
Abstract
Background
Hormesis is an adaptive response of a biological system to low dose of stressors. It exerts beneficial effects to enable the cells and organisms to sustain the unfavourable conditions. Mild heat stress is one of the widely studied hormetic agents having antiageing and lifespan prolonging effects. In order to study the effect of mild heat stress on the primary culture of mouse prefrontal cerebrocortical neurons with reference to ageing-associated degenerative alterations the present investigations were carried out.
Study design
The prefrontal cerebrocortical neurons of E17 day mouse embryo were cultured on poly-l-lysine coated coverslips and fed with neurobasal medium supplemented with B-27 at 37 °C and 5% CO2. The neurons were divided into two groups: control group and mild heat stress group. The neurons from the control group were incubated at 37 °C without any heat stress. The neurons from the mild heat stress group were subjected to hyperthermic stress of 38 °C for 30 min on 2nd, 4th and 6th day of seeding.
Methods
On the 3rd, 5th and 7th day of incubation, viability was studied by calcein-AM and propidium iodide assay and cell death assay was carried out by lactate dehydrogenase (LDH) assay. The surviving neurons were enumerated on 10th, 15th and 20th day of incubation by live cell imaging. All microscopic studies were carried out by observer blinding.
Results
It was observed that there was higher percentage of viable neurons and lower percentage of degenerating neurons in the mild heat stress group than the control. The difference was highly significant (p < 0.001).
Conclusion
Mild heat stress (38 °C for 30 min on every alternate day up to 6 days of incubation) exerts hormetic effects on the primary culture of mouse prefrontal cerebrocortical neurons by delaying the degenerative alterations.
Similar content being viewed by others


Background
Hormesis is an adaptive response of the biological system to cope up with transient unfavourable conditions (Murry et al., 1986). During the past two decades, the number of publications in the field of hormesis in ageing and longevity is increased exponentially (Calabrese & Baldwin, 2002; Arumugam et al., 2006; Ji et al., 2010; Calabrese et al., 2016a) that the concept of hormesis may be used as an alternative therapy in near future. Temperature stress exerts deleterious effects on the cellular metabolism. However, mild heat stress exerts beneficial effects (Rattan 1998). Calabrese (2016b, c) demonstrated that short term exposure to heat stress results in beneficial effects, whereas, long term exposure causes deleterious effects. When the cells are subjected to heat stress, they respond by expressing heat shock proteins (HSPs): one of the evolutionarily conserved intracellular defence mechanisms. HSPs act as molecular chaperones that prevent misfolding of proteins (Whitley et al., 1999; Sørensen et al., 2003; Banerjee Mustafi et al., 2009). The present investigation addresses the effect of mild heat stress on the primary culture of mouse prefrontal cerebrocortical neurons with reference to their viability and age-associated cytological alterations in the morphology of neurons undergoing ageing in vitro.
Material and methods
In the present investigations, E17 embryos (17th day gestational age) of Swiss albino mouse Mus musculus were used. All experimental procedures were carried out in accordance with the guidelines of CPCSEA (Committee for the Purpose of Control and Supervision of Experimental Animals, Ministry of Environment, Forest and Climate change, Government of India) and the protocol was approved by the Institutional Animal Ethics Committee (1825/PO/EReBi/S/15/CPCSEA).
Primary culture of mouse prefrontal cerebrocortical neurons
All the procedures were carried out in aseptic conditions in laminar airflow. The prefrontal cerebral cortices were dissected from mouse E17 embryos under a stereoscopic dissecting microscope in chilled PBS (pH 7.4) containing 40 μg/ml gentamycin. The pieces of tissue were minced in Ca-Mg free Hank’s Balanced Salt Solution (Himedia, TL1108) containing 40 μg/ml gentamycin. The minced content was incubated with 0.25% trypsin for 10 min at 37 °C. Trypsin activity was terminated by 1X soybean trypsin inhibitor (Himedia, TCL068) at 37 °C for 5 min. The neurons were dissociated from trypsinized prefrontal cerebral cortices by trituration with fire-polished Pasteur pipette. After viability testing by trypan blue dye exclusion method, 50,000 live cells per coverslip were seeded on poly-l-lysine coated 18 mm glass coverslips. The cells were incubated at 37 °C and 5% CO2 for 30 min in the CO2 incubator (New Brunswick an Eppendorf company, Galaxy 48R, 41823) for adhesion. Four coverslips were placed in a 60 mm tissue culture dish. The cells were fed with neurobasal medium (Gibco, 15630-106) supplemented with 0.2% B-27 (Gibco, 17504-044), 40 μg/ml Gentamycin (Abbott), 25 mM HEPES (Himedia, TL1108) and 1% Glutamax (Gibco, 35050-061). The culture was maintained at 37 °C and 5% CO2 in the CO2 incubator. Two-third of the old culture medium was replaced with fresh medium on every third day.
