
Abstract
Background
Precise serotyping of pneumococci is essential for vaccine development, to better understand the pathogenicity and trends of drug resistance. Currently used conventional and molecular methods of serotyping are expensive and time-consuming, with limited coverage of serotypes. An accurate and rapid serotyping method with complete coverage of serotypes is an urgent necessity. This study describes the development and application of a novel technology that addresses this need.
Methods
Polymerase chain reaction (PCR) was performed, targeting 1061 bp cpsB region, and the amplicon was subjected to sequencing. The sequence data was analyzed using the National Centre for Biotechnology Information database. For homologous strains, a second round of PCR, sequencing, and data analysis was performed targeting 10 group-specific genes located in the capsular polysaccharide region. Ninety-one pneumococcal reference strains were analyzed with PCRSeqTyping and compared with Quellung reaction using Pneumotest Kit (SSI, Denmark).
Results
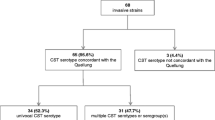
A 100% correlation of PCRSeqTyping results was observed with Pneumotest results. Fifty-nine reference strains were uniquely identified in the first step of PCRSeqTyping. The remaining 32 homologous strains out of 91 were also uniquely identified in the second step.
Conclusion
This study describes a PCRSeqTyping assay that is accurate and rapid, with high reproducibility. This assay is amenable for clinical testing and does not require culturing of the samples. It is a significant improvement over other methods because it covers all pneumococcal serotypes, and it has the potential for use in diagnostic laboratories and surveillance studies.
Similar content being viewed by others


Background
Streptococcus pneumoniae, found in the upper respiratory tract of healthy children and adults, causes a range of infections including meningitis, septicemia, pneumonia, sinusitis, and otitis media. Children < 2 years of age and adults aged ≥65 years of age are particularly susceptible [1]. According to the Morbidity and Mortality Weekly Report, April 26 2013 [2], an estimated 14.5 million cases of serious pneumococcal disease (including pneumonia, meningitis, and sepsis) occur each year in children aged <5 years worldwide, which has resulted in approximately 500,000 deaths, mostly in low- and middle-income developing countries.
The high morbidity and mortality caused by pneumococci are not clearly understood. The pathogenicity of pneumococci has been linked to various virulence factors such as capsule, cell wall and its component polysaccharides, pneumolysin, PspA, complement factor H-binding component, autolysin, neuraminidase, peptide permeases, hydrogen peroxide, and IgA1 protease [3,4,5]. Capsular polysaccharide (CPS) is the primary virulence factor, and is also used to categorize, S. pneumoniaeinto more than 90 different serotypes [6,7,8]. Capsule is important for the survival of bacteria at infection site as it provides resistance to phagocytosis [9].
Pneumococcal CPS is generally synthesized by the Wzx/Wzy-dependent pathway, except for types 3 and 37, which are produced by the synthase pathway [10, 11]. Most genes required for synthesis of capsule are within the capsule polysaccharide synthesis (cps) operon, which ranges from 10 kb (serotype 3) to 30 kb (serotype 38). Cps operon is flanked by dexB in 5′ end and aliA at 3′ end. Neither of these participates in capsule synthesis. The 5′-end of the CPS loci starts with regulatory and processing genes wzg, wzh, wzd, and wze (also known as cpsABCD), which are conserved with high sequence identity in all serotypes, followed by the central region consisting of serotype specific genes [12, 13].
Pneumococcal serotyping is necessary for epidemiological and vaccine impact studies. It also aids in understanding the pathogenicity of the organism and closely monitors for the emergence of non-vaccine strains, replacement serotypes, and new serovars [14, 15]. Widespread use of pneumococcal vaccines has led to replacement with serotypes that are not included in the vaccines. Continuous monitoring of serotypes is therefore essential for epidemiological surveillance and long-term vaccine impact studies [16,17,18,19,20].
