
Abstract
Background
Senior–Løken syndrome (SLS) and Bardet–Biedl syndrome (BBS) are ciliopathies. SLS is characterized by retinitis pigmentosa (RP) and familial nephronophthisis, leading to end-stage kidney disease, while BBS is characterized by six major symptoms: RP, polydactyly, obesity, genital abnormalities, learning difficulties, and renal defects. Ciliopathies have been diagnosed on a phenotypic basis, but diagnosis can now be established by genetic testing, using techniques such as next-generation sequencing. Here, we report a patient clinically diagnosed with SLS but diagnosed with BBS 13 years later using next-generation sequencing.
Case presentation
The patient was diagnosed with RP at the age of 6 years. She had some difficulty in social interactions and pre-obesity, but no polydactyly. At the age of 8 years, she was diagnosed with chronic kidney disease, anemia, and liver dysfunction. Kidney and liver biopsy revealed renal tubule cysts, tubule membrane disruption, and liver fibrosis. Therefore, SLS was diagnosed but no NPHP1 mutations were detected. Peritoneal dialysis was started at the age of 9 years, and she underwent kidney transplantation with a graft from her father at the age of 13 years. At the age of 21 years, she again underwent genetic testing for most of the mutations associated with ciliopathy. This revealed a homozygous frameshift mutation in intron 11 of SDCCAG8.
Conclusions
Mutations in SDCCAG8 are known to be causative of SLS and BBS without polydactyly. The fact that the patient had RP, some difficulty in social interactions, pre-obesity, juvenile nephronophthisis, liver fibrosis, bronchial asthma, and otitis media suggested BBS. However, some of these are not specific symptoms for BBS and she had few typical symptoms of BBS. Therefore, a definitive diagnosis of BBS was difficult without genetic analysis. Because many more patients with SDCCAG8 mutations have extrarenal manifestations like the current patient than standard nephronophthisis patients, careful monitoring of extrarenal manifestations is needed to improve patient management.
Similar content being viewed by others

Background
Senior–Løken syndrome (SLS) and Bardet–Biedl syndrome (BBS) are ciliopathies. SLS is characterized by retinitis pigmentosa (RP) and familial nephronophthisis (NPHP), which leads to end-stage kidney disease. BBS is characterized by six major symptoms: RP, polydactyly, obesity, genital abnormalities, learning difficulties, and renal defects. Although some variations in secondary symptoms have been reported, typical secondary symptoms of BBS are as follows: developmental delay, ataxia, poor coordination, speech defects, brachydactyly or syndactyly, dental defects, olfactory deficits, craniofacial dysmorphism, diabetes mellitus, congenital heart disease, hypertension, liver abnormalities, bronchial asthma, otitis, and rhinitis. A patient with more than four major symptoms, or three major symptoms with more than two secondary symptoms, can be definitively diagnosed with BBS [1, 2]. Thus far, ciliopathies have been diagnosed on a phenotypic basis; however, diagnosis can now be established by genetic testing using techniques such as next-generation sequencing. Here, we report a patient clinically diagnosed with SLS but diagnosed with BBS 13 years later using next-generation sequencing.
Case presentation
The female patient was born at term and exhibited a normal birth weight with no apparent congenital malformation. Her parents were consanguineous cousins with no major health problems. From childhood, the patient became easily exhausted and suffered from bronchial asthma, chronic bronchitis, atopic dermatitis, and secretory otitis media, which required middle ear ventilation at the age of 5 years. Since then, she experienced poor eyesight in the evening and was diagnosed with RP at the age of 6 years. When she was 8 years old, her Rohrer index was 152 indicating pre-obesity, and she experienced right cervical lymphadenopathy and underwent a blood test because of her extremely poor general status. Her inflammatory response was very low; additional blood test results were as follows: urea nitrogen, 70.9 mg/dL; creatinine, 3.0 mg/dL; hemoglobin, 7.4 g/dL; aspartate transaminase, 137 IU/L; and alanine aminotransferase, 115 IU/L. Urinary analysis revealed a specific gravity of 1.010 and a urinary beta-2 microglobulin level of > 16,000 μg/L, together with microhematuria and microproteinuria; this indicated end-stage renal failure, anemia, and liver dysfunction. Kidney and liver biopsy procedures revealed renal tubular cysts, tubular membrane disruption, and liver fibrosis. Therefore, SLS was diagnosed, but no NPHP1 mutations were detected.