Mild heat stress (MHS) treatment
The neurons were divided into two groups, a control group and the mild heat stress (MHS) group. The control group neurons were incubated at 37 °C without any heat stress. The neurons from the experimental group were subjected to hyperthermic stress of 38 °C for 30 min on the 2nd, 4th and 6th day of seeding the cells. The pilot studies demonstrated that the cerebrocortical prefrontal neurons were able to tolerate the hyperthermic stress of 38 °C for 30 min on every alternate day. However, exposure to higher temperatures (38.5, 39, 39.5 and 40 °C) resulted in cell death. Similarly, exposure to 38 °C for 60 min on every day and every alternate day also caused the death of the neurons. The prefrontal cerebrocortical neurons survived in vitro up to three weeks. In the pilot studies, exposure to MHS was carried out in the first, second and third week. The beneficial effects of MHS were seen when it was applied in the first week. Therefore, for the present study, mild heat stress of 38 °C for 30 min at an interval of 48 h, (i.e., every alternate day) was finalised.
Study of viability by calcein–AM and propidium iodide fluorescence-based method
The percent viability from the control group and the mild heat stress group was carried out on the 3rd, 5th and 7th day of incubation by calcein–AM and propidium iodide fluorescence method described by Giordano et al. (2011). The neurons were treated with calcein–AM and propidium iodide stain, incubated at 37 °C and 5% CO2 for 15 min and washed three times with PBS, observed under fluorescence microscope at 495/515 nm excitation and emission wavelength.
Intracellular esterase of live cells converts the non-fluorescent calcein-AM (acetoxymethyl ester) to the highly fluorescent calcein, which is retained within the live cells and produces an intense green fluorescence. The plasma membrane of live cell is impermeable to DNA-binding agent propidium iodide which only enters dead cells. Thus, cells emitting green fluorescence are live and cells emitting orange fluorescence are dead.
Study of cell death by measurement of lactate dehydrogenase (LDH) in the culture medium
Lactate dehydrogenase (LDH) is a cytoplasmic enzyme. Plasma membranes of live cells do not allow the release of this enzyme in the culture medium. However, the enzyme is leaked out of the dead cells. Therefore, the measurement of LDH content in the culture medium is a biochemical tool for quantifying the number of dead cells (Decker and Lohmann-Matthes 1988). The standard graph of LDH content in the culture medium vs the number of dead cells was plotted. The graph was linear. On the 3rd, 5th and 7th day of incubation, 1 ml of culture medium from experimental and control group were transferred to respective test tubes. 1 ml of 2X LDH assay buffer (containing iodonitrotetrazolium chloride, phenazine methosulfate, NAD+, lactic acid) was added, mixed well and incubated at 25 °C for 30 min in a complete dark condition. After incubation, 1 ml stop solution (1 M acetic acid) was added and the absorbance was measured at 520 nm on spectrophotometer against blank. The number of dead neurons from both the groups was determined using the standard graph.
Study of in vitro survival capacity of neurons
In vitro survival capacity of neurons from control and MHS group was studied by live cell imaging. Dead neurons show loss of typical neuronal morphology and exhibit apoptotic bodies. The neurons having typical neuronal morphology, i.e., cyton, dendrites, axon hillock and intact axon were counted on the 10th, 15th and 20th day of culture in both the groups.
Imaging and analysis
The results of the viability and morphological changes were captured by live cell imaging (ProgRes®, Jenoptik). The whole area of the culture was captured in different snapshots. Analysis was carried out by observer blinding.
