
Abstract
Background
Some congenital heart conditions are very rare. In a climate of limited resources, a viewpoint could be advanced that identifying diagnostic criteria for such conditions and, through empiricism, effective treatments should suffice and that extensive mechanistic research is unnecessary. Taking the rare but dangerous short QT syndrome (SQTS) as an example, this article makes the case for the imperative to study such rare conditions, highlighting that this yields substantial and sometimes unanticipated benefits.
Genetic forms of SQTS are rare, but the condition may be under-diagnosed and carries a risk of sudden death. Genotyping of SQTS patients has led to identification of clear ion channel/transporter culprits in < 30% of cases, highlighting a role for as yet unidentified modulators of repolarization. For example, recent exome sequencing in SQTS has identified SLC4A3 as a novel modifier of ventricular repolarization. The need to distinguish “healthy” from “unhealthy” short QT intervals has led to a search for additional markers of arrhythmia risk. Some overlap may exist between SQTS, Brugada Syndrome, early repolarization and sinus bradycardia. Genotype-phenotype studies have led to identification of arrhythmia substrates and both realistic and theoretical pharmacological approaches for particular forms of SQTS. In turn this has increased understanding of underlying cardiac ion channels. In silico and pharmacological data have highlighted risks with abbreviation of refractoriness accompanied by local dispersion of repolarization, and this urges caution with the deployment of K+ channel activation as a novel antiarrhythmic approach. The association between abbreviated QTc intervals and primary carnitine deficiency, particularly in patients with concomitant cardiomyopathy illustrates a link between metabolism and electrogenesis, in which the correct identification of causation could, in some cases, lead to dietary intervention that may obviate the need for antiarrhythmic or heart failure drugs.
Conclusions
As illustrated here for the SQTS, the detailed study of rare disorders is both directly beneficial for the treatment/management of affected patients and for increasing the understanding of associated underlying cardiac physiology and pharmacology. The pursuit of underlying gene mutations can lead to unanticipated new links between particular genes and cardiac electrophysiology, opening new avenues for research and potential therapeutic intervention.
Similar content being viewed by others


Introduction
The ability to link individual cases of particular inherited cardiac conditions to specific gene mutations is valuable in establishing causality and potential treatments, although it is recognized that the identification of gene variants of unknown significance can be problematic [1]. At its best, however, the integration of clinical and basic science can reveal causality, underlying mechanism(s) and provide a rational basis for therapeutic interventions.
The prevalence of different inherited and congenital heart conditions varies greatly. In a climate of limited resources, a viewpoint could be advanced that for extremely rare cardiac conditions extensive mechanistic research is excessive; identifying reliable diagnostic criteria and, through empiricism, effective treatments should suffice. Here, we present evidence refuting such a view. Drawing on the example of the rare congenital arrhythmia disorder short QT syndrome (SQTS), we illustrate the benefits to be gained from detailed study of very rare heritable cardiac conditions.
Abbreviated cardiac repolarization and short QT syndrome
It has long been known that species of kangaroo that are prone to sudden cardiac death (SCD) exhibit abbreviated ventricular action potentials and QT intervals on the ECG [2, 3]. In 1993 a link was first made between shorter than average QT intervals (< 400 ms) in humans and an increased risk (2.4 fold) for SCD [4]. Abnormally short QT intervals have been recorded immediately before and after episodes of ventricular tachycardia (VT) and ventricular fibrillation (VF) [5, 6]. Approximately 35% of male patients with idiopathic ventricular fibrillation exhibit somewhat shortened QT intervals [7]. These observations point towards a link between abbreviated repolarization and serious ventricular arrhythmia. A significant correlation has also been identified between shortened QT intervals and incidence of atrial fibrillation (AF), independent of other variables [8]. Analysis from 281,277 subjects from the Copenhagen ECG Study has reported a “J”-shaped association between QT interval and risk of AF, with significantly increased hazard ratio for QTc intervals lower than the first percentile [9]. The association between abbreviated repolarization and VT/VF and AF suggests that it should be instructive to study the basis of arrhythmogenesis in circumstances where causality can be clearly established. Accelerated repolarization linked to single gene mutations provides such opportunities.
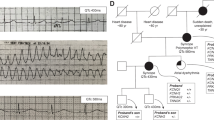
As a distinct congenital condition, the Short QT Syndrome (SQTS) is a relatively young entity, having been first reported in 2000 [10]. The SQTS is characterized by accelerated ventricular repolarization, leading to abbreviation of the rate-corrected QT (QTc) interval on the ECG, with poor rate adaptation of the QT interval such that it remains short even at low heart rates [11,12,13]. These changes can occur in the absence of obvious structural heart disease. The ECGs of affected patients often show narrow, tall T waves, especially on the precordial leads [11,12,13] (Fig. 1). Importantly, there is a strong association between the SQTS and both atrial and ventricular arrhythmias and of sudden death [11,12,13]. The pathogenicity of the condition is well illustrated by the fact that approximately 40% of SQTS cases first present as cardiac arrest [14].

Example SQTS ECG. ECG recording from young adult male who had experienced out-of-hospital arrest. Heart rate at time of ECG measurement 62 min− 1, with QT and QTc intervals respectively of 322 and 326 ms (below the cut-off level in ESC diagnostic criteria [24]). Note tall T waves in precordial leads. ST segment elevation probably reflects early repolarization
Since 2004, mutations have been identified in 8 ion channel subunits (SQT1–7, SQT9 in Table 1) and in the SLC4A3 encoded anion exchanger 3 (AE3; SQT8 in Table 1), with functional data that have demonstrated causality for 7 of the 8 ion channel genes implicated [11, 13, 15]. Broadly speaking the ion channel mutations identified are gain-of-function mutations to potassium channels (SQT1–3) or loss-of-function mutations to sodium or calcium channel subunits (SQT4–7 and, putatively, SQT9) [11, 13, 15]. Patients with the metabolic disorder primary carnitine deficiency (which arises due to loss of function mutations to SLC225A gene encoding the OCTN2 carnitine transporter) can also exhibit abbreviated QTcintervals and tall T waves, with additional structural abnormalities, in particular dilated cardiomyopathy [16,17,18].
Complexities in the diagnosis of the SQTS
Short QT intervals appear to be rare in the general population, with a prevalence of QTc intervals of < 320 ms of ~ 0.1% [19, 20]. The prevalence of the SQTS seems also to be low [13, 15]. Around 200 cases of SQTS have been reported in the literature [21]. One recent analysis listed 27 probands with genetic mutation [22], whilst another considered a total of 132 reported cases [15]. In the nearly two decades since the identification of congenital SQTS there has not been an accumulation of cases to mirror that of the long QT syndrome. There are several points to make in this regard, however.
First, the cut-off between a healthy but abbreviated QT interval and a pathological short QTc interval is not entirely clear. This has led to development of diagnostic criteria combining clinical electrophysiological measurements (QTc and Jpoint-Tpeak) intervals with patient and family clinical history [23]. More recently, simplified diagnostic criteria have emerged from the European Society of Cardiology (ESC): a positive diagnosis can be made with a QTc interval of ≤340 ms [24], or with a longer QTc interval of ≤360 ms, if there is evidence also of one or more of: a familial history of SQTS; a confirmed pathogenic mutation; a family history of sudden death below 40 years of age; survival from ventricular tachycardia (VT) or fibrillation (VF) in the absence of structural heart disease [24].
Second, there is the issue of measuring accurately the QTc interval. The correction method used may influence the derived value and hence diagnosis and it is therefore important that clinical measurement of the QT interval are made at a low/resting rate [25]. Measurements at or near to a low resting heart rate are likely to be particularly valuable given the impaired rate adaptation of the QTc interval in SQTS patients. Furthermore, as has been highlighted by others, a “spot” ECG may be insufficient to identify SQTS, as surface ECG abnormalities can be intermittent [26, 27]. The use of 24 h Holter monitoring or repeated ECGs during hospitalization may be preferable [26, 27]. Additional potential markers for SQTS that have been identified include PQ segment depression on the ECG (seen in > 80% of 64 patients studied [28] and in a further recent case report [29]) a refractory period cut-off of 200 ms in the right ventricular outflow tract during invasive testing (whilst pacing at a cycle length of 500–600 ms) [30].
Third, the atrial tachyarrhythmia in SQTS patients presenting with AF can make it difficult to measure the QT interval accurately and some SQTS cases could be missed in such a scenario. A useful analogy can be made here with familial atrial fibrillation caused by a gain-of-function mutation (S140G) to the major KCNQ1 subunit of cardiac “IKs” channels [31]. In vitro and in silico biophysical data indicate that, aside from its propensity to cause AF, this KCNQ1 mutation should also abbreviate ventricular APs and increase susceptibility to ventricular arrhythmia [32,33,34]; however the initial report of this mutation in AF did not find QTc abbreviation, noting that irregular ventricular beats could impact QT interval representation and accuracy of QTc correction [31].
Fourth, there is evidence that in some instances SQTS may be an ‘overlap syndrome’, concurrent with Brugada [35, 36], early repolarization syndrome [37] or sick sinus syndrome [15] phenotypes, which may complicate primary diagnosis. Fifth, the yield of successful gene culprits from genotyping suspected SQTS cases is rather low (less than 25–30%) [13, 23]. Given that genetic screening generally includes a wide range of ion channel candidates, this highlights a limit in our current understanding of factors controlling cardiac repolarization.
