
Abstract
Background
Some patients with congenital heart defects are considered inoperable because of severe pulmonary arterial hypertension (PAH) at birth, and some of these patients are followed for a long period. Recently, PAH-specific drugs have been developed to treat PAH. These drugs might make it possible to operate in adult patients with congenital heart defects who were previously considered inoperable. However, it is unclear which cases are appropriate for treatment and how they should be managed.
Case presentation
We describe a 32-year-old woman who was diagnosed with a ventricular septal defect, patent ductus arteriosus, and coarctation of the aorta, as well as severe PAH at birth. The attending physician at the time decided not to perform total repair. We re-evaluated the patient’s haemodynamics at 32 years of age. Her mean pulmonary artery pressure was 105 mmHg, pulmonary vascular resistance was 14.5 Wood units and pulmonary blood flow-to-systemic blood flow ratio (Qp/Qs) was 2.8. However, she had good vasoreactivity to oxygen. Vasoreactivity testing with 100% oxygen showed decreases in the following measures: the mean pulmonary artery pressure was 88 mmHg, pulmonary vascular resistance was 3.8 Wood units, and Qp/Qs was 10.8. We repaired the cases of coarctation of the aorta, patent ductus arteriosus and ventricular septal defect without the occurrence of a pulmonary hypertension crisis after surgery by using PAH-specific drugs. Furthermore, the continuous use of PAH-specific drugs improved the patient’s symptoms and the 6-min walk distance and decreased the mean pulmonary artery pressure and pulmonary vascular resistance. Her mean pulmonary artery pressure was 22 mmHg, and her pulmonary vascular resistance was 4.7 Wood units at the one-year follow-up.
Conclusions
With perioperative use of PAH-specific drugs, CHDs can be repaired without the occurrence of a pulmonary hypertension crisis after surgery in patients with a good vasoreactivity response to oxygen, even if the PVR is high. Medical treatment and surgery can therefore treat PAH and improve patients’ exercise capacity over time.
Similar content being viewed by others

Background
Some patients with congenital heart defects (CHDs) are considered inoperable because of severe pulmonary arterial hypertension (PAH) at birth, and some of these patients are followed over a long period. Recently, PAH-specific drugs have been developed to treat PAH [1]. These drugs might make it possible to operate in adult patients with CHD who were previously considered inoperable. However, it is unclear which cases are appropriate for treatment and how they should be managed. We present a patient who underwent repair of a ventricular septal defect (VSD), patent ductus arteriosus (PDA), and coarctation of the aorta (CoA) and had severe PAH that was managed by PAH-specific drugs before and after surgery.
Case presentation
A 32-year-old woman was diagnosed with VSD, PDA, and CoA, as well as severe PAH at birth, in another hospital. The attending physician at the time decided not to perform repair for VSD and CoA because the patient also had severe PAH and Eisenmenger syndrome. The patient reported experiencing dyspnoea upon exertion at the age of 32 years and was administered home oxygen therapy and macitentan 10 mg/day. She was referred to our hospital for an evaluation and treatment for VSD, CoA, and PAH. The patient was diagnosed with World Health Organization functional class II PAH. Her heart rate was 93 bpm, blood pressure was 114/58 mmHg, and SpO2 was 84% in the upper limbs and 68% in the lower limbs under 2 L oxygen. The cardiovascular examination revealed an increased intensity of P2 and a pansystolic murmur in the third right intercostal space upon auscultation. She had a clubbed finger. Her 6 min walk distance was 360 m with a minimal SpO2 value of 60% in the upper limbs under 2 L oxygen. The blood examination showed elevated B-type natriuretic peptide (149 pg/mL) and polycythaemia levels (haemoglobin level, 19.9 g/dL). Chest radiography revealed an enlargement of the pulmonary artery (Fig. 1a). Transthoracic echocardiography showed a perimembranous-type VSD (17 × 21 mm) with a bidirectional shunt and a tricuspid regurgitation pressure gradient of 125 mmHg (Fig. 2a and b, three of the additional movie files show this finding in more detail [see Additional files 1.mpeg, 2.mpeg, 3.mpeg, 4.mpeg and 5.mpeg]). The left ventricular ejection fraction was 57%, and the tricuspid annular plane systolic excursion value was 21 mm. Computed tomography showed the presence of CoA immediately distal to the left common carotid artery and PDA (Fig. 3a and b). Right heart catheterization (RHC) revealed a mean pulmonary artery pressure (PAP) of 105 mmHg, a pulmonary vascular resistance (PVR) of 14.5 Wood units, a pulmonary blood flow-to-systemic blood flow ratio (Qp/Qs) of 2.8, and a PVR systemic vascular resistance ratio (Rp/Rs) of 0.36. Vasoreactivity testing with 100% oxygen showed decreases in the following measures: the mean PAP was 88 mmHg, PVR was 3.8 Wood units, Qp/Qs was 10.8, and Rp/Rs was 0.07 (Table 1).