Peritoneal dialysis was begun at the age of 9 years, and she underwent kidney transplantation, with a graft from her father, at the age of 13 years. Before this, from the age of 12 years, she received growth hormone therapy because of her extremely short height (130.4 cm, − 3.5 SD). However, at the age of 13 years, luteinizing hormone-releasing hormone agonist therapy was started to increase growth. She exhibited no morphological abnormalities in her genital organs and menarche occurred at the age of 15 years; the development of secondary sex characteristics was rather slow but almost normal. No obvious developmental delay or learning disabilities were evident. Although her behavior was rather childish and caused some difficulties in appropriate social interactions, she learned Braille and obtained a national qualification as a massage practitioner. Hypothyroidism and mild hypertension had been evident since she was an elementary school student, but she did not have diabetes mellitus, central nervous system problems, or anosmia.
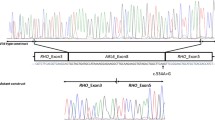
At the age of 21 years, we obtained written informed consent for genetic testing from the patient and her parents, and this was approved by the ethics board of Osaka Medical College (approval number 1867-01). She underwent genetic testing for most of the mutations associated with ciliopathy; these new tests revealed a homozygous mutation in intron 11 of SDCCAG8 (c.1357-9G>A), which caused splice site changes leading to a frameshift mutation (Fig. 1). The incidence rate of this mutation is extremely rare: 1/66,556 alleles in a genetic database of subjects in North America [3], and no alleles in a genetic database of subjects in Japan [4]. Because the SDCCAG8 mutation can be causative of both SLS and BBS [1, 2, 5,6,7,8], we noted her possibility of having BBS. Considering her RP, pre-obesity, difficulty in appropriate social interactions, juvenile NPHP, liver fibrosis, bronchial asthma, and otitis media, we diagnosed BBS. Her mother was initially confused by this new diagnosis, but eventually appreciated that the difficulties experienced in her daughter’s upbringing were partially caused by the phenotype of BBS.

Genetic testing. This patient has a missense mutation in intron 11, resulting in the addition of eight bases (shown in red font)
Discussion and conclusions
We experienced challenges in our attempt to diagnose a patient with BBS, largely because this is a very rare disease. Moreover, most of us are not experts in the diagnosis of BBS and we have not treated other patients with BBS before. The primary diagnostic criteria for BBS are phenotypic in nature, but blindness and end-stage kidney disease affect the interpretation of other diagnostic phenotypes. For example, blindness may affect a patient’s social characteristics and learning abilities, thereby making it difficult for them to read or befriend other children; moreover, kidney disease can lead to obesity in association with malnutrition, overeating and/or drinking, and a lack of exercise. Therefore, it is difficult to know whether these symptoms are caused by blindness, kidney disease, or overall manifestations of BBS. Moreover, some forms of symptomatic treatment may make phenotypes more difficult to clearly discern. For instance, when growth hormone therapy was administered to our patient, it may have interfered with obesity, while luteinizing hormone-releasing hormone agonist therapy could have made some genital abnormalities less prominent. Furthermore, BBS is a progressive disease, and most cases are diagnosed in older patients.
Mutations in SDCCAG8 (NPHP10/BBS16) are causative of both SLS and BBS without polydactyly. However, SDCCAG8 mutations are quite rare, with only 1–3% of BBS cases exhibiting a causative mutation in SDCCAG8 [1, 2, 5]. In this case, we first clinically diagnosed the patient’s disease as SLS [6,7,8]. However, after next-generation sequencing revealed a mutation in SDCCAG8, we reviewed her phenotype and finally diagnosed BBS. The childish character, difficulty in social interactions, and pre-obesity (body mass index, 27) the patient had suffered from since childhood are major symptoms of BBS in addition to RP and NPHP. Additionally, she displayed some secondary symptoms including liver fibrosis, bronchial asthma, and otitis media. However, these symptoms are not limited to BBS. Moreover, patients harboring a mutation in SDCCAG8 lack polydactyly which is a major symptom of BBS [5]. Thus, a differential diagnosis of BBS would not have been possible without genetic analysis.