Finally, as highlighted by others, the majority of studies of the general population have centred on adults and this may underestimate the occurrence of short QT intervals in the early years of life, during which fatal arrhythmias may occur [21]. Irrespective of its incidence and prevalence, the SQTS is highly pathogenic [13, 14], making it important to understand the basis, mechanisms for arrhythmia susceptibility and treatment potential for identified SQTS variants. The following sections illustrate the mutual benefits of combining the pursuit of clinical and pre-clinical basic science research in this rare condition.
Establishing causality
Whilst bioinformatic tools such as Polyphen [38] and SIFT [39] can provide valuable estimates of likely pathogenicity of identified mutations, the only way definitively to establish causality, and certainly mechanism, is to demonstrate altered gene product function. Indeed, it has been highlighted for the long QT syndrome that results of in silico bioinformatics predictions of pathogenicity and functional testing of mutations can be at variance with one another [40]. For nearly all of the ion channel gene mutations implicated in the SQTS, detailed cellular electrophysiology study using patch clamp electrophysiology has been performed. Such recordings have demonstrated the gain-of-function consequences of SQT1-SQT3 K+ mutations [15, 41,42,43,44,45,46,47,48,49,50,51,52,53,54,55] and loss-of-function consequences of Na and Ca channel subunit mutations [36, 56,57,58,59]. With an historical lack of genotypically accurate mammalian models of the SQTS, computational modelling has been employed to determine the consequences of altered channel function for repolarization. Such work is time-consuming but has value in enabling unequivocal demonstration of a causal link between specific mutations and in explaining how particular functional changes underlie disease phenotypes.
Synergy between clinical and preclinical studies in understanding genotype-phenotype links
It has recently been reported that among genotyped variants with mutations to K+ channel or Ca2+ channel subunits: 55.5% of patients had mutations to hERG, 11.1% had mutations to KCNQ1, 14.8% had mutations to KCNJ2, 11.1% had mutations to CACNA1C, and 3.7% had mutations to each of CACNB2 and CANAD1 [22]. Penetrance and expressivity varies between mutations [13], with an overall penetrance of ~ 82% [15]. Correlations between phenotype and genotype have been made in vitro and in silico.
The first genotyped SQTS (SQT1) variant was found to be due to an asparagine to lysine (N588K) change in the external S5-Pore linker region of the hERG channel [41]. N588K accounts for ~ 18.5% of genotyped SQTS cases and exhibits 100% penetrance [22]. The S5-Pore linker region of the hERG channel is implicated in the uniquely rapid inactivation kinetics of the hERG channel [60,61,62] and the N588K mutation was found to shift inactivation profoundly towards more positive voltages [42, 43]. These findings have added to the available evidence for the role of the S5-Pore linker in hERG/IKr channel inactivation and the critical role of inactivation in shaping the contribution of IKr to ventricular repolarization [63, 64]. Computational modelling has demonstrated causality between this inactivation change and abbreviated repolarization [65,66,67]. Moreover, in multicellular ventricular simulations incorporating regional heterogeneity of IKr channel expression, the N588K mutation was found to increase transmural membrane potential heterogeneity (δV) during AP repolarization and also APD dispersion within localised regions of the ventricular wall [67]. Such changes may underlie the increased T wave amplitude seen in patients. Figure 2 summarises proarrhythmic consequences of the N588K-hERG mutation. A second mutation in the hERG channel pore (S631A) that produces a similarly marked shift in hERG/IKr inactivation [68, 69] has been identified in an SQTS family [70]. It produces comparable effects on hERG during ventricular repolarization to those reported for N588K [47].

A schematic diagram of the main pro-arrhythmic mechanisms of the N588K-hERG short QT syndrome (SQTS) mutation identified by experiments and modelling. The upper panel shows experimental voltage and action potential (AP) clamp characterisations of wild type (WT) and N588K mutant hERG currents at the ion channel level. The N588K mutation was found to increase greatly the current over the physiological range of membrane potentials due to impaired inactivation [43] causing increased maximal IhERG to occur earlier during a simulated human ventricular AP waveform [148]. These changes were integrated into a mathematical models of the human ventricular AP in which the N588K mutation was shown to shorten the action potential duration (APD) whilst increasing the transmural dispersion of repolarisation (TDR) across the ventricular wall [67, 132]. At the tissue level this had the effect of shortening the QT interval whilst increasing the amplitude of the T wave, as has been observed clinically in SQTS patients. When integrated into a human left ventricular wedge model with realistic geometry, the combined pro-arrhythmic substrate of shortened AP with increased heterogeneity of repolarisation due to the N588K-hERG mutation rendered ventricular tachycardia/fibrillation (VT/VF) inducible. Modified with permission from [132]
The T618I hERG mutation is the most commonly occurring ion channel mutation thus far observed in genotyped patients, accounting for 25.9% of genotyped cases SQTS [22]. It is strongly associated with arrhythmia, but produces a smaller abbreviation of the QTc interval than does the N588K mutation [22]. This may reflect a more modest effect of the mutation on voltage dependent inactivation kinetics of hERG [44, 45]. However, not all studies of T618I have reported an inactivation voltage shift and a negative shift in voltage dependent activation and faster activation time-course have also been suggested [22]. Curiously, in contrast with the N588K mutation, no T618I carriers have exhibited atrial fibrillation [22, 71]. Intriguingly, the I560T mutation, located towards the top of the S5 transmembrane segment of the hERG channel, appears to produce broadly similar changes to hERG current to those seen in some studies of T618I [44, 45] and has been associated with atrial fibrillation and flutter [15]. Further work is therefore required to uncover the basis for the difference in AF susceptibility between T618I and these other mutations.
The E50D [23] and R1135H hERG mutations have been reported in isolated cases of SQTS [46]. R1135H is particularly notable because it has been associated with a mixed SQTS/Brugada phenotype. The mutation’s principal effects were to increase current magnitude and slow the deactivation kinetics of hERG channels [46]. Computer simulations have shown that slowed hERG channel deactivation can result in fewer channels closing during diastole, increasing the recruitment of hERG channels early in successive APs and increasing the likelihood of all-or-none repolarization in the right ventricle [35]. A subsequent study has reported several hERG mutations in Brugada syndrome patients that increase hERG current without modifying inactivation kinetics that are also associated with QT interval shortening [72]. The R164C mutation was found in a 58 year old man with coved-type ST segment elevation and a QTc interval of 342 ms. No genetic data were available for other family members, although he had an uncle who had undergone sudden cardiac death [72]. The W927G mutation was found in a 32 year old man who lost consciousness and experienced VF detected by an external defibrillator [72]. He had saddleback ST segment elevation in lead V1 and coved ST elevation at the third intercostal position. His QTc interval was 350 ms. Both mutations increased hERG current density and produced a modest left-ward shift in voltage dependent activation kinetics [72].
What this body of information collectively indicates is that, depending on the particular effects of a gain-of-function hERG mutation on channel function, the resulting phenotype may be SQTS alone or, more subtly, that of mixed SQTS-Brugada syndrome. Very recent work has reported a heterozygous missense hERG mutation (K801T) that produces a moderate gain-of-function in a family with early repolarization syndrome and sudden death [73]. Affected family members had shorter QTc intervals (384 ± 12 ms) than ethnically matched controls (415 ± 9 ms) [73]. This is consistent with the notion that there may be a continuum on which the different abbreviated repolarization disorders lie.
SQT2 typically shows an earlier age of manifestation than does SQT1 [15]. Several KCNQ1 mutations (V307L, V141M, R295H, F279I) have now been identified in SQTS patients [48,49,50,51,52]. Similar to the situation outlined above for hERG, the phenotypic consequences of SQT2 mutations depend on the nature of the channel current/kinetics modification. For example, for the V307L mutation (the first KCNQ1 mutation to be identified in SQTS2) residing within the P-loop of the KCNQ1 channel, mutant channels (when co-expressed with KCNE1 to recapitulate cardiac “IKs” channels) exhibits left-ward shifted and accelerated voltage dependent activation and moderately slowed deactivation kinetics [48, 50]. This mutation led to abbreviated QT interval and an episode of aborted sudden death at age 70 [48], with causality established in silico [74, 75]. The V141M mutation, in the S1 segment of the KCNQ1 protein, led to a phenotype that was detectable in utero as bradycardia and irregular rhythm [49]. This mutation has now been associated with multiple reports of a mixed AF and sinus bradycardia phenotype [15, 49, 76, 77]. This mutation results in an instantaneous component of IKs current and initial modelling (using a human ventricular model and rabbit sinoatrial node model) showed abbreviation of ventricular APs and abolition of pacemaking [49]. Our recent human atrial and sinus node simulations have shown that the distinct changes to IKs produced by the two mutations can account for their different phenotypes in respect of sinus bradycardia [78]. Only the V141M mutation exerted a marked effect on simulated sinoatrial node pacemaking rate, due to its greater effect in increasing IKs over the diastolic membrane potential range (the increased IKs resulting in a slowed diastolic depolarisation) [78]. The V141M mutation also produced a greater effect on atrial AP duration than did V307L and whilst both mutations shortened atrial tissue excitation wavelength for re-entry, V141M led to more stationary spiral wave dynamics than did V307L [78].