Chest radiography. a Enlargement of the pulmonary artery before surgery. b 1 year after surgery

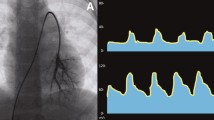
Transthoracic echocardiography. a Perimembranous-type VSD in the parasternal long-axis view before surgery. b Tricuspid regurgitation before surgery with a peak pressure gradient of 125 mmHg. c 1 year after surgery. d Tricuspid regurgitation at 1 year after surgery with a peak pressure gradient of 36 mmHg

Contrast-enhanced computed tomography. a Before surgery. VSD and CoA (yellow arrow). b The three-dimensional reconstruction of contrast-enhanced computed tomography images. CoA (yellow arrow) and PDA (green arrow). c After surgery. AO, aorta; LV, left ventricle; RV, right ventricle; LA, left atrium; RA, right atrium; VSD, ventricular septal defect; PDA, patent ductus arteriosus
We repaired the CoA by extended end-to-end anastomosis and closed the VSD with a patch. Immediately after surgery, the mean PAP was 33 mmHg under nitric oxide (NO) inhalation (20 ppm). We resumed macitentan immediately and added selexipag. We also added intravenous epoprostenol because withdrawal from NO was difficult. Adding epoprostenol enabled us to withdraw NO and subsequently administer tadalafil. After we increased the dose of epoprostenol to 10 ng/kg/min, we substituted epoprostenol with subcutaneous treprostinil. The patient’s length of stay in the intensive care unit was 43 days. An indwelling catheter-related thrombus formed at 14 days postoperatively, and cardiac tamponade occurred 35 days postoperatively. The hospitalization period was 86 days. RHC performed 2 months postoperatively showed a mean PAP of 27 mmHg and a PVR of 5.5 Wood units (with the patient on macitentan 10 mg/day, tadalafil 40 mg/day, selexipag 3.6 mg/day, and treprostinil 10 ng/kg/min) (Table 1). We reduced the doses of selexipag and treprostinil gradually. Chest radiography performed at 1 year postoperatively showed slight enlargement of the pulmonary artery (Fig. 1b). Transthoracic echocardiography performed at 1 year postoperatively revealed an estimated right ventricular systolic pressure of 36 mmHg (Fig. 2c and d, two of the additional movie files show this finding in more detail [see Additional files 4.mp4 and 5.mp4). The left ventricular ejection fraction was 60%, and the tricuspid annular plane systolic excursion value was 18 mm. RHC performed 1 year postoperatively showed a mean PAP of 22 mmHg and a PVR of 4.7 Wood units (with the patient on macitentan 10 mg/day, tadalafil 40 mg/day, and treprostinil 5 ng/kg/min) (Table 1). Her case of dyspnoea upon exertion was resolved. Her 6 min walk distance was 450 m with a minimal SpO2 value of 96% in the upper and lower limbs under 2 L oxygen. She regained the ability to live independently, while she required the support of her parents before surgery.
Additional file 4: Video 4. Transthoracic echocardiography in the parasternal long-axis view after surgery.
Additional file 5: Video 5. Transthoracic echocardiography in the parasternal short-axis view after surgery.
Discussion and conclusions
CHDs are diagnosed at birth or in early childhood, and whether surgery is indicated is usually discussed. Most cases of CHDs with severe PAH diagnosed 20–30 years ago were deemed inoperable because effective drugs for PAH were not available. However, over the past two decades, PAH-specific drugs have been developed. Epoprostenol was first clinically applied for the treatment of idiopathic PAH in 1995. Subsequently, endothelin receptor antagonists, phosphodiesterase-5 inhibitors, and soluble guanylate cyclase stimulators have been used clinically [1]. These drugs improve exercise capacity, haemodynamics, and survival in patients with PAH [2] and may change the treatment strategy for adult CHD–PAH patients considered inoperable at childhood.
Cases of PAH associated with CHD are divided into four clinical groups: (i) Eisenmenger syndrome; (ii) PAH associated with a prevalent systolic/pulmonary shunt; (iii) PAH with small/coincidental defects; and (iv) PAH after defect correction [3]. The optimal treatment strategy for PAH associated with a prevalent systolic/pulmonary shunt is controversial. Some guidelines are based on criteria of defect closures that depend on the PVR, Qp/Qs, and Rp/Rs values and the patient’s reactivity to oxygen [3,4,5]. However, these criteria may no longer be applicable in current practice because they were determined on the basis of evidence acquired before PAH-specific drugs were developed. Recently, a ‘treat-and-repair’ strategy has been proposed for CHD patients who initially have a systolic/pulmonary shunt but high PVR [6,7,8]. With this strategy, patients are initially treated with PAH-specific drugs and then repair is performed by the closure of the CHD. In the present case, a high Qp/Qs at baseline and a dramatic reduction in PVR with 100% oxygen were observed, indicating that the level of pulmonary artery reactivity was maintained. Thus, the perioperative use of PAH-specific drugs can prevent pulmonary hypertension crises. Hence, we decided to repair the VSD and CoA. Although combination therapy with four PAH-specific drugs (macitentan, tadalafil, selexipag, and epoprostenol) was required immediately after surgery, we were able to decrease the number of medications administered and doses of these medications.
