develop only after recurrence, if it happens. It may be suggested that the secretary ability of such extra GI tumors can be an acquired phenomenon that may develop at any time in its course or may be linked to recurrence. On the contrary, patients with classical gut carcinoma may present with the unique symptoms of the carcinoid syndrome right from the start, particularly if liver metastases are present (Givi et al. 2006). Fortunately, those secretory tumors respond well to somatostatin and its analogs (Kunz et al. 2013).
Surgery is the best treatment for carcinoid tumors in general, and in simulation with bronchial carcinoids, we may argue in favor of conservative resection which may be repeated (Caplin et al. 2015). This local resection may be very aggressive and may include operations like segmental colon resection, hemicolectomy, low anterior or abdominoperineal resections of the rectum, or even liver resection and tubo-ovariectomy (Metwally et al. 2016). In such situations, open surgery is preferred to laparoscopic one as it gives better exploration and easier handling. Evaluation of the patient’s heart condition is mandatory in all cases as heart disease commonly complicates carcinoid syndrome and may interfere with safe general anesthesia (Mehta et al. 1999). The prognosis for any treated carcinoid patient with progressive or recurrent disease is poor. Deciding on further treatment depends on many factors, including prior treatment, site of recurrence, and individual patient considerations. Attempts at re-resecting slow-growing tumors are worthy of consideration, since a successful further reduction of tumor volume may provide long-term palliation.
Carcinoid tumors of the breast are rare, usually mistaken for the much more common classical ductal carcinoma, and are treated in the same way including chemo and radiotherapies with fair response (Fine et al. 2014) (Fig. 1). Repeated local recurrences in spite of previous successful management may direct the attention to the possible presence of carcinoid. Also, one should be attentive of the existence—though rare—of breast tumor that is metastases from a carcinoid tumor at a distant site and may be confused with a primary one. This may pose a diagnostic challenge as it can closely mimic conventional breast cancer (Geyer et al. 2010). Careful attention to clinical features and the use of auxiliary immunohistochemical studies can help in achieving a correct diagnosis. Somatostatin receptor scintigraphy with 111Indium-labeled pentetreotide is useful in confirming the neuroendocrine mass and excluding distant metastases (Saint-Mar et al. 2004). Paraganglionmata in general are extremely rare and usually affect middle-aged males, with the thorax as the preferred site. They are asymptomatic except if they secrete neuropeptides particularly one of the catecholamines causing hypertension (Richter et al. 2011). Distinctions between these tumors and carcinoids are difficult and are possible only by monitoring the difference in histology particularly with specific staining and also by their biologic behavior (Googe et al. 1988). The two cases reported in this study were not typical, as they affected old females and the tumors were intraabdominal in the left paravertebral regions, and one of them is close to the suprarenal gland. Their diagnosis as carcinoid was only possible by immunostaining, and one of them demonstrated carcinoid syndrome symptoms following aggressive local recurrence that necessitated local debulking and somatostatin therapy. Ovarian carcinoids are usually diagnosed as a histological surprise unless carcinoid syndrome symptoms are present. Surgery is the standard management with or without somatostatin analogs therapy if required.
Conclusion
Carcinoid tumors are described as cancers in slow motion, and though aggressive or recurrent forms are reported, the outlook for most patients is more hopeful than it used to be. The biggest impediment to making the diagnosis is not thinking of or even considering it because of its infrequency. Once thought of, the diagnosis usually can be confirmed easily by immunostaining or determining the level of urine 5-HIAA.
References
Broaddus RR, Herzog CE, Hicks MJ (2003) Neuroendocrine tumors (carcinoid and neuroendocrine carcinoma) presenting at extra-appendiceal sites in childhood and adolescence. Arch Pathol Lab Med 127(9):1200–1203
Caplin ME, Baudin E, Ferolla P et al (2015) Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann Oncol 26(8):1604–1620
Carling RS, Degg TJ, Allen KR et al (2002) Evaluation of whole blood serotonin and plasma and urine 5-hydroxyindole acetic acid in diagnosis of carcinoid disease. Ann Clin Biochem 39(Pt 6):577–582
Dreijerink KM, Roijers JF, van der Luijt RB et al (2000) Multiple endocrine neoplasia type 1: recent developments and guidelines for DNA diagnosis and periodic clinical monitoring. Ned Tijdschr Geneeskd 144(51):2445–2449
Fine RL, Gulati A, Tsushima D, et al. New, rationally-based chemotherapy combination appears highly effective in patients with treatment-resistant neuroendocrine tumors. Presented at: 2014 Symposium Highlights Treatment Advances for Multiple GI Cancers [press release]. Alexandria; 2014. https://www.asco.org/about-asco/press-center/news-releases/2014-symposium-highlights-treatment-advances-multiple-gi. Accessed Dec 2017.