SQT3 involves mutations to KCNJ2 which encodes the Kir2.1 channel which contributes to inwardly rectifying K+ current, IK1 [13]. The observed Kir2.1 mutations are D172N, M301K, E299V, K346T [53,54,55, 79]. The first reported SQT3 mutation was D172N, which was associated with a distinct T wave morphology in which not only was T wave height increased, but the T waves exhibited marked asymmetry [53]. This SQT variant is a clear instance where identifying the biophysical changes to the underlying channel has explained the resultant T wave changes. IK1 usually exhibits marked voltage-dependent rectification so that it contributes little repolarizing current during the ventricular AP plateau, but contributes markedly to the terminal repolarization phase of the AP. In D172N-Kir2.1 channels, the processes underlying rectification of Kir2.1 current are impaired, leading to a selective increase in outward current [53, 80]. When these changes were incorporated in ventricular AP simulations they resulted in an abrupt acceleration of the final repolarization stage of the AP, and in tissue simulations this recapitulated the T wave asymmetry seen clinically [53, 81].
The M301K mutation leads to a greater attenuation of Kir2.1 current rectification, resulting in a contribution of IK1 over a wider range of voltages than is normally the case and thus to a greater QTc shortening than the D172N mutation [54]. The E299V mutation produces a still greater decrease in Kir2.1 current rectification and so a larger current at positive voltages, with simulations demonstrating greater AP abbreviation [55]. Thus, the degree to which KCNJ2 gain-of-function mutations alter AP repolarization can be linked to the severity of the underlying mutant’s effects on Kir2.1 channel function and the range of voltages over which outward Kir2.1 current is augmented.
SQT4–6 variants involve loss of function mutations to genes encoding L-type Ca channel subunits (CACNA1C, CACNB2b, CACNA2D1) [36, 56, 57, 59]. In each case, the observed mutations lead to reductions in L-type Ca current in vitro. Patients exhibit a mixed Brugada/SQTS phenotype, combining changes in ST segment morphology (with or without drug testing) with QTc abbreviation [36, 56, 57, 59]. For example, in the first study associating CACNA1C with Brugada/SQTS, the 44 year-old male patient with the A93V mutation presented with marked ST segment elevation in ECG lead V1, saddle-back elevation in lead 2 and a prominent J wave in lead 3. His QTc interval was 360 ms. His mother had experienced syncope and undergone sudden death [36]. The proband with G490R mutation identified in the same study presented with AF and an abbreviated QTc of 346 ms. ST segment elevation in leads V1 and V2 was exacerbated by ajmaline [36]. In electrophysiological testing, extrastimuli induced monomorphic VT. L-type current from recombinant channel expression experiments was suppressed across a wide range of voltages by both mutations [36]. Mutations in L-type Ca2+ channel subunit genes are linked to Brugada and Early Repolarization Syndrome [82] and it has been suggested that Brugada/early repolarization and QTc shortening phenotypes may be part of a continuum of manifestations of decreased L-type Ca current [59]. Putative SQT7 has been reported to involve a mutation to SCN5A which leads to a R689H substitution in the Nav1.5 protein which carries cardiac Na current [58]. The index patient had a Brugada-like ECG concurrent with a short QT interval and his father had died suddenly aged 39. There are conflicting biophysical data on this mutation: some data suggest a loss of function (consistent with a Brugada/SQTS phenotype [58], whilst other data suggest an increase in late Na current (which would prolong repolarization) [83]. It is possible that the clinical manifestation of effects of this mutation may depend on additional as yet to be identified factors.
What is reasonable to conclude from consideration of the relationship between preclinical and clinical findings is that examination of the precise consequences of identified mutations helps establish causality and in some instances explains the distinct nature of the clinical phenotype. In turn, the clinical information helps establish the importance to different phases of cardiac repolarization of particular biophysical changes to the affected ion channels.
Evidence for novel modifiers of the QT interval from SQTS studies
The dominant cardiac Nav1.5 channel isoform is encoded by SCN5A, but growing evidence has suggested that Nav1.8 (encoded by SCN10A) may influence cardiac conduction and SCN10A mutations have been identified in Brugada Syndrome [84]. Recently, a case of sudden death in J wave syndrome associated with short QT interval (QTc of 303 ms) was found to be linked to an SCN10A mutation which led to a G805V substitution in the transmembrane domain of Nav1.8 [85]. The patient had been treated by the COX-2 inhibitor etoricoxib for rheumatoid arthritis and, as prostaglandin E2 influences Nav1.8 trafficking, it is possible that there was a pharmacogenomic component to the altered repolarization in this case [85]. Whilst functional work is needed to verify the loss-of-function effect of the G805V mutation, this case appears to extend the evidence for involvement of SCN10A in aberrant cardiac repolarization and to highlight the potential for trafficking modulation and pharmacogenomics as components of SQTS.
Exome screening of two SQTS families with no mutations in candidate ion channels has recently identified a missense (R370H) mutation in a conserved motif of the AE3 anion transporter encoded by the SLC4A3 gene, which was absent in healthy controls [86]. Expression studies of recombinant wild-type and mutant AE3 protein revealed reduced surface localisation of the mutant variant along with reduced HCO3− transport. A causal link with QTc abbreviation has been identified through knockdown of slc4a3 in zebrafish embryos; this abbreviated repolarization and resulted in a rise in intracellular pH in zebrafish embryo hearts [86]. Additionally, intracellular alkalization and reduced intracellular chloride concentration ([Cl−]i) shortened rabbit QT interval and ventricular AP duration [86]. This work has demonstrated that altered function of an anion transporter can lead to abbreviated ventricular repolarization, though the electrogenic processes that effect accelerated repolarization remain to be determined.
A second transporter that has been implicated in the SQTS is the SLC22A5 encoded OCTN2 sodium-dependent carnitine transporter [18]. Oxidation of long chain fatty acids (LCFAs) is important for cardiac energy metabolism and L-carnitine is a key cofactor for the transport of LFCAs into mitochondria in cardiac myocytes. Sarcolemmal OCTN2 is important for carnitine entry to occur. In primary carnitine deficiency (PCD), SLC22A5 mutations lead to malfunction of OCTN2, resulting in impaired LCFA oxidation and accumulation in the cell cytosol [87, 88]. PCD is characterized by low intracellular and plasma carnitine and reduced renal carnitine reabsorption. PCD patients can develop a progressive cardiomyopathy and arrhythmias [89]. A SQTS phenotype has been identified in some patients with PCD, leading to the suggestion that PCD should be considered as a potential cause of SQTS, particularly when concurrent with dilated cardiomyopathy [16,17,18]. A causal link between carnitine deficiency and abbreviated ventricular repolarization and arrhythmia has been demonstrated in a mouse model of PCD [16]. It is critical that, where present, PCD is identified in SQTS patients as it can be treated by dietary L-carnitine supplementation [16, 17], potentially obviating the need for antiarrhythmic pharmacology or antiarrhythmic device implantation. The underlying ventricular ionic current changes that lead to QT shortening in PCD remain to be established. In vitro data raise the possibility that hERG/IKr modulation may be involved [16, 90]. However, this cannot account for accelerated repolarization in the mouse carnitine deficiency model, as mice do not rely on hERG/IKr for ventricular repolarization [18, 91]. Consequently, further work is required to understand the basis of QTc shortening in PCD.
The identification of links between both SLC4A3 and SLC22A5 mutations and the SQTS indicates that understanding of the cellular mechanisms controlling ventricular repolarization has been incomplete, providing fertile ground for further research.
Insights from preclinical SQTS studies into arrhythmia substrates with abbreviated repolarization
Insight into SQTS arrhythmia mechanisms has come from experiments using pharmacological K+ channel activation as a surrogate for gain-of-function K+ channel mutations and in silico simulations of the effects of SQTS mutations [13, 92]. The association between abbreviated QT intervals and both ventricular and atrial fibrillation highlighted earlier in this article means that insights gained from such studies may also have wider relevance to settings of fibrillation associated with abbreviated repolarization. Consistent with this, the SQTS has been suggested to provide a paradigm for increasing understanding of the roles of K+ channels in (ventricular) fibrillation [93]. The SQT3 variant provides a valuable illustration in this regard. It has been known for some time that the magnitude of outward current carried by IK1 influences stability of high frequency rotors, with a larger outward current producing greater APD shortening and rotor stabilization [93, 94]. Concordant with this notion, incorporation of the effects of the D172N Kir2.1 mutation into human ventricle models led not only to shorter APD and effective refractory period (ERP), but altered excitability (reducing excitability at low excitation rates and enhancing excitability at high rates) and increased tissue vulnerability to 2D/3D re-entry [53, 81]. In both 2D and 3D tissue models this led to ability of tissue to support reentrant spiral/scroll waves with increased dominant frequency compared to wild-type tissue [81]. Subsequent simulations compared the ability of D127N and E299V mutations to abbreviate ventricular APD. These found a greater effect of E299V due its markedly weaker rectification properties. This led to absence of a clear ST segment [55] and activation delay of the right ventricle leading to QRS alterations and to an increased susceptibility to re-entry in an atrial tissue model [55]. Comparison of effects of the two mutations in atrial tissue models has subsequently shown a greater shortening of wavelength of re-entry for E299V than D172N and increased susceptibility to sustained spiral wave re-entry in both conditions [95]. In 3D tissue simulations, stable spiral wave re-entry was seen for both mutations in heterozygous expression conditions mimicking those of patients, but with greater meander for the E299V condition [95]. Overall, the simulation data indicate that mutation-induced increased outward IK1 has strong potential to increase susceptibility to pro-fibrillatory re-entry.