We consider this study to have the following strengths. First, the patient showed a good response to oxygen in the vasoreactivity test. Second, PAH-specific drugs were administered before surgery to prevent a pulmonary hypertension crisis from occurring after surgery. Third, the PAH-specific drugs were continued after surgery.
In conclusion, this case demonstrates that with the perioperative use of PAH-specific drugs, CHDs can be repaired without the occurrence of a pulmonary hypertension crisis after surgery in patients with a good vasoreactivity response to oxygen, even if the PVR is high. Medical treatment and surgery can therefore treat PAH and improve exercise capacity over time. The accumulation of additional cases and investigations are warranted regarding perioperative management and long-term efficacy.
Availability of data and materials
Data sharing is not applicable to this article, as no datasets were generated or analysed during the current study.
Abbreviations
- CHD:
-
Congenital heart defect
- PAH:
-
Pulmonary arterial hypertension
- VSD:
-
Ventricular septal defect
- PDA:
-
Patent ductus arteriosus
- CoA:
-
Coarctation of aorta
- SpO2 :
-
Saturation of percutaneous oxygen
- PAP:
-
Pulmonary artery pressure
- PVR:
-
Pulmonary vascular resistance
- Qp/Qs:
-
Pulmonary blood flow-to-systemic blood flow ratio
- Rp/Rs:
-
Pulmonary vascular resistance systemic vascular resistance ratio
- NO:
-
Nitric oxide
References
Akagi S, Matsubara H, Nakamura K, Ito H. Modern treatment to reduce pulmonary arterial pressure in pulmonary arterial hypertension. J Cardiol. 2018;72:466–72.
Akagi S, Nakamura K, Miyaji K, Ogawa A, Kusano KF, Ito H, et al. Marked hemodynamic improvements by high-dose epoprostenol therapy in patients with idiopathic pulmonary arterial hypertension. Circ J. 2010;74:2200–5.
Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2015;46(4):903.
Warnes CA, Williams RG, Bashore TM, Child JS, Connolly HM, Dearani JA, et al. ACC/AHA 2008 guidelines for the Management of Adults with congenital heart disease: a report of the American College of Cardiology/American Heart Association task force on practice guidelines (writing committee to develop guidelines on the management of adults with congenital heart disease). Circulation. 2008;118:e714–833.
Baumgartner H, Bonhoeffer P, De Groot NM, de Haan F, Deanfield JE, Galie N, et al. Task force on the Management of Grown-up Congenital Heart Disease of the European Society of C, Association for European Paediatric C, guidelines ESCCfP. ESC guidelines for the management of grown-up congenital heart disease (new version 2010). Eur Heart J. 2010;31:2915–57.
Kijima Y, Akagi T, Takaya Y, Akagi S, Nakagawa K, Kusano K, et al. Treat and repair strategy in patients with atrial septal defect and significant pulmonary arterial hypertension. Circ J. 2016;80:227–34.
Akagi S, Kasahara S, Sarashina T, Nakamura K, Ito H. Treat-and-repair strategy is a feasible therapeutic choice in adult patients with severe pulmonary arterial hypertension associated with a ventricular septal defect: case series. Eur Heart J Case Rep. 2018;2(2):yty033.
Constantine A, Dimopoulos K. Evaluating a strategy of PAH therapy pre-treatment in patients with atrial septal defects and pulmonary arterial hypertension to permit safe repair (“treat-and-repair”). Int J Cardiol. 2019;291:142–4.
Acknowledgements
We thank Sarah Williams, PhD, from Edanz Group (www.edanzediting.com) for editing a draft of this manuscript.
Funding
No funding was obtained by either author for completion/research of this case report.
Author information
Authors and Affiliations
Contributions
SA was involved in diagnosing and treating the patient for PAH and wrote the manuscript. SK performed the surgery. TA made the initial patient diagnosis. KN and HI edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was not required for this case report.
Consent for publication
Patient consent was received for the use of anonymised images and the presentation of this case report. The consent form is available upon request.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Video 1.
Transthoracic echocardiography in the parasternal long-axis view before surgery.
Additional file 2: Video 2.
Transthoracic echocardiography in the parasternal long-axis view with color Doppler before surgery.
Additional file 3: Video 3.
Transthoracic echocardiography in the parasternal short-axis view before surgery.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Akagi, S., Kasahara, S., Akagi, T. et al. Medical and surgical management of a pulmonary hypertensive adult patient with unrepaired complex congenital heart disease: a case report. J Congenit Heart Dis 4, 2 (2020). https://doi.org/10.1186/s40949-020-00033-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40949-020-00033-z