Several phenotypic and genotypic methods are currently used to identify pneumococcal group and type. The phenotypic serotyping methods of capsular swelling reaction, latex agglutination and coagglutination tests are costly, require skilled personnel, and cannot detect all serotypes. Genotypic typing methods that assess genome variation include sequential multiplex polymerase chain reaction (PCR), sequential real-time PCR, restriction fragment length polymorphism (RFLP), microarray, sequetyping, and matrix-assisted lazer desorption ionization-time of flight (MALDI-TOF) analysis. In addition to general applicability and a high discriminatory power, these genotypic assays are economical, detect pneumococci directly from the clinical specimen, and detect emerging serovars, replacement strains, and vaccine escape recombinants [21]. However, many of these methods are multistep, intricate, and do not discriminate all serotypes [22,23,24,25,26].
It is crucial to develop a robust, simple method with complete serotype coverage for serotype detection and pneumococcal serogroup/serotype surveillance [27]. Herein, the authors describe an innovative serotyping approach that relies on sequencing of assembly genes located in the capsular operon to identify all pneumococcal serotypes.
Methods
Reference strains
There were 91 reference serotype strains of S. pneumoniae obtained from Staten Serum Institute, Copenhagen, Denmark (Table 1).
Clinical isolates
There were 28 clinical isolates of S. pneumoniae selected from isolates submitted to Central Research Laboratory, KIMS Hospital, Bangalore (Table 2). They were isolated from blood (n = 23), cerebrospinal fluid (CSF) (n =3) and pleural fluid (n = 2).
Media and culture conditions
Strains were stored in skim milk, tryptone, glucose, and glycerol (STGG) media at −80 °C. They were cultured on 5% sheep blood agar (Chromogen, Hyderabad) for 18–24 hrs at37°C with 5% CO2. The isolates were characterized as S. pneumoniae by colony morphology, alpha hemolysis, bile solubility, and optochin susceptibility.
Serotyping
Quellung reaction was performed using Pneumotest kit and type-specific antisera (SSI, Denmark), as recommended by the manufacturer.
PCRSeqTyping
PCRSeqTyping assay was performed in two steps. Step I involved PCR amplification and sequencing of the cpsB gene from genomic DNA. There were 91 serotypes that were divided into non-homologous group (Group I, 59 serotypes) and homologous group (Group II, 32 serotypes) based on the cpsB sequence data. The homologous group was further subdivided into 10 subgroups based on the sequence homology. The second step involved PCR and sequencing of each homology group by using specific primers in order to identify the unique serotypes.
Nucleic acid extraction
Genomic DNA was extracted from bacterial strains using QIAamp DNA mini kit (Qiagen, Germany), as per the manufacturer’s protocol.
PCR amplification
PCR reaction was performed using the primers designed by Leung et al. [26] with modifications. Primers used in the study were cps1-FP (5′-GCAATGCCAGACAGTAACCTCTAT-3)′, cps2-RP (5′-CCTGCCTGCAAGTCTTGATT-3′) and cps-2538-RP (5′-CTTTACCAACCTTTGTAATCCAT-3′). The reaction mixture was modified to contain 50–100 ng of genomic DNA, 0.75 units XT-5 polymerase (3 U/μl, Merck, which is a mixture of thermo stable enzymes Taq DNA polymerase and proof-reading [PR] polymerase), 1X XT5A-Assay buffer, 1 μl deoxynucleoside triphosphates (dNTPs, 2.5 mM each [Fermentas, United States]), 1 μl forward primer (100 ng/μl), 1 μl of reverse primer mix (100 ng/μl). The final reaction volume was made up to 25 μl with DNase/RNase-free distilled water (Gibco, United States). Thermal cycling was performed in GeneAmp PCR system 9700 (Applied Biosystems, United States) under the following conditions: 94 °C for 5 min, followed by 35 amplification cycles of 94 °C for 30 s, 50 °C for 30 s, and 72 °C for 1 min and final extension at 72 °C for 5 min. The PCR products were separated by electrophoresis on 1.2% agarose gel for 45 min at 80 V in 1X Tris-acetate EDTA buffer. Ethidium bromide-stained DNA products were visualized under ultraviolet (UV) illumination and size of the DNA products was determined by using a 1–kb DNA molecular size marker (Fermentas).