Simulations of the N588K hERG mutation have shown a strong propensity towards re-entry consequent to attenuation of voltage-dependent inactivation [67]. The mutation produced a marked shortening of APD and the ERP and localised increases in δV facilitated vulnerability to uni-directional block and re-entry. In 2D and 3D tissue models this led to a reduced substrate size for re-entry, prolonging the lifespan of induced spiral or scroll waves and supporting an increased dominant frequency of excitation [67]. Interestingly, temporal vulnerability was decreased for N588K hERG at many locations across the ventricular wall, but the significant decrease in minimal substrate size required to sustain reentry was sufficient to increase markedly spatial vulnerability [67]. Similar observations have been made from simulations of KCNQ1 and KCNJ2 mutations associated with genetic forms of AF [34, 96]. This highlights that effects on indices of both temporal and spatial vulnerability need to be evaluated in order to understand the overall effects of arrhythmia causing mutations. Simulations of the I560T hERG mutation, which produces a more modest attenuation of inactivation than N588K, have also shown susceptibility to sustained spiral wave re-entry [15]. Simulations of KCNQ1 mutations in SQT2 have shown abbreviation of ERP and reduction of the tissue substrate required to support reentry, for both ventricular and atrial tissue [74, 75, 78].
Computer simulations incorporating electromechanical coupling have predicted a decrease in the phasic Ca2+ transient that leads to ventricular contraction, with APD shortening due to SQTS K+ channel mutations [97]. Subsequent speckle-tracking echocardiography and Doppler imaging has shown a measurable decrease in left ventricular contraction and greater mechanical dispersion in individuals with SQTS [98, 99].
Arrhythmogenesis in the SQTS has also been studied by using K+ channel openers to reproduce accelerated repolarization, albeit without the ability to reproduce effects of specific K+ channel gene mutations. In studies of the canine ventricular wedge or rabbit perfused heart, the KATP channel activator pinacidil abbreviated repolarization and the ERP, augmented transmural dispersion of repolarization and increased susceptibility to polymorphic VT or VF [100,101,102]. Utilization of the hERG activator PD118057 also abbreviated the QT interval and refractoriness whilst increasing transmural dispersion of repolarization and predisposition to arrhythmia in ventricular wedge preparations [103]. Application of the same hERG activator to perfused canine atrial preparations also shortened AP and ERP duration, whilst increasing spatial dispersion of repolarization and vulnerability to arrhythmia provocation [104].
The only genotypically accurate mammalian experimental models of (hERG-linked) SQTS have been recently reported. Two of these are cardiomyocyte lines derived from induced pluripotent stem cells (iPSCs) made from fibroblasts from two SQT1 patients with (i) the N588K [105] and (ii) T618I [106] hERG mutations. As expected, myocytes from these lines exhibited abbreviated repolarization and increased IKr compared to a control line and, interestingly, also showed increased hERG/KCNH2 expression [105, 106]. Unlike normal adult ventricular myocytes, iPSC-derived myocytes tend to exhibit an immature phenotype of spontaneous activity and spontaneous Ca2+ transients from N588K-myocytes also exhibited after-depolarization like events, which were exacerbated by application of the cholinergic agonist carbachol [105]. Whether or not such events are likely to occur in myocytes with a more adult phenotype remains to be established. However, their observation points intriguingly to a potential arrhythmia trigger in SQTS under conditions of vagal stimulation. In the T618I hERG study, comparison between the myocyte line derived from the patient with a line with I618➔T genetic correction showed alterations in functional expression of other ion channels, including Na+ and L-type Ca2+ channels [106].
Very recently, a transgenic rabbit model of SQT1 has been reported in which the human N588K hERG mutation has been expressed [107]. SQT1 transgenic animals exhibited shortened QTc intervals, altered QTc-rate adaptation and abbreviated ventricular and atrial APs and ERP. Alterations in T wave height were not reported [107]. Increased regional AP dispersion was seen in SQT1 rabbits (consistent with reported localised alterations in δV seen in silico [67]). In perfused heart experiments, VT/VF was readily inducible in SQT1 hearts [107]. Diastolic relaxation was enhanced in SQT1 hearts, but changes in systolic function were not seen [107], contrasting with systolic changes reported in patients and electromechanical simulations [97, 98]. Moderate electrical remodeling involving IKs, IK1 and L-type ICa was observed [107]. Significantly, similar to patient and simulation studies, quinidine normalized ventricular repolarization [107]. The generation of the first mammalian genetic model of the SQTS represents a significant advance for the field. A model precisely recapitulating the situation for N588K hERG patients would involve heterozygous expression of the analogous N➔K mutation in rabbit erg. In the absence of such a model, it remains to be established whether overexpression of a human transgene results in differences from the patient situation of heterozygous expression of mutated endogenous IKr.
Implications from preclinical SQTS studies for the therapeutic deployment of K+ channel activators
There has been interest in the development and potential use of K+ channel activators as novel antiarrhythmic agents, particularly in the setting of pathologically prolonged repolarization, where deployment of a K+ channel activator may act to normalize repolarization [108,109,110]. The presence of endogenous KATP channels comprised of Kir 6.x and SUR2A subunits, which only conduct significant current when cellular ATP falls, may exert some protective effects for the heart in situations of cardiac ischaemia or hypoxia, although it can lead to AP triangulation and associated arrhythmia risk [109, 111]. There is some evidence that administration of KATP channel activators in patients with LQTS mutations exerts beneficial effects on repolarization [112,113,114]. On the other hand, the successful deployment of KATP channel activation by pinacidil to produce experimental models of the SQTS [100,101,102] suggests that some caution is warranted in the use of such a strategy. If excessive ventricular APD shortening occurs as a result of KATP channel activation the result may be proarrhythmic. Similarly, the use of the hERG activator PD118057 to model ventricular and atrial arrhythmic substrates in the SQTS [103, 104] highlights that augmenting IKr from a baseline of normal repolarization also carries risk. Consistent with this, Lu and colleagues have highlighted potential risks of QT shortening drugs in a study in which hERG activators (mallotoxin and NS1643) and KATP activators (levcromokalin and nicorandil) significantly abbreviated the QT interval and elicited ventricular fibrillation in isolated perfused rabbit hearts [115]. Additionally, a recent study has shown marked proarrhythmic effects of KATP and hERG activators on guinea-pig hearts at high concentrations and has suggested the use of J-Tpeak interval and JT area as biomarkers of arrhythmia risk with QT shortening drugs [116]. Such observations do not preclude entirely the development or therapeutic deployment of K channel activators; agents that abbreviate repolarization whilst also prolonging the post-repolarization refractory period (PRRP) may in some circumstances be beneficial [117]. What the data from pharmacological QT shortening teach us is that the most likely circumstances for beneficial deployment would be those in which baseline ventricular repolarization is already delayed and that care would be needed to avoid excessive ventricular AP shortening and the unintentional production of an SQTS phenotype. Moreover, the effectiveness of KATP activators in recapitulating an SQTS phenotype would perhaps make it not surprising if gain-of-function mutations to KATP channel subunits were to be identified in the future in clinical SQTS cases.
Insights from preclinical pharmacological studies of the SQTS
Due to the high risk of sudden death in the SQTS [118] the use of implantable cardioverter defibrillators (ICDs) is warranted, particularly in patients who have survived cardiac arrest or who have document episodes of ventricular arrhythmia [24]. However, the changes in T wave height in SQTS ECGs can lead to T wave oversensing and inappropriate ICD shocks [11, 119,120,121]. Young patients in particular have a high rate of inappropriate ICD shocks [118]. Also, ICDs do not normalize repolarization or the arrhythmia substrate and so additional, pharmacological, approaches are desirable to help normalize repolarization and reduce arrhythmia burden.