Observer blinding
Before live cell imaging and viability testing, the culture plates from the control group and the experimental group were coded. The confidentiality of the code was maintained. The images were named as per the code. The counting of live and dead cells was carried out using these images.
Statistical analysis
The investigations were carried out in six trials. Each trial was performed in triplicate sets. The results were calculated as arithmetic mean ± standard deviation of the observations of six trials. The level of significance between the two groups was estimated by ‘unpaired t test’.
Results
The results of the viability study by calcein-AM and propidium iodide are shown in Figs. 1 and 2a. On the 3rd day, in the control group 74.97 ± 0.99% neurons were viable, while in the MHS group 79.40 ± 1.25% neurons were viable (Fig. 1a and b respectively). On the 5th day, in the control group, 53.86 ± 1.94% neurons were viable, while in the MHS group this number was 69.45 ± 1.20% (Fig. 1c and d respectively). On the 7th day, in the control group, the viability was 36.75 ± 1.58%, whereas, in the MHS group it was 60.17 ± 1.81% (Fig. 1e and f respectively). The viability in the MHS group was higher than the control. The difference was highly significant (p < 0.001).

Fluorescence micrograph of the primary cultured mouse prefrontal cerebrocortical neurons stained by Calcein-AM and propidium iodide. Green fluorescence indicates functional esterase activity in the live cells, and orange fluorescence indicates the staining of nuclei in dead cells by propidium iodide. Note that a higher number of viable neurons (green fluorescence) in the mild heat stress group and a greater number of dead neurons (orange fluorescence) in the control group

Effect of mild heat stress on the primary culture of mouse prefrontal cerebrocortical neurons with reference to a: percent viability (stained by calcein–AM and propidium iodide); b: cell death (measured by release of LDH in the culture medium by LDH assay) and c: maintenance of neuronal morphology. Values are arithmetic mean ± SD (n = 6), *** indicates that the difference is highly significant (p < 0.001)
The results of the LDH assay are shown in Fig. 2b. The death of neurons in both groups was directly proportional to the days of incubation. However, neurons subjected to MHS exhibited significantly lower mortality (p < 0.001). In the control group, on the 3rd day of incubation 26.98 + 2.34%, neurons were dead, whereas, in experimental group it was 20.8 + 1.6%. On the 5th day, control group showed 48.85 + 2.07% dead neurons and in the MHS group they were 29.98 ± 1.18%. On the 7th day of in vitro life, in the control group 68.54 ± 3.1%, neurons were dead, whereas, in the MHS group it was 41.56 ± 5.08%.
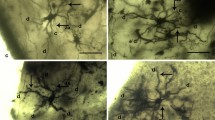
The number of neurons showing intact and typical neuronal morphology on the 10th, 15th and 20th days of culture is shown in Fig. 2c. In the control group, such neurons were significantly lower than the MHS group (p < 0.001). In the control group, on the 10th day of incubation, culture medium was fully occupied by suspended apoptotic bodies (Fig. 3a). Such apoptotic bodies were scanty in the culture medium of MHS group (Fig. 3d). The neurodegenerative alterations like nuclear fragmentation and condensation, cytoplasmic vacuolation, axonal and dendritic degeneration and formation of apoptotic bodies were common in both groups but more prevalent in control.

Effect of mild heat stress on the morphological alterations in the primary culture of mouse prefrontal cerebrocortical neurons undergoing senescence in vitro. Fragmented nuclei (fn), apoptotic body (ab), suspended apoptotic body (sab), vacuolated cyton (vc), dendrites (d), cyton (c), nucleus (n), axon (a), axonal degeneration (ad), degenerating cyton (dc)
Discussion
In the present study, the viability of the neurons in the MHS group was significantly higher as compared to the control group. In vitro neuronal death in early periods of incubation is due to damage caused by cell dissociation and abrupt shift from in vivo to in vitro condition. Those neurons which sustained in vitro condition grew and developed synapses. In the present investigations, the results of the microscopic study and LDH assay exhibited significantly higher number of viable neurons in the MHS group than the control (Fig. 2). This might be due to the stimulation of stress-responsive adaptive pathways on exposure to mild heat stress. This is in accordance with Rattan et al. (2004) who demonstrated that repeated MHS has antiageing hormetic effects on human fibroblasts undergoing ageing in vitro. Fonager et al. (2002) demonstrated that cultured human fibroblasts subjected to repeated mild heat-shock exhibited a significant increase in the levels of heat shock proteins: Hsp27 and Hsp70 throughout their replicative lifespan that led to delayed ageing. Primary culture of cortical progenitor cells of rat E14.5 embryos when exposed to 38.5 °C for 4 days exhibited an increase in viability, neurosphere diameter and expression of heat shock proteins (Hossain et al., 2017).