Sequencing and data analysis
PCR products were purified using QIA quick PCR purification kit (Qiagen, Germany) following manufacturer’s protocol. Purified PCR products were subjected to sequencing, employing the Big Dye Sequence Terminator kit V3.1 (Applied Biosystems) and analyzed on ABI 3730 XL Genetic Analyzer (Applied Biosystems). Sequencing was performed in one direction using forward primer (cps1), 5′-GCA ATG CCA GAC AGT AAC CTC TAT-3′ and Long Seq Module (ABI). DNA sequences that were obtained were analyzed for sequence similarity using GenBank database (http://www.ncbi.nlm.nih.gov/blast) and then assigned to serotype [26]. Serotype of the cpsB nucleotide sequence was determined from GenBank with the highest BLAST bit score of > 99% sequence identity with the query ‘amplicon nucleotide sequence’.
Homology group assignment and PCRSeqTyping
Homology groups
Amplifiable serotypes that shared identical interceding sequences (e.g. sequences for serotypes 2 and 41A, 7B, and 40) were grouped into 10 different groups based on their homology by in silico analysis of cpsB region. Individual primer sets were designed for each subgroup. Sequetyping data obtained in Step I was used to assign the homologous strains into subgroups (Fig. 1). Serotypes were considered homologous when the highest bit score was shared between two or more serotypes (i.e. the same amount of nucleotide variation between query and database sequences), and then assigned to one of the 10 groups (Table 3).

Homology group assignment for 91 pneumococcal serotypes
For homologous strains, a second round of PCR was performed using group specific primers as specified in Table 3. PCR products were subjected to sequencing reaction. The nucleotide sequence data was used to assign the serotype.
Results
PCRSeqTyping results for reference strains
The 91 pneumococcal serotype reference strains (sourced from SSI) were tested with PCRSeqTyping protocol. All 91 strains were amplified using the modified method. In Step I of amplification and sequencing, 59 strains of the non-homologous group (Group I) were correctly assigned to their respective serotype. There were 32 strains (Group II) identified along with their homologous type. The homologous types were correctly assigned to their respective type in Step II by performing a second round of amplification using group specific primers and sequencing. Quellung reaction performed using Pneumotest kit (SSI), in parallel with PCRSeqTyping, showed 100% concordant results (Table 1).
The results were further evaluated by blinded testing of PCRSeqtyping. Samples were evaluated randomly by assigning codes. Quellung reaction data showed no discrepancies between serotypes assigned by Quellung and PCRSeqTyping for all reference strains.
PCRSeqTyping results for clinical isolates
Twenty eight pneumococcal isolates tested in the study were from children <5 years with invasive pneumococcal disease. The predominant serotypes were 1, 6B, 19A, 19 F, 14 and 7 F (Table 2). PCRSeqTyping results and serotyping results by Quellung reaction were in concordance, without any discrepancies. Among 28 isolates, 25 isolates were assigned to their serotype with the first step of PCRSeqTyping. Three isolates belonging to the homologous group were subsequently identified with the second step of PCRSeqTyping.
Discussion
There is a renewed interest in pneumococcal capsular typing techniques, as a result of an increased complexity in the management of pneumococcal disease and the widespread use of pneumococcal vaccines [8]. The ability to differentiate pneumococcal strains efficiently is essential to track the emerging serovars, and for epidemiological investigations. The limitations of the Quellung serotyping method, many DNA-based typing protocols, PCR, restriction fragment length polymorphisms, hybridization assays, microarrays and sequencing for S. pneumoniae are well known. Different PCR strategies, namely multiplex PCR, sequential PCR, serotype-specific PCR, and real time multiplex PCR [25, 28–36] targeting serotype-specific regions of cps could detect only 22 serotypes uniquely, and 48 serotypes along with their homologous types [37, 38]. Despite the fact these methods cover imited serotypes, PCR is a widely used technique, which avoids the use of serological reagents and requires specific expertise to conduct.
Methods using multiple restriction enzymes and long cps fragments [39, 40] for PCR make the amplification difficult and inconsistent. Another protocol based on sequencing of regulatory region of cps [30, 31] shows poor resolution with cross reactivity of serotypes. An approach targeting serotype-specific glycosyl transferase genes [6] was only tested for serogroup 6 and serotype 19 F. The cross reactivity of serotypes, along with the requirement for a higher number of primers, and poor resolution limits their wide usage.