(Hydro) quinidine has proved effective in a number of SQTS patients and at present is probably the pharmacological treatment of choice [13, 92]. SQTS cohort findings have shown a high level of effectiveness of hydroquinidine in preventing life-threatening proarrhythmic events during long term follow up [122]. In studies of SQT1 patients with the N588K hERG mutation quinidine but not sotalol retained antiarrhythmic effectiveness, prolonging repolarization, ventricular ERP and protecting against VF [41, 123]. Sotalol and other class III drugs that selectively inhibit hERG/IKr show a strong dependence on intact hERG channel inactivation to block the channel effectively [124,125,126,127,128,129]. The N588K mutation significantly impairs sotalol block of hERG channels [41, 123], whilst quinidine is comparatively little affected [126, 128]. The S631A mutation, also recently found in an SQTS family, retains sensitivity to quinidine too [47, 124, 129]; from this one can predict that quinidine should retain effectiveness in patients with this mutation. Disopyramide, a second class Ia antiarrhythmic agent, also retains effectiveness against N588K and S631A channels [125, 128, 129] and has been tested successfully in SQT1 patients and a patient with unknown genotype [130, 131]. Computational analysis of quinidine and disopyramide in the setting of N588K-linked SQT1 has demonstrated that normative effects on repolarization arise from the drug’s IKr blocking action, whilst ERP prolongation results from a combination of IKr block and actions on INa [132]. The fact that the N588 residue lies outside the hERG channel pore means that the N588K mutation provides a means of interrogating inactivation dependence of hERG blockade. Since its discovery in SQTS, this has been exploited in mechanistic studies of a range of cardiac and non-cardiac drugs in addition to quinidine and disopyramide, including amiodarone, astemizole, cisapride, dofetilide, doxepin, E-4031, flecainide, ivabradine, lignocaine, ranolazine and terfenadine [127, 129, 133,134,135,136].
In the ventricular wedge model of SQT1 using the hERG activator PD118057, quinidine effectively prolonged QT interval and ERP suggestive that it would be effective in other settings with augmented IKr [103]. Considering known effects of hERG inactivation modulation on drug sensitivity of hERG, it is likely that for those SQT1 variants with profoundly altered inactivation, quinidine and disopyramide would be reasonable first line antiarrhythmic choices, whilst sotalol may be ineffective. One notable potential exception to quinidine effectiveness in SQT1 is the T618I mutation, for which in vitro data suggest retained effectiveness of quinidine [44, 45]. However, whilst quinidine prolonged QTc interval it did not prevent ventricular arrhythmias in all recipients [22]. This suggests that further work is needed to understand the relationship between in vitro and in vivo pharmacology of this SQT mutation. Bepridil was found to be effective in a case of T618I linked VF unresponsive to other treatment and the in vitro study of interactions between T618I-hERG and bepridil to elucidate this effect is likely to be instructive.
Quinidine has been reported to exert beneficial effects in non-SQT1 variants of the SQTS [36, 71], though with stronger effects in SQT1 than non-SQT1 variants [71]. In a recent simulation study of atrial arrhythmia substrates in SQT2, simulated quinidine application increased atrial ERP for both V307L and V141M KCNQ1 mutations. However, it was not effective at restoring APD in the V141M setting [78] though it decreased the dominant frequency of excitation for both mutations, consistent with a potential role for rate if not rhythm control. The in silico observations regarding lack of effectiveness of quinidine for rhythm control with V141M correspond well to clinical findings in which recurrent AF associated with the V141M KCNQ1 mutation failed to respond to antiarrhythmic agents including quinidine [77]. There are no selective IKs inhibitors in clinical use, but preclinical experiments have suggested that the IKs inhibitor HMR-1556 actually exhibits enhanced affinity for V141M KCNQ1 and, in principle, would be expected to mitigate effects on repolarization of this mutation [137]. Simulations of the effects of the V307L mutation on ventricular cell and tissue electrophysiology suggest that ~ 60% IKs inhibition would be likely to normalize repolarization in this SQT2 variant [75]. The location of the V307 residue is in the binding region for canonical, chromanol based IKs inhibitors and IKs incorporating the V307L mutation exhibits reduced sensitivity to chromanol 293B [138]. The antimalarial drug mefloquine is an effective IKs inhibitor [139] and has been shown to block recombinant V307L-KCNQ1 + KCNE1 (recapitulating IKs) [50]. This highlights a potential value for non-canonical IKs inhibitors and establishing the mefloquine binding site on KCNQ1 may be helpful in respect of potential future IKs inhibitor development.
There are currently no selective IK1 inhibitors in clinical use and because the role of IK1 is normally confined to terminal repolarization selective IK1 blockade might in any event be of limited value for non-SQT3 variants of the SQTS [13]. There is experimental and simulation evidence, however, that IK1 inhibition might in principle be of value for SQT3. The antimalarial drug chloroquine inhibits native IK1 from ventricular myocytes, with a preference for the outward over inward current component [140]. Chloroquine has been shown to be able to inhibit effectively D172N Kir2.1 mutant channels and, in simulations, to prolong repolarization and ventricular ERP [80, 141, 142] Auxiliary effects on IKr may contribute to the drug’s effect [141, 142]. The E299 residue lies in the binding site for chloroquine [143] so it is quite likely that the drug would not be effective against the E299V SQT3 variant, whilst mutation of M301 did not impair chloroquine block [143] and so it may be effective against the M301K mutation. Recent work has discovered a pentamidine analogue PA-6 that shows inhibitory selectivity for IK1 [144] and reduces D172N-Kir2.1 current [145]. Our recent simulation work has shown in respect of atrial electrophysiology that 50% inhibition of IK1 alone was sufficient to prevent re-entry in E299V but not D17N mutant conditions; on the other hand, simulated combined IK1+ IKr inhibition was effective at terminating reentry for both mutations [95]. Interestingly, however, partial inhibition of IK1 precluded complete excitation of the pulmonary vein area in E299V conditions, whereas 50% inhibition of IKr alone was sufficient to terminate reentry. This highlights both the complexity of mutation effects and the value of exploring these in silico. The ultra-rapid delayed rectifier K+ current, IKur, has received considerable attention as a potential atrio-selective Class III antiarrhythmic target [146, 147]. In SQT3 simulations, 50% inhibition of IKur decreased the dominant frequency of excitation for both D172N and E299V mutations, but did not terminate re-entry. When this effect was combined with 50% inhibition of IKr reentry was terminated for both mutations. This highlights a potentially effective Class III combination effect against this SQT variant.
From a therapeutic standpoint the overall pharmacological findings in respect of SQTS suggest that some drug interventions may be of value in one genotype but of no benefit (or perhaps even hazardous) in another. Moreover, it is clear that the use of a drug that carries possible side effects with no predicted therapeutic benefit is poor medicine and wasteful of scarce resources. The data from preclinical studies can inform judicious drug selection and development of novel treatment strategies.
Conclusion
There is clear synergy between characterization of the clinical phenotype of a rare genetic syndrome like the SQTS, genotyping and preclinical studies that can establish causality, provide mechanistic insights into the basis of clinically observed effects and a rational basis for the deployment of existing pharmacological treatments or ‘proof of concept’ for the pursuit of novel approaches. As evidenced for the SQTS, efforts to understand the underlying basis of particular clinical phenotypes can in turn provide insight into the role of particular biophysical properties of identified ion channels in controlling or modifying the underlying biological process – in this case cardiac repolarization. In turn this can lead to a rational use of medications which may modify the ion channel in a beneficial way and potentially improve prognosis. The comparatively low success rate in targeted genotyping of SQTS cases highlights that there is probably still much that we still do not understand about modifiers of cardiac repolarization; it follows that the identification of hitherto unknown associations between particular genes and the syndrome promises to drive a broader and deeper understanding of this essential process.
Abbreviations
- AF:
-
Atrial fibrillation
- AP:
-
Action potential
- APD:
-
Action potential duration
- Ca2+ :
-
Calcium
- ERP:
-
Effective refractory period
- hERG :
-
Human ether-à-go-go related gene
- I K1 :
-
Inward rectifier K+ current
- I Kr :
-
Rapid delayed rectifier K+ current
- I Ks :
-
Slow delayed rectifier K+ current
- I Na :
-
Fast sodium current
- K+ :
-
Potassium ion
- Kir2.1:
-
Pore forming subunit of channels mediating IK1
- LQTS:
-
Long QT syndrome
- PCD:
-
Primary carnitine deficiency
- PRRP:
-
Post repolarization refractory period
- SQT:
-
Short QT
- SQTS:
-
Short QT syndrome
- VF:
-
Ventricular fibrillation
- VT:
-
Ventricular tachycardia
References
Ackerman MJ. Genetic purgatory and the cardiac channelopathies: exposing the variants of uncertain/unknown significance issue. Heart Rhythm. 2015;12(11):2325–31.
Campbell TJ. Characteristics of cardiac action potentials in marsupials. J Comp Physiol B. 1989;158(6):759–62.
Rezakhani A, Webster JD, Atwell RB. The electrocardiogram of the eastern grey kangaroo (Macropus giganteus). Aust Vet J. 1986;63(9):310–2.
Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QT interval variables from 24 hour electrocardiography and the two year risk of sudden death. Br Heart J. 1993;70(1):43–8.
Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med. 1990;227(3):211–3.
Fei L, Camm AJ. Shortening of the QT interval immediately preceding the onset of idiopathic spontaneous ventricular tachycardia. Am Heart J. 1995;130(4):915–7.
Viskin S, Zeltser D, Ish-Shalom M, Katz A, Glikson M, Jutso D, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls? Heart Rhythm. 2004;1(5):587–91.
Poglajen G, Fister M, Radovancevic B, Vrtovec B. Short QT interval and atrial fibrillation in patients without structural heart disease. J Am Coll Cardiol. 2006;47:1905–7.