Severe developmental delay or obesity is thought to be rare in cases of BBS with a SDCCAG8 mutation. Additionally, considering the absence of polydactyly and the patient’s age at the time of sight loss and the development of end-stage renal disease, her phenotype was compatible with this diagnosis [5, 7]. We suspect that many SLS patients harboring a mutation in SDCCAG8 may actually suffer from BBS, so are likely to remain misdiagnosed. Therefore, we urge clinicians to carefully monitor SLS patients to determine if BBS-associated complications emerge. Furthermore, as additional cases of SDCCAG8 mutations are reported, BBS professionals can stratify characteristics associated with SDCCAG8 mutation phenotypes; in contrast, BBS patients could be sub-classified based on mutations rather than on phenotypes.
In conclusion, we describe a Japanese female patient with ciliopathy harboring a mutation in SDCCAG8. At first, she was diagnosed with SLS, but subsequent genetic testing and detailed phenotypic observations strongly supported a re-diagnosis of BBS. Because only a few cases of SDCCAG8 mutations have been reported previously of which none exhibit polydactyly, it is difficult for general pediatricians to confidently diagnose BBS. We propose that general pediatricians or physicians should perform genetic testing of NPHP patients, with informed consent of the patient and/or parents, and that those with SDCCAG8 mutations should be carefully monitored to determine if they develop SLS or BBS-associated complications. Reporting of additional cases will help clarify phenotypic characteristics associated with SDCCAG8 mutations and may determine whether there is a need for new groupings based on genetic mutations rather than phenotypes. These changes will contribute to improved management of patients with ciliopathies.
Mini-review
NPHP and NPHP-related ciliopathies (NPHP-RC)
NPHP is a ciliopathy characterized by the presence of small cysts in the cortico-medullary border legion of the kidney and histologically progressive glomerulosclerosis and tubulointerstitial nephritis. NPHP almost always causes end-stage kidney disease and is the cause of 5–10% of all end-stage kidney diseases in pediatric patients. Although most cases of NPHP are thought to be caused by a single mutation, the causative mutation has only been detected in 40% of all NPHP patients with the remainder being unidentified. Approximately 10–20% of NPHP patients have extrarenal symptoms such as SLS and BBS, so are diagnosed with NPHP-RC [6, 8,9,10,11]. Because a single mutation associated with a disease in NPHP-RC often causes another disease, a definitive diagnosis is difficult for many general pediatricians. Therefore, some experts regard NPHP-RC as a comprehensive disease.
SLS
SLS is characterized by NPHP and RP and accounts for 10% of NPHP and NPHP-RC cases [6, 8,9,10,11]. SLS patients who are not blind from birth are described as having the late-onset form of disease. Such patients typically have night blindness then develop blindness during school age. Juvenile nephronophthisis is common, and end-stage kidney disease occurs by the age of 13 on average.
BBS
BBS is defined as six major symptoms: RP, polydactyly, obesity, genital abnormalities, learning difficulties, and renal defects, and several secondary symptoms also occur. A patient with more than four major symptoms, or three major symptoms with more than two secondary symptoms, can be definitively diagnosed with BBS [1, 2]. Of the major symptoms, polydactyly is a symbolic symptom of BBS but this occurs in only 63–81% of all BBS patients [1]. Obesity and learning difficulty are not specific for BBS. Genital abnormalities are symbolic for BBS boys because most are sterile, while BBS girls sometimes have genital anomalies but menarche is not typically early or late. BBS is therefore often difficult to diagnose in girls because they lack two typical symptoms of BBS.
SDCCAG8
SDCCAG8, encoding serologically defined colon cancer antigen 8, is also known as NPHP10 or BBS16. A mutation in SDCCAG8 is causative of both SLS and BBS [1, 2, 5,6,7,8]. However, only 1–3% of all BBS patients harbor SDCCAG8 mutations [1, 2, 5, 7, 8].