With the characterization of the cps locus of 92 serotypes [13], Leung et al. [26] developed sequetyping protocol using single primer pair, which binds in all pneumococcal serotypes. Recently, several research groups [27, 41,42,43] have published their results using sequetyping assay. Limitations of the sequetyping protocol were as follows: (i) only 84 serotypes out of 92 were predicted to be amplified by in silico analysis; (ii) cross-reacting serotypes (30/84) belonging to homologous groups could not be uniquely identified; and (iii) considering the central 732 bp region of the cpsB amplicon which could be sequenced, only 46 of 54 serotypes could be sequetyped.
In the first step of this study’s modified approach, successful amplification of all 91 serotypes was achieved with the addition of a new reverse primer to amplify 25A, 25 F and 38 serotypes specifically. Additionally, XT-5 polymerase used in the PCR amplification reactions contains Taq DNA polymerase and Pfu enzyme. This enzyme blend utilizes the powerful 5′-3′ polymerase activity of Taq DNA polymerase and the 3′-5′ exonuclease-mediated proof-reading activity of PR polymerase, resulting in high fidelity PCR products [44]. PCR annealing temperature of 50 °C and extension time of 1 min were found to be optimal for amplification of cpsB gene of all 91 strains.
The serotypes were grouped into homologous (32) and non-homologous (59) based on cpsB sequence. Non-homologous types were identified uniquely. The 32 homologous strains were further subdivided into 10 groups (HG 1–10) based on their sequence similarity. Homology group-specific primers were designed and evaluated for their ability to differentiate between strains. HG primers were designed to be able to assign the serotype accurately with second step of PCR and sequencing.
The limitation of using 732 bp region of cpsB amplicon in sequetyping assay, resulting in prediction of 46 of 54 serotypes, was overcome with the use of Long Seq module. Approximately 1.0 kb quality reads in a single sequencing reaction were obtained with modification. This resulted in providing good quality reads up to the end of the PCR template, identifying cross-reacting serotypes (15B/15C, 7 F/7A, 18B/18C, 9 L/9 N, 15B/C, 17 F/33C, 18B/C, 7A/F, 12A/46, 6C/6D) which have a single SNP in the cpsB region.
A 100% concordance of serotype results of PCRSeqTyping and Quellung testing was seen for the 28 clinical isolates. Moving forward, the study will be extended for serotyping a larger number of clinical isolates and clinical samples. The limitation of the protocol will be in quantification and serotype identification in multiple carriage; however, studies are underway to address these issues. For multiple carriage, the PCR amplicon obtained in the first step will be subcloned into T/A cloning vector and the individual clones will be sequenced for assigning the specific serotype. As the corresponding cpsB gene sequence of the recently discovered serotypes 6E, 6 F, 6G, 6H, 11E, 20A, 20B and 23B1 [45,46,47] were unavailable at the time of the study design, they will be included in future studies.
In the study’s center, the typing cost with Pneumotest Kit (SSI, Denmark) was US$35/isolate, while PCRSeqTyping cost was US$10 for Group I (non-homologous strains) and US$15 for Group II (homologous strains). With the easy availability of outsourced sequencing services, the accurate and reliable PCRSeqTyping test can be adopted in a regular microbiology laboratory, even without the sequencing facility.
This modified typing method has several advantages over other reported methods. It involves techniques with a workflow that many microbiology laboratories can easily implement. The high throughput PCRSeqTyping method features good discriminatory power, reproducibility, and portability, making it suitable for epidemiological studies. The assay has the flexibility of incorporating additional primers for the characterization of emerging serotypes. An added advantage of this method is that raw data from experiments can be reanalyzed upon the addition of new entries to the serotyping database.
Conclusion
PCRSeqTyping assay is a cost-effective alternative to currently available phenotypic and molecular typing methods. The method is simple to perform, robust, and economical. It can identify all 91 serotypes specifically and uniquely.