Nielsen JB, Graff C, Pietersen A, Lind B, Struijk JJ, Olesen MS, et al. J-shaped association between QTc interval duration and the risk of atrial fibrillation: results from the Copenhagen ECG study. J Am Coll Cardiol. 2013;61(25):2557–64.
Gussak I, Brugada P, Brugada J, Wright RS, Kopecky SI, Chaitman BR, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99–102.
Maury P, Extramiana F, Sbragia P, Giustetto C, Schimpf R, Duparc A, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis. 2008;101(11–12):779–86.
Giustetto C, Di MF, Wolpert C, Borggrefe M, Schimpf R, Sbragia P, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J. 2006;27(20):2440–7.
Hancox JC, Whittaker DG, Du C, Stuart AG, Zhang H. Emerging therapeutic targets in the short QT syndrome. Expert Opin Ther Targets. 2018;22(5):439–51.
Mazzanti A, Kanthan A, Monteforte N, Memmi M, Bloise R, Novelli V, et al. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol. 2014;63(13):1300–8.
Harrell DT, Ashihara T, Ishikawa T, Tominaga I, Mazzanti A, Takahashi K, et al. Genotype-dependent differences in age of manifestation and arrhythmia complications in short QT syndrome. Int J Cardiol. 2015;190:393–402.
Roussel J, Labarthe F, Thireau J, Ferro F, Farah C, Roy J, et al. Carnitine deficiency induces a short QT syndrome. Heart Rhythm. 2016;13(1):165–74.
Perin F, Rodriguez-Vazquez Del Rey MDM, Carreras-Blesa C, Rrabal-Fernandez L, Jimenez-Jaimez J, Tercedor L. Dilated Cardiomyopathy With Short QT Interval Suggests Primary Carnitine Deficiency. Rev Esp Cardiol (Engl Ed). 2018;71(12):1074–75.
Hancox JC. Primary carnitine deficiency as a potential cause of short QT syndrome. Cardiovasc Res Med. 2018;2(1):10–2.
Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation. 2007;116(7):714–20.
Dhutia H, Malhotra A, Parpia S, Gabus V, Finocchiaro G, Mellor G, et al. The prevalence and significance of a short QT interval in 18,825 low-risk individuals including athletes. Br J Sports Med. 2016;50(2):124–9.
Mazzanti A, Underwood K, Nevelev D, Kofman S, Priori SG. The new kids on the block of arrhythmogenic disorders: short QT syndrome and early repolarization. J Cardiovasc Electrophysiol. 2017;28(10):1226–36.
Hu D, Li Y, Zhang J, Pfeiffer R, Gollob MH, Healey J, et al. The phenotypic Spectrum of a mutation hotspot responsible for the short QT syndrome. JACC: Clin Electrophysiol. 2017;J3(7):727–43.
Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol. 2011;57(7):802–12.
Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the Management of Patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36(41):2793–867.
Providencia R, Karim N, Srinivasan N, Honarbakhsh S, Vidigal Ferreira MJ, Goncalves L, et al. Impact of QTc formulae in the prevalence of short corrected QT interval and impact on probability and diagnosis of short QT syndrome. HEART. 2018;104(6):502–8.
Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol. 2009;25(4):e133–5.
Terlemez S, Cil E, Kula S, Oguz AD, Tunaoglu FS. A diagnosis that escapes our attention: short QT syndrome. Gazi Med J. 2018;29(3):246–8.
Tulumen E, Giustetto C, Wolpert C, Maury P, Anttonen O, Probst V, et al. PQ segment depression in patients with short QT syndrome: a novel marker for diagnosing short QT syndrome? Heart Rhythm. 2014;11(6):1024–30.
Wakatsuki D, Iso Y, Mase H, Kurata M, Kyuno E, Shimojima H, et al. Sudden cardiac arrest during marathon training in a young adult with short QT syndrome. Int J Cardiol Heart Vasc. 2018;18:101–3.
Rollin A, Gandjbakhch E, Giustetto C, Scrocco C, Fourcade C, Monteil B, et al. Shortening of the short refractory periods in short QT syndrome. J Am Heart Assoc. 2017;31:6(6).
Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299(5604):251–4.
Kharche S, Adeniran I, Stott J, Law P, Boyett MR, Hancox JC, et al. Pro-arrhythmogenic effects of the S140G KCNQ1 mutation in human atrial fibrillation - insights from modelling. J Physiol. 2012;590(Pt 18):4501–14.
El Harchi A, Zhang H, Hancox JC. The S140G KCNQ1 atrial fibrillation mutation affects 'I (KS)' profile during both atrial and ventricular action potentials. J Physiol Pharmacol. 2010;61(6):759–64.
Hancox JC, Kharche S, El Harchi A, Stott J, Law P, Zhang H. In silico investigation of a KCNQ1 mutation associated with familial atrial fibrillation. J Electrocardiol. 2014;47(2):158–65.
Wilders R, Verkerk AO. Role of the R1135H KCNH2 mutation in Brugada syndrome. Int J Cardiol. 2009;25.
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115(4):442–9.
Watanabe H, Makiyama T, Koyama T, Kannankeril PJ, Seto S, Okamura K, et al. High prevalence of early repolarization in short QT syndrome. Heart Rhythm. 2010;7(5):647–52.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;76(1):7.20.1–7.20.41.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Paquin A, Ye D, Tester DJ, Kapplinger JD, Zimmermann MT, Ackerman MJ. Even pore-localizing missense variants at highly conserved sites in KCNQ1-encoded Kv7.1 channels may have wild-type function and not cause type 1 long QT syndrome: do not rely solely on the genetic test company's interpretation. HeartRhythm Case Rep. 2018;4(2):37–44.
Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–5.
Cordeiro JM, Brugada R, Wu YS, Hong K, Dumaine R. Modulation of IKr inactivation by mutation N588K in KCNH2: a link to arrhythmogenesis in short QT syndrome. Cardiovasc Res. 2005;67:498–509.
McPate MJ, Duncan RS, Milnes JT, Witchel HJ, Hancox JC. The N588K-HERG K+ channel mutation in the 'short QT syndrome': mechanism of gain-in-function determined at 37°C. Biochem Biophys Res Comm. 2005;334:441–9.
Sun Y, Quan XQ, Fromme S, Cox RH, Zhang P, Zhang L, et al. A novel mutation in the KCNH2 gene associated with short QT syndrome. J Mol Cell Cardiol. 2011;50(3):433–41.
El Harchi A, Melgari D, Zhang YH, Zhang H, Hancox JC. Action potential clamp and pharmacology of the variant 1 short QT syndrome T618I hERG K+ channel. PLoS One. 2012;7(12):e52451.
Itoh H, Sakaguichi T, Ashihara T, Ding WG, Nagaoka I, Oka Y, et al. A novel KCNH2 interval as a modifier for short QT interval. Int J Cardiol. 2009;137(1):83–5.
Butler A, Zhang Y, Stuart AG, Dempsey CE, Hancox JC. Action potential clamp characterization of the S631A hERG mutation associated with short QT syndrome. Physiol Rep. 2018 Sep;6(17):e13845.
Bellocq C, van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004 May 25;109(20):2394–7.
Hong K, Piper DR, Diaz-valdecantos A, Brugada J, Oliva A, Burashnikov E, et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. 2005;68(3):433–40.
El Harchi A, McPate MJ, Zhang YH, Zhang H, Hancox JC. Action potential clamp and mefloquine sensitivity of recombinant 'IKS' channels incorporating the V307L KCNQ1 mutation. J Physiol Pharmacol. 2010;61(2):123–31.
Wu ZJ, Huang Y, Fu YC, Zhao XJ, Zhu C, Zhang Y, et al. Characterization of a Chinese KCNQ1 mutation (R259H) that shortens repolarization and causes short QT syndrome 2. J Geriatr Cardiol. 2015;12(4):394–401.
Moreno C, Oliveras A, de la CA BC, Munoz C, Salar E, et al. a new KCNQ1 mutation at the S5 segment that impairs its association with KCNE1 is responsible for short QT syndrome. Cardiovasc Res. 2015;107(4):613–23.
Priori SG, Pandit SV, Rivolta I, Berenfeld O, Ronchetti E, Dhamoon A, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96(7):800–7.
Hattori T, Makiyama T, Akao M, Ehara E, Ohno S, Iguchi M, et al. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of Kir2.1 currents. Cardiovasc Res. 2012;93(4):666–73.
Deo M, Ruan Y, Pandit SV, Shah K, Berenfeld O, Blaufox A, et al. KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc Natl Acad Sci U S A. 2013;110(11):4291–6.
Chen Y, Barajas-Martinez H, Zhu D, Wang X, Chen C, Zhuang R, et al. Erratum to: novel trigenic CACNA1C/DES/MYPN mutations in a family of hypertrophic cardiomyopathy with early repolarization and short QT syndrome. J Transl Med. 2017;15(1):101.
Chen Y, Barajas-Martinez H, Zhu D, Wang X, Chen C, Zhuang R, et al. Novel trigenic CACNA1C/DES/MYPN mutations in a family of hypertrophic cardiomyopathy with early repolarization and short QT syndrome. J Transl Med. 2017;15(1):78.