Therefore, SLS and BBS patients carrying a SDCCAG8 mutation lack any consistent pattern of symptoms except that none have polydactyly. Although only 10–20% of NPHP patients have extrarenal symptoms, most NPHP patients with SDCCAG8 mutations have multiple extrarenal symptoms (Table 1). Therefore, careful monitoring of extrarenal symptoms by general physicians is needed. The concepts of SLS, BBS, and NPHP-RC patients carrying SDCCAG8 mutations are shown in Fig. 2.

Disease concepts of SLS, BBS, and NPHP-RC patients carrying SDCCAG8 mutations. Major symptoms of SLS, BBS, and NPHP-RC patients carrying SDCCAG8 mutations are described. No polydactyly has been reported among NPHP-RC patients with SDCCAG8 mutations. Asthma and otitis media are common symptoms of ciliopathy and are frequently reported in NPHP-RC patients with SDCCAG8 mutations. NPHP, nephronophthisis
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- SLS:
-
Senior–Løken syndrome
- BBS:
-
Bardet–Biedl syndrome
- RP:
-
Retinitis pigmentosa
- NPHP:
-
Nephronophthisis
- NPHP-RC:
-
Nephronophthisis-related ciliopathies
References
Khan SA, Muhammad N, Khan MA, et al. Genetics of human Bardet-Biedl syndrome, an updates. Clin Genet. 2016;90:3–15.
Suspitsin EN, Imyanitov EN. Bardet-Biedl syndrome. Mol Syndromol. 2016;7:62–71.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Higasa K, Miyake N, Yoshimura J, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61:547–53.
Schaefer E, Zaloszyc A, Lauer J, et al. Mutations in SDCCAG8/NPHP10 cause Bardet-Biedl syndrome and are associated with penetrant renal disease and absent polydactyly. Mol Syndromol. 2010;1:273–81.
Arts HH, Knoers NV. Current insights into renal ciliopathies: what can genetics teach us? Pediatr Nephrol. 2013;28:863–74.
Otto EA, Hurd TW, Airik R, et al. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet. 2010;42:840–50.
Chaki M, Hoefele J, Allen SJ, et al. Genotype-phenotype correlation in 440 patients with NPHP-related ciliopathies. Kidney Int. 2011;80:1239–45.
Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol. 2017. https://doi.org/10.1101/cshperspect.a028191.
Wolf MT. Nephronophthisis and related syndromes. Curr Opin Pediatr. 2015;27:201–11.
Sugimoto K, Miyazawa T, Enya T, et al. Clinical and genetic characteristics of Japanese nephronophthisis patients. Clin Exp Nephrol. 2016;20:637–49.
Halbritter J, Porath JD, Diaz KA, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132:865–84. https://doi.org/10.1007/s00439-013-1297-0.
Braun DA, Schueler M, Halbritter J, et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. 2016;89:468–75. https://doi.org/10.1038/ki.2015.317.
Watanabe Y, Fujinaga S, Sakuraya K, et al. Tohoku J Exp Med. 2019;249:29–32. https://doi.org/10.1620/tjem.249.29.
Acknowledgements
We thank Sarah Williams, Ph.D., from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
YF and AA wrote an initial draft of the manuscript. All of the authors collected, analyzed, and interpreted the data and revised the manuscript critically. All of the authors approved the final version of the manuscript and were responsible for all aspects of the work, ensuring that questions related to its accuracy or integrity were appropriately investigated and resolved. All of the authors read and agree with the submission of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
For genetic testing, written informed consent was obtained from the patient (approval number of the ethics board of Osaka Medical College, 1867-01). For this type of case report, formal consent from a local ethics committee is not required.
Consent for publication
We offered the patient and her mother the opportunity to read the manuscript and obtained their written consent for publication. The manuscript and the informed consent form were approved by the ethics board of Osaka Medical College (approval number 2017-OMC-1).
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fujii, Y., Matsumura, H., Shirasu, A. et al. Genetic analysis diagnosed Bardet–Biedl syndrome in a patient with a clinical diagnosis of Senior–Løken syndrome. Ren Replace Ther 6, 46 (2020). https://doi.org/10.1186/s41100-020-00294-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41100-020-00294-9