Geyer HL, Viney J, Karlin N (2010) Metastatic carcinoid presenting as a breast lesion. Curr Oncol 17(6):73–77
Givi B, Pommier SJ, Thompson AK, Diggs BS, Pommier RF (2006) Operative resection of primary carcinoid neoplasms in patients with liver metastases yields significantly better survival. Surgery 140(6):891–897 discussion 897-8
Googe PB, Ferry JA, Bhan AK et al (1988) Comparison of paraganglioma, carcinoid tumor, and small-cell carcinoma of the larynx. Arch Pathol Lab Med. 112(8):809–815
Greenblatt DY, Kunnimalaiyaan M, Chen H (2007) Raf-1 activation in gastrointestinal carcinoid cells decreases tumor cell adhesion. Am J Surg 193(3):331–335 discussion 335
Guadagno E, De Rosa G, Del Basso De Caro M (2016) Neuroendocrine tumours in rare sites: differences in nomenclature and diagnostics—a rare and ubiquitous histotype. J Clin Pathol 69:563–574
Hemminki K, Li X (2001) Familial carcinoid tumors and subsequent cancers: a nation-wide epidemiologic study from Sweden. Int J Cancer 94(3):444–448
Hlatky R, Suki D, Sawaya R (2004) Carcinoid metastasis to the brain. Cancer. 101(11):2605–2613
Igaz P (2009) MEN1 clinical background. Adv Exp Med Biol 668:1–15
King MD, Young DG, Hann IM, Patrick WJ (1985) Carcinoid syndrome: an unusual cause of diarrhoea. Arch Dis Child 60(3):269–271
Kulke MH, Mayer RJ (1999) Carcinoid tumors. N Engl J Med 340(11):858–868
Kunz PL, Reidy-Lagunes D, Anthony LB et al (2013) Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 42(4):557–577
Mehta AC, Rafanan AL, Bulkley R et al (1999) Coronary spasm and cardiac arrest from carcinoid crisis during laser bronchoscopy. Chest 115(2):598–600
Metwally H, Elalfy A, Awny S et al (2016) Primary ovarian carcinoid: a report of two cases and a decade registry. J Egypt Natl Cancer Inst 28:267–275
Modlin IM, Lye KD, Kidd M (2003) A 5-decade analysis of 13,715 carcinoid tumors. Cancer 97:934–959
Richter A, Halm HF, Lerner T et al (2011) Long-term follow-up after en bloc resection and reconstruction of a solitary paraganglioma metastasis in the first lumbar vertebral body: a case report. J Med Case Rep 5:45
Rindi G, Klöppel G, Alhman H et al (2006) European Neuroendocrine Tumor Society (ENETS). TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch 449:33-37
Robertson RG, Geiger WJ, Davis NB (2006) Carcinoid tumors. Am Fam Physician 74(3):429–434
Saint-Mar O, Cogliandolo A, Pozzo A et al (2004) A primary pancreatic carcinoid tumour with unusual clinical complaints: a case report. World J Surg Oncol 2:3
Soga J (2003) J Exp Clin Cancer Res 22(4):517–530
Volpe A, Willert J, Ihnken K et al (2000) Metastatic appendiceal carcinoid tumor in a child. Med Pediatr Oncol 34(3):218–220
Acknowledgement
Authors are grateful to Professor A. Hendawi (Department of Pathology, Faculty of Medicine Cairo University) for pathologically assessing the removed specimens and biopsy materials including immunohistochemistry.
Funding
The study was neither done under grant nor funding.
Availability of data and materials
datasets that were generated and analysed during the current study and supporting its findings can be found in the surgical patients’ archives at the Cairo University Hospitals, Egypt.
Both authors have no limitations in sharing data with others.
Author information
Authors and Affiliations
Contributions
GMS suggested the study, collected the cases, participated in surgical management, and prepared the manuscript for publication. KGM was responsible for the data collection and data analysis, participation in surgical management, and also for the linguistic revision of the manuscript. KGM is the correspondent author. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Study was approved by the Ethical Committee, Department of General Surgery – Faculty of Medicine, Cairo University, Egypt. The approval included the authorization of the 6 patients to endorse the study and to make their data available for future work or publication.
Consent for publication
Both authors arranged to upload the article for publication to the Bulletin of the National Research Centre. They also have got a consent from all patients included in the study for same item. (All were adults).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Moustafa, K.G., Saied, G.M. Primary carcinoid tumors at atypical sites: surgery is mandatory and size determines survival—an Egyptian study. Bull Natl Res Cent 43, 36 (2019). https://doi.org/10.1186/s42269-019-0075-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-019-0075-0