Abbreviations
- Cps:
-
Capsular polysaccharide
- DNA:
-
DeoxyRibo Nucleic Acid
- EDTA:
-
Ethylenediaminetetraacetic acid
- MALDI-TOF:
-
Matrix Assisted Laser Desorption Ionization - Time of Flight
- PCR:
-
Polymerase chain reaction
- RFLP:
-
Restriction fragment length polymorphism
- SSI:
-
Staten Serum Institute
- STG:
-
Serotype/group
- STGG:
-
Skim milk, tryptone, glucose, and glycerol
References
Pneumococcal Disease. Atlanta: Centers for Disease control and prevention. Available from https://www.cdc.gov/pneumococcal/clinicians/streptococcus-pneumoniae.html. Accessed 23 Mar 2016.
Progress in Introduction of Pneumococcal Conjugate Vaccine — Worldwide, 2000–2012. Morb Mortal Wkly Rep. 2013: 62(16);308–311. Available from: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm6216a4.htm
Velasco EL, Verheul AFM, Verhoef J, Snippe H. Streptococcus pneumoniae: Virulence Factors, Pathogenesis, and Vaccines. Microbiol Rev. 1995;59:591–603.
Shenoy AT, Orihuela CJ. Anatomical site-specific contributions of pneumococcal virulence determinants. Pneumonia. 2016;8:7.
Normark BH, Tuomanen EI. The Pneumococcus: Epidemiology, Microbiology, and Pathogenesis. Cold Spring HarbPerspect Med. 2013;3:a010215.
Tomita Y, Okamoto A, Yagi T, Hasegawa Y, Ohta M. Capsulartype prediction by phylogenetic tree of glycosyltransferase gene polymorphism in Streptococcus pneumoniae. Open Access Bioinforma. 2011;3:67–73.
Kim JO, Weiser JN. Association of intrastrain phase variation in quantity of capsular polysaccharide and teichoic acid with the virulence of Streptococcus pneumonia. J Infect Dis. 1998;177:368–77.
Ashu EE, Jarju S, Dione M, Mackenzie G, Ikumapayi UN, Manjang A, et al. Population structure, epidemiology and antibiotic resistance patterns of Streptococcus pneumoniae serotype 5: prior to PCV-13 vaccine introduction in Eastern Gambia. BMC Infect Dis. 2016;16:33.
Elberse KE, van de Pol I, Witteveen S, van der Heide HG, Schot CS, van Dijk A, et al. Population structure of Invasive Streptococcus pneumoniae in the Netherlands in the Pre-vaccination era assessed by MLVA and capsular sequence typing. PLoS ONE. 2011;6:e20390.
Llull D, Munoz R, Lopez R, Garcia E. A single gene (tts) located outside the cap locus directs the formation of Streptococcus pneumoniae type 37 capsular polysaccharide. Type 37 pneumococci are natural, genetically binary strains. J Exp Med. 1999;190:241–51.
Cartee RT, Forsee WT, Jensen JW, Yother J. Expression of the Streptococcus pneumoniae type 3 synthase in Escherichia coli. Assembly of type 3 polysaccharide on a lipid primer. J Biol Chem. 2001;276:48831–9.
Varvio SL, Auranen K, Arjas E, Makela PH. Evolution of the Capsular Regulatory GenesinStreptococcus pneumonia. J Infect Dis. 2009;200:1144–51.
Bentley SD, Aanensen DM, Mavroidi A, Saunders D, Rabbinowitsch E, Collins M, et al. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2006;2:e31.
Vernet G, Saha S, Satzke C, Burgess DH, Alderson M, Maisonneuve JF, et al. Laboratory-based diagnosis of pneumococcal pneumonia: state of the art and unmet needs. ClinMicrobiol Infect. 2011;17 (Suppl 3):1–13.
Jauneikaite E, Jefferies JMC, VereChurton NW, Pin Lin RT, Hibberd ML, Clarke SC. Genetic diversity of Streptococcus pneumoniae causing meningitis and sepsis in Singapore during the first year of PCV7 implementation. Emerging Microbes and Infect. 2014;3:e39.