Hong K, Hu J, Yu J, Brugada R. Concomitant Brugada-like and short QT electrocardiogram linked to SCN5A mutation. Eur J Hum Genet. 2012;20(11):1189–92.
Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur Heart J. 2011;32(9):1077–88.
Liu J, Zhang M, Jiang M, Tseng GN. Structural and functional role of the extracellular S5-P linker in the HERG potassium channel. J Gen Physiol. 2002;120:723–37.
Dun W, Jiang M, Tseng GN. Allosteric effects of mutations in the extracellular S5-P loop on the gating and ion permeation properties of the hERG potassium channel. Pflugers Arch. 1999;439(1–2):141–9.
Torres AM, Bansal PS, Sunde M, Clarke CE, Bursill JA, Smith DJ, et al. Structure of the HERG K+ channel S5P extracellular linker: role of an amphipathic alpha-helix in C-type inactivation. J Biol Chem. 2003;278(43):42136–48.
Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, et al. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J. 1998;74:230–41.
Hancox JC, Levi AJ, Witchel HJ. Time course and voltage dependence of expressed HERG current compared with native 'rapid' delayed rectifier K current during the cardiac ventricular action potential. Pflugers Archiv - Eur J Physiol. 1998;436:843–53.
Zhang H, Hancox JC. In silico study of action potential and QT interval shortening due to loss of inactivation of the cardiac rapid delayed rectifier potassium current. Biochem Biophys Res Commun. 2004;322(2):693–9.
Itoh H, Horie M, Ito M, Imoto K. Arrhythmogenesis in the short-QT syndrome associated with combined HERG channel gating defects: a simulation study. Circ J. 2006;70(4):502–8.
Adeniran I, McPate MJ, Witchel HJ, Hancox JC, Zhang H. Increased vulnerability of human ventricle to re-entrant excitation in hERG-linked variant 1 short QT syndrome. PLoS Comput Biol. 2011;7(12):e1002313.
Zou A, Curran ME, Keating MT, Sanguinetti MC. Single HERG delayed rectifier K+ channels expressed in Xenopus oocytes. Am J Phys. 1997;272:H1309–14.
Schoenherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol. 1996;493(3):635–42.
Akdis D, Saguner AM, Medeiros-Domingo A, Schaller A, Balmer C, Steffel J, et al. Multiple clinical profiles of families with the short QT syndrome. Europace. 2017;20(FI1):f113–21.
Giustetto C, Schimpf R, Mazzanti A, Scrocco C, Maury P, Anttonen O, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol. 2011;58(6):587–95.
Wang QI, Ohno S, Ding WG, Fukuyama M, Miyamoto A, Itoh H, et al. Gain-of-function KCNH2 mutations in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2014;25(5):522–30.
Cheng YJ, Yao H, Ji CC, Chen XM, Fan J, Liu LJ, et al. A heterozygous missense hERG mutation associated with early repolarization syndrome. Cell Physiol Biochem. 2018;51(3):1301–12.
Zhang H, Kharche S, Holden AV, Hancox JC. Repolarisation and vulnerability to re-entry in the human heart with short QT syndrome arising from KCNQ1 mutation--a simulation study. Prog Biophys Mol Biol. 2008;96(1–3):112–31.
Adeniran I, Whittaker DG, El HA, Hancox JC, Zhang H. In silico investigation of a KCNQ1 mutation associated with short QT syndrome. Sci Rep. 2017;7(1):8469.
Villafane J, Fischbach P, Gebauer R. Short QT syndrome manifesting with neonatal atrial fibrillation and bradycardia. Cardiology. 2014;128(3):236–40.
Righi D, Silvetti MS, Drago F. Sinus bradycardia, junctional rhythm, and low-rate atrial fibrillation in short QT syndrome during 20 years of follow-up: three faces of the same genetic problem. Cardiol Young. 2016;26(3):589–92.
Whittaker DG, Colman MA, Ni H, Hancox JC, Zhang H. Human atrial arrhythmogenesis and Sinus Bradycardia in KCNQ1-Linked Short QT Syndrome: Insights From Computational Modelling. Front Physiol. 2018;9:1402. https://doi.org/10.3389/fphys.2018.01402.
Ambrosini E, Sicca F, Brignone MS, D'Adamo MC, Napolitano C, Servettini I, et al. Genetically induced dysfunctions of Kir2.1 channels: implications for short QT3 syndrome and autism-epilepsy phenotype. Hum Mol Genet. 2014;23(18):4875–86.
El Harchi A, McPate MJ, Zhang YH, Zhang H, Hancox JC. Action potential clamp and chloroquine sensitivity of mutant Kir2.1 channels responsible for variant 3 short QT syndrome. J Mol Cell Cardiol. 2009;137:83–5.
Adeniran I, El HA, Hancox JC, Zhang H. Proarrhythmia in KCNJ2-linked short QT syndrome: insights from modelling. Cardiovasc Res. 2012;94(1):66–76.
Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpon E, Hu D, Desai M, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm. 2010;7(12):1872–82.
Sottas V, Rougier JS, Jousset F, Kucera JP, Shestak A, Makarov LM, et al. Characterization of 2 genetic variants of Na(v) 1.5-arginine 689 found in patients with cardiac arrhythmias. J Cardiovasc Electrophysiol. 2013;24(9):1037–46.
Zimmer T, Haufe V, Blechschmidt S. Voltage-gated sodium channels in the mammalian heart. Glob Cardiol Sci Pract. 2014;2014(4):449–63.
Di SG, Palumbo P, Castellana S, Mastroianno S, Biagini T, Palumbo O, et al. Sudden cardiac death in J wave syndrome with short QT associated to a novel mutation in Nav 1.8 coding gene SCN10A: first case report for a possible pharmacogenomic role. J Electrocardiol. 2018;51(5):809–13.
Thorsen K, Dam VS, Kjaer-Sorensen K, Pedersen LN, Skeberdis VA, Jurevicius J, et al. Loss-of-activity-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat Commun. 2017;8(1):1696.
Tamai I, Ohashi R, Nezu J, Yabuuchi H, Oku A, Shimane M, et al. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem. 1998;273(32):20378–82.
Iwata D, Kato Y, Wakayama T, Sai Y, Kubo Y, Iseki S, et al. Involvement of carnitine/organic cation transporter OCTN2 (SLC22A5) in distribution of its substrate carnitine to the heart. Drug Metab Pharmacokinet. 2008;23(3):207–15.
Fu L, Huang M, Chen S. Primary carnitine deficiency and cardiomyopathy. Korean Circ J. 2013;43(12):785–92.
Ferro F, Ouille A, Tran TA, Fontanaud P, Bois P, Babuty D, et al. Long-chain acylcarnitines regulate the hERG channel. PLoS One. 2012;7(7):e41686.
Nerbonne JM, Nichols CG, Schwarz TL, Escande D. Genetic manipulation of cardiac K(+) channel function in mice: what have we learned, and where do we go from here? Circ Res. 2001;89(11):944–56.
Perike S, McCauley MD. Molecular insights into short QT syndrome. J Innovat Cardiac Rhythm Mgmt. 2018;9:3065–70.
Cerrone M, Noujaim S, Jalife J. The short QT syndrome as a paradigm to understand the role of potassium channels in ventricular fibrillation. J Intern Med. 2006;259:24–38.
Samie FH, Berenfeld O, Anumonwo J, Mironov SF, Udassi S, Beaumont J, et al. Rectification of the background potassium current: a determinant of rotor dynamics in ventricular fibrillation. Circ Res. 2001;89(12):1216–23.
Whittaker DG, Ni H, Harchi AE, Hancox JC, Zhang H. Atrial arrhythmogenicity of KCNJ2 mutations in short QT syndrome: insights from virtual human atria. PLoS Comput Biol. 2017;13(6):e1005593.
Kharche S, Garratt CJ, Boyett MR, Inada S, Holden AV, Hancox JC, et al. Atrial proarrhythmia due to increased inward rectifier current (I(K1)) arising from KCNJ2 mutation - a simulation study. Prog Biophys Mol Biol. 2008;96:112–31.
Adeniran I, Hancox JC, Zhang H. In silico investigation of the short QT syndrome, using human ventricle models incorporating electromechanical coupling. Front Physiol. 2013;4:166.
Frea S, Giustetto C, Capriolo M, Scrocco C, Fornengo C, Benedetto S, et al. New echocardiographic insights in short QT syndrome: more than a channellopathy? Heart Rhythm. 2015;12(10):2096–105.
Frea S, Pidello S, Giustetto C, Scrocco C, Gaitan F. Author's reply to "altered in vivo systolic function in the short QT syndrome anticipated in silico". Heart Rhythm. 2015;12(9):e115–6.
Extramiana F, Antzelevitch C. Amplified transmural dispersion of repolarization as the basis for arrhythmogenesis in a canine ventricular-wedge model of short-QT syndrome. Circulation. 2004;110(24):3661–6.
Frommeyer G, Ellermann C, Dechering DG, Kochhauser S, Bogeholz N, Guner F, et al. Ranolazine and Vernakalant Prevent Ventricular Arrhythmias in an Experimental Whole-Heart Model of Short QT Syndrome. J Cardiovasc Electrophysiol. 2016;27(10):1214–19.