The degenerative alternations in the nucleus such as nuclear fragmentation and condensation seen in the phase contrast microscopic studies may be due to the decline in DNA repair and onset of apoptosis, thereby activation of intranucleosomal endonuclease activity (Famulski et al., 1999; Lombard et al., 2005; Gorbunova et al., 2007). The cytoplasmic vacuoles seen in degenerating neurons appear as a response to change in the cellular environment. Ohkuma and Poole (1981) demonstrated that the accumulation of amyloid-beta (Aβ) peptides and increased concentration of lipophilic compounds in the lysosome caused cytoplasmic vacuolation in mouse peritoneal macrophages. Gotz et al. (2006) and Wirths et al. (2006) demonstrated that axonal and dendritic degeneration in the neurons is due to increased accumulation of Aβ peptides and neurofibrillary tangles (NFTs). In our earlier studies, it was found that neurons from MHS group had a lower accumulation of NFTs and Aβ peptides (Mane et al., 2018). In the present investigations, the MHS group exhibited significantly higher number of neurons with typical neuronal morphology having intact dendrites, axon hillock and axon. This indicates that the MHS slows down the process of neurodegeneration. Rattan (1998) demonstrated the maintenance of young morphology of fibroblasts in repeated heat shock cultures of human fibroblasts.
Hyperthermia is practised since time immemorial to treat pain and inflammation by locally applying the heating pads at the site of pain. Our study did not intend to apply hyperthermic stress to the human brain. However, our observations suggest that mild heat stress strengthens the survival capacity of neurons in vitro and slows down the neurodegenerative alterations.
Conclusion
Mild heat stress (exposure to 38 °C for 30 min on 2nd, 4th and 6th day of culture) exerts beneficial effects to slow down the process of neurodegeneration in the primary culture of mouse prefrontal cerebrocortical neurons.
Availability of data and materials
All data are available upon request.
Abbreviations
- LDH:
-
Lactate dehydrogenase
- HSP:
-
Heat shock protein
- HEPES:
-
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
References
Arumugam, T. V., Gleichmann, M., Tang, S. C., & Mattson, M. P. (2006). Hormesis/preconditioning mechanisms, the nervous system and aging. Ageing research reviews, 5(2), 165–178.
Banerjee Mustafi, S., Chakraborty, P. K., Dey, R. S., & Raha, S. (2009). Heat stress upregulates chaperone heat shock protein 70 and antioxidant manganese superoxide dismutase through reactive oxygen species (ROS), p38MAPK, and Akt. Cell Stress Chaperones, 14, 579–589.
Calabrese, E. J., Dhawan, G., Kapoor, R., Iavicoli, I., & Calabrese, V. (2016a). HORMESIS: a fundamental concept with widespread biological and biomedical applications. Gerontology, 62(5), 530–535.
Calabrese, E. J. (2016b). Preconditioning is hormesis part II: How the conditioning dose mediates protection: Dose optimization within temporal and mechanistic frameworks. Pharmacol. Res., 110, 265–275.
Calabrese, E. J. (2016c). Preconditioning is hormesis part I: Documentation, dose-response features and mechanistic foundations. Pharmacol. Res., 110, 242–264.
Calabrese, E. J., & Baldwin, L. A. (2002). Defining hormesis. Hum. Exp. Toxicol., 21, 91–97.
Decker, T., & Lohmann-Matthes, M. L. (1988). A quick and simple method for the quantitation of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J. Immunol. Methods, 115, 61–69.
Famulski, K. S., Macdonald, D., Paterson, M. C., & Sikora, E. (1999). Activation of a low pH-dependent nuclease by apoptotic agents. Cell Death Differ., 6, 281–289.