Hausdorff WP, Bryant J, Paradiso PR, Siber GR. Which pneumococcal serogroups cause the most invasive disease: implications for conjugate vaccine formulation and use, part I. Clin Infect Dis. 2000;30:100–21.
Johnson HL, Deloria-Knoll M, Levine OS, Stoszek SK, FreimanisHance L, Reithinger R, et al. Systematic evaluation of serotypes causing invasive pneumococcal disease among children under five: the pneumococcal global serotype project. PLoS Med. 2010;7:e1000348. doi:10.1371/journal.pmed.1000348.
Weinberger DM, Malley R, Lipsitch M. Serotype replacement in disease after pneumococcal vaccination. Lancet. 2011;378:1962–73.
Obaro SK, Adegbola RA, Banya WA, Greenwood BM. Carriage of pneumococci after pneumococcal vaccination. Lancet. 1998;348:271–2.
Habib M, Porter BD, Satzke C. Capsular Serotyping of Streptococcus pneumoniaeusing the Quellung Reaction. J Vis Exp. 2014;84:e51208.
Bonofiglio L, Gardella N, Mollerach M. In: Magdeldin S, editor. Application of Molecular Typing Methods to the Study of Medically Relevant Gram-Positive Cocci, Gel Electrophoresis - Advanced Techniques. 2012. ISBN 978-953-51-0457-5.
Gonzalez TB, Rivera-Olivero IA, Sisco MC, Spadola E, Herman PW, de Waard JH. PCR deduction of invasive and colonizing pneumococcal serotypes from Venezuela: a critical appraisal. J Infect DevCtries. 2014;8:469–73. doi:10.3855/jidc.3274.
Slinger R, Hyde L, Moldovan I, Chan F, Pernica M. Direct Streptococcus pneumoniae real-time PCR serotyping from pediatric parapneumoniceffusions. BMC Pediatr. 2014;14:189.
Raymond F, Boucher N, Allary R, Robitaille L, Lefebvre B, et al. Serotyping of Streptococcus pneumoniaeBased on Capsular Genes Polymorphisms. PLoS ONE. 2013;8:e76197. doi:10.1371/journal.pone.0076197.
Pai R, Gertz RE, Beall B. Sequential multiplex PCR approach for determining capsular serotypes of Streptococcus pneumoniae isolates. J Clin Microbiol. 2006;44:124–31.
Leung MH, Bryson K, Freystatter K, Pichon B, Edwards G, Charalambous BM, et al. Sequetyping: serotyping Streptococcuspneumoniae by a single PCR sequencing strategy. J Clin Microbiol. 2012;50:2419–27.
Jin P, Wu L, Oftadeh S, Kudinha T, Kong F, Zeng Q. Using a practical molecular capsular serotype prediction strategy to investigate Streptococcus pneumoniaeserotypedistribution and antimicrobial resistance in Chinese local hospitalized children. BMC Pediatr. 2016;16:53.
Lawrence ER, Griffiths DB, Martin SA, George RC, Hall LM. Evaluation of semiautomated multiplex PCR assay for determination of Streptococcus pneumoniae serotypes and serogroups. J Clin Microbiol. 2003;41:601–7.
Jiang SM, Wang L, Reeves PR. Molecular characterization of Streptococcus pneumoniae type 4, 6B, 8, and 18C capsular polysaccharide gene clusters. Infect Immun. 2001;69:1244–55.
Kong F, Gilbert GL. Using cpsA-cpsB sequence polymorphisms and serotype-/group-specific PCR to predict 51 Streptococcus pneumoniae capsular serotypes. J Med Microbiol. 2003;52:1047–58.
Kong F, Wang W, Tao J, Wang L, Wang Q, Sabananthan A, et al. A molecular-capsular-type prediction system for 90 Streptococcus pneumoniae serotypes using partial cpsA-cpsB sequencing and wzy- or wzx-specific PCR. J Med Microbiol. 2005;54:351–6.
Rubin LG, Rizvi A. PCR-based assays for detection of Streptococcus pneumoniae serotypes 3, 14, 19 F and 23 F in respiratory specimens. J Med Microbiol. 2004;53:595–602.