Frommeyer G, Weller J, Ellermann C, Kaese S, Kochhauser S. Lange PS, et al. Clin Exp Pharmacol Physiol: Antiarrhythmic properties of ivabradine in an experimental model of Short-QT-Syndrome; 2017;44(9):941–45.
Patel C, Antzelevitch C. Cellular basis for arrhythmogenesis in an experimental model of the SQT1 form of the short QT syndrome. Heart Rhythm. 2008;5(4):585–90.
Nof E, Burashnikov A, Antzelevitch C. Cellular basis for atrial fibrillation in an experimental model of short QT1: implications for a pharmacological approach to therapy. Heart Rhythm. 2010;7(2):251–7.
El-Battrawy I, Lan H, Cyganek L, Zhao Z, Li X, Buljubasic F, et al. Modeling short QT syndrome using human-induced pluripotent stem cell-derived cardiomyocytes. J Am Heart Assoc. 2018;24:7(7).
Guo F, Sun Y, Wang X, Wang H, Gong T, Chen X, et al. Patient specific and gene corrected induced pluripotent stem-cell derived cardiomyocytes elucidate single cell phenotype of short QT syndrome. Circ Res. 2019;124(1):66–78.
Odening KE, Bodi I, Franke G, Rieke R, de MA R, Perez-Feliz S, et al. Transgenic short-QT syndrome 1 rabbits mimic the human disease phenotype with QT/action potential duration shortening in the atria and ventricles and increased ventricular tachycardia/ventricular fibrillation inducibility. Eur Heart J. 2018;40(10):842–53.
Szabo G, Farkas V, Grunnet M, Mohacsi A, Nanasi PP. Enhanced Repolarization Capacity: New Potential Antiarrhythmic Strategy Based on hERG Channel Activation. Curr Med Chem. 2011.
Grunnet M. Repolarization of the cardiac action potential. Does an increase in repolarization capacity constitute a new anti-arrhythmic principle? Acta Physiol (Oxf). 2010 Feb;198(Suppl 676):1–48.
Testai L, Cecchetti V, Sabatini S, Martelli A, Breschi MC, Calderone V. Effects of K openers on the QT prolongation induced by HERG-blocking drugs in Guinea-pigs. J Pharm Pharmacol. 2010;62(7):924–30.
Tamargo J, Caballero R, Gomez R, Valenzuela C, Delpon E. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004;62(1):9–33.
Chinushi M, Kasai H, Tagawa M, Washizuka T, Hosaka Y, Chinushi Y, et al. Triggers of ventricular tachyarrhythmias and therapeutic effects of nicorandil in canine models of LQT2 and LQT3 syndromes. J Am Coll Cardiol. 2002;40(3):555–62.
Aizawa Y, Uchiyama H, Yamaura M, Nakayama T, Arita M. Effects of the ATP-sensitive K channel opener nicorandil on the QT interval and the effective refractory period in patients with congenital long QT syndrome. Investigator Group for K-channel openers and arrhythmias. J Electrocardiol. 1998;31(2):117–23.
Shimizu W, Kurita T, Matsuo K, Suyama K, Aihara N, Kamakura S, et al. Improvement of repolarization abnormalities by a K+ channel opener in the LQT1 form of congenital long-QT syndrome. Circulation. 1998;97(16):1581–8.
Lu HR, Vlaminckx E, Hermans AN, Rohrbacher J, Van AK, Towart R, et al. Predicting drug-induced changes in QT interval and arrhythmias: QT-shortening drugs point to gaps in the ICHS7B guidelines. Br J Pharmacol. 2008;154(7):1427–38.
Qiu B, Wang Y, Li C, Guo H, Xu Y. Utility of the JT peak interval and the JT area in determining the Proarrhythmic potential of QT-shortening agents. J Cardiovasc Pharmacol Ther. 2019;24(2):160–17.
Grunnet M, Hansen RS, Olesen SP. hERG1 channel activators: a new anti-arrhythmic principle. Prog Biophys Mol Biol. 2008;98(2–3):347–62.
Villafane J, Atallah J, Gollob MH, Maury P, Wolpert C, Gebauer R, et al. Long-Term Follow-Up of a Pediatric Cohort With Short QT Syndrome. J Am Coll Cardiol. 2013;61(11):1183–91.
Schimpf R, Wolpert C, Gaita F, Giustetto C, Borggrefe M. Short QT syndrome. Cardiovasc Res. 2005;67(3):357–66.
Schimpf R, Wolpert C, Bianchi F, Giustetto C, Gaita F, Bauersfeld U, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol. 2003;14(12):1273–7.
Schimpf R, Bauersfeld U, Gaita F, Wolpert C. Short QT syndrome: successful prevention of sudden cardiac death in an adolescent by implantable cardioverter-defibrillator treatment for primary prophylaxis. Heart Rhythm. 2005;2(4):416–7.
Mazzanti A, Maragna R, Vacanti G, Kostopoulou A, Marino M, Monteforte N, et al. Hydroquinidine prevents life-threatening arrhythmic events in patients with short QT syndrome. J Am Coll Cardiol. 2017;70(24):3010–5.
Hong K, Bjeerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J Cardivas Electophysiol. 2005;16:394–6.
Lees-Miller JP, Duan Y, Teng GQ, Duff HJ. Molecular determinant of high affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol. 2000;57:367–74.
Paul AA, Witchel HJ, Hancox JC. Inhibition of HERG potassium channel current by the class 1a antiarrhythmic agent disopyramide. Biochem Biophys Res Comm. 2001;280:1243–50.
Wolpert C, Schimpf R, Giustetto C, Antzelevitch C, Cordeiro JM, Dumaine R, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electophysiol. 2005;16:54–8.
Perrin MJ, Kuchel PW, Campbell TJ, Vandenberg JI. Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-à-go-go-related gene channels. Mol Pharmacol. 2008;74:1443–52.
McPate MJ, Duncan RS, Witchel HJ, Hancox JC. Disopyramide is an effective inhibitor of mutant HERG K+ channels involved in variant 1 short QT syndrome. J Mol Cell Cardiol. 2006;41:563–6.
McPate MJ, Duncan RS, Hancox JC, Witchel HJ. Pharmacology of the short QT syndrome N588K-hERG K+ channel mutation: differential impact on selected class I and class III antiarrhythmic drugs. Br J Pharmacol. 2008;155:957–66.
Schimpf R, Veltmann C, Giustetto C, Gaita F, Borgreffe M, Wolpert C. In vivo effects of mutant HERG K+ channel inhibition by disopyramide in patients with a short QT-1 syndrome: a pilot study. J Cardiovasc Electophysiol. 2007;18:1157–60.
Mizobuchi M, Enjoji Y, Yamamoto R, Ono T, Funatsu A, Kambayashi D, et al. Nifekalant and disopyramide in a patient with short QT syndrome: evaluation of pharmacological effects and electrophysiological properties. Pace - Pacing and Clinical Electrophysiology. 2008;31:1229–32.
Whittaker DG, Ni H, Benson AP, Hancox JC, Zhang H. Computational analysis of the mode of action of Disopyramide and quinidine on hERG-linked short QT syndrome in human ventricles. Front Physiol. 2017;8:759. https://doi.org/10.3389/fphys.2017.00759
Duncan RS, McPate MJ, Ridley JM, Gao Z, James AF, Leishman DJ, et al. Inhibition of the HERG potassium channel by the tricyclic antidepressant doxepin. Biochem Pharmacol. 2007;74:425–37.
Melgari D, Zhang Y, El Harchi A, Dempsey CE, Hancox JC. Molecular basis of hERG potassium channel blockade by the class Ic antiarrhythmic flecainide. J Mol Cell Cardiol. 2015;86:42–53.
Du C, Zhang Y, El Harchi A, Dempsey CE, Hancox JC. Ranolazine inhibition of hERG potassium channels: drug-pore interactions and reduced potency against inactivation mutants. J Mol Cell Cardiol. 2014 May 27;74C:220–30.
Melgari D, Brack KE, Zhang C, Zhang Y, El HA, Mitcheson JS, et al. hERG potassium channel blockade by the HCN channel inhibitor bradycardic agent ivabradine. J Am Heart Assoc. 2015:4(4).
Campbell CM, Campbell JD, Thompson CH, Galimberti ES, Darbar D, Vanoye CG, et al. Selective targeting of gain-of-function KCNQ1 mutations predisposing to atrial fibrillation. Circ Arrhythm Electrophysiol. 2013;6(5):960–6.
Lerche C, Bruhova I, Lerche H, Steinmeyer K, Wei AD, Strutz-Seebohm N, et al. Chromanol 293B binding in KCNQ1 (Kv7.1) channels involves electrostatic interactions with a potassium ion in the selectivity filter. Mol Pharmacol. 2007;71(6):1503–11.
Kang J, Chen XL, Wang L, Rampe D. Interactions of the antimalarial drug mefloquine with the human cardiac potassium channels KvLQT1/minK and HERG. J Pharmacol Exp Ther. 2001;299(1):290–6.
Sanchez-Chapula JA, Salinas-Stefanon E, Torres-Jacome J. Avides-Haro DE, Navarro-Polanco RA. Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. J Pharmacol Exp Ther. 2001;297