Fonager, J., Beedholm, R., Clark, B. F., & Rattan, S. I. (2002). Mild stress-induced stimulation of heat-shock protein synthesis and improved functional ability of human fibroblasts undergoing aging in vitro. Exp. Gerontol., 37, 1223–1228.
Giordano, G., Hong, S., Faustman, E.M., & Costa, L.G. (2011) Measurements of cell death in neuronal and glial cells. In Methods in Molecular Biology (Clifton, N.J.). pp. 171–178.
Gorbunova, V., Seluanov, A., Mao, Z., & Hine, C. (2007). Changes in DNA repair during aging. Nucleic Acids Res., 35, 7466–7474.
Gotz, J., Ittner, L. M., & Kins, S. (2006). Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer’s disease? J. Neurochem., 98, 993–1006.
Hossain, M. E., Matsuzaki, K., Katakura, M., Sugimoto, N., Al Mamun, A., Islam, R., Shido, O. (2017). Direct exposure to mild heat promotes proliferation and neuronal differentiation of neural stem/progenitor cells in vitro. PLoS One, 12, e0190356.
Ji, L. L., Dickman, J. R., Kang, C., & Koenig, R. (2010). Exercise-induced hormesis may help healthy aging. Dose-Response, 8(1), 73–79.
Lombard, D. B., Chua, K. F., Mostoslavsky, R., Franco, S., Gostissa, M., & Alt, F. W. (2005). DNA repair, genome stability, and aging. Cell, 120, 497–512.
Mane, N. R., Gajare, K. A., & Deshmukh, A. A. (2018). Mild heat stress induces hormetic effects in protecting the primary culture of mouse prefrontal cerebrocortical neurons from neuropathological alterations. IBRO reports, 5, 110–115.
Murry, C. E., Jennings, R. B., & Reimer, K. A. (1986). Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation, 74(5), 1124–1136.
Ohkuma, S., & Poole, B. (1981). Cytoplasmic vacuolation of mouse peritoneal macrophages and the uptake into lysosomes of weakly basic substances. J. Cell Biol., 90, 656–664.
Rattan, S. (1998). Repeated mild heat shock delays ageing in cultured human skin fibroblasts. IUBMB Life, 45, 753–759.
Rattan, S. I. S., Eskildsen-Helmond, Y. E. G., & Beedholm, R. (2004). Molecular mechanisms of anti-aging hormetic effects of mild heat stress on human cells. Nonlinearity Biol. Toxicol. Med., 2, 154014204904643.
Sørensen, J. G., Kristensen, T. N., & Loeschcke, V. (2003). The evolutionary and ecological role of heat shock proteins. Ecol. Lett., 6, 1025–1037.
Whitley, D., Goldberg, S. P., & Jordan, W. D. (1999). Heat shock proteins: A review of the molecular chaperones. J. Vasc. Surg., 29, 748–751.
Wirths, O., Weis, J., Szczygielski, J., Multhaup, G., & Bayer, T. A. (2006). Axonopathy in an APP/PS1 transgenic mouse model of Alzheimer’s disease. Acta Neuropathol., 111, 312–319.
Acknowledgements
The authors acknowledge the Department of Science and Technology, New Delhi for the financial assistance under DST PURSE phase II.
Funding
The purchase of consumables was supported by DST PURSE phase II program.
Author information
Authors and Affiliations
Contributions
AAD and KAG were responsible for the idea, the design of the study, writing, and revising the manuscript. AAD trained NRM to carry out the experimental procedures. AAD coded the cultures for observer blinding. NRM performed the experiments, live cell imaging and the statistical analysis. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocol of the present study was approved by the Institutional Animal Ethics Committee. The experimental protocol was carried out according to the guidelines of CPCSEA (Committe for the Purpose of Control and Supervision of Experimental Animals, Ministry of Environment, Forest and Climate change, Government of India).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mane, N.R., Gajare, K.A. & Deshmukh, A.A. Hormetic effects of mild heat stress on the primary culture of mouse prefrontal cerebrocortical neurons. JoBAZ 81, 25 (2020). https://doi.org/10.1186/s41936-020-00158-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41936-020-00158-y