O’Halloran DM, Cafferkey MT. Multiplex PCR for identification of seven Streptococcus pneumoniae serotypes targeted by a 7-valent conjugate vaccine. J Clin Microbiol. 2005;43:3487–90.
Billal DS, Hotomi M, Tasnim S, Fujihara K, Yamanaka N. Evaluation of serotypes of Streptococcus pneumoniae isolated from otitis media patients by multiplex polymerase chain reaction. ORLJ Otorhinolaryngol Relat Spec. 2006;68:135–8.
Park IH, Park S, Hollingshead SK, Nahm MH. Genetic basis for the new pneumococcal serotype, 6C. Infect Immun. 2007;75:4482–9.
Brito DA, Ramirez M, de Lencastre H. Serotyping Streptococcus pneumoniaeby multiplex PCR. J Clin Microbiol. 2003;41:2378–84.
Singleton RJ, et al. Invasive pneumococcal disease caused by nonvaccine serotypes among Alaska native children with high levels of 7-valent pneumococcal conjugate vaccine coverage. JAMA. 2007;297:1784–92.
Brugger SD, Frey P, Aebi S, Hinds J, Muhlemann K. Multiple colonization with S. pneumoniaebefore and after introduction of the seven valent conjugated pneumococcal polysaccharide vaccine. PLoS One. 2010;5:e11638. doi:10.1371/journal.pone.0011638.
Lawrence ER, et al. Evaluation of serotype prediction by cpsA-cpsB gene polymorphism in Streptococcus pneumoniae. J Clin Microbiol. 2000;38:1319–23.
Batt SL, Charalambous BM, McHugh TD, Martin S, Gillespie SH. Novel PCR-restriction fragment length polymorphism method for determining serotypes or serogroups of Streptococcus pneumoniae isolates. J Clin Microbiol. 2005;43:2656–61.
Dube FS, van Mens SP, Robberts L, Wolter N, Nicol P, Mafofo J, et al. Comparison of a Real-Time Multiplex PCR and Sequetyping Assay for Pneumococcal Serotyping. PLoS ONE. 2015;10:e0137349.
Satzke C, Dunne EM, Porter BD, Klugman KP, Mulholland EK, et al. The PneuCarriage Project: A Multi-Centre Comparative Study to Identify the Best Serotyping Methods for Examining Pneumococcal Carriage in Vaccine Evaluation Studies. PLoS Med. 2015;12:e1001903.
Zhenzhen D, Erqing Z, Wei G, Kaihu Y, Sangjie Y, Yonghong Y. Analysis of serotype results of 94 Streptococcus pneumoniae isolates with partial cpsA-cpsB serotype prediction system. J Appl Clin Pediatr. 2015;12:934–7.
Cline J, Braman JC, Hogrefe HH. PCR fidelity of Pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996;24:3546–51.
Calix JJ, Porambo RJ, Brady AM, Larson TR, Yother J, Abeygunwardana C, Nahm MH. Biochemical, Genetic, and Serological Characterization of Two Capsule Subtypes among Streptococcus pneumoniae Serotype 20 Strains. J Biol Chem. 2012;287(33):27885–94.
Geno KA, Gilbert GL, Song JY, Skovsted IC, Klugman KP, Jones C, et al. Pneumococcal Capsules and Their Types: Past, Present, and Future. Clin Microbiol Rev. 2015;28:871–99.
Oliver MB, van der Linden MP, Küntzel SA, Saad JS, Nahm MH. Discovery of Streptococcus pneumoniae Serotype 6 Variants with Glycosyltransferases Synthesizing Two Differing Repeating Units. J Biol Chem. 2013;288(36):25976–85.
Acknowledgements
Not applicable.
Funding
No funding agencies involved.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Authors’ contributions
GN – concept, designing the experiment, executing, data analysis and writing the manuscript. RKL – Guided the experimentation process and execution, reviewed the manuscript. FG and VG assisted during experimentation. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nagaraj, G., Ganaie, F., Govindan, V. et al. Development of PCRSeqTyping—a novel molecular assay for typing of Streptococcus pneumoniae . Pneumonia 9, 8 (2017). https://doi.org/10.1186/s41479-017-0032-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41479-017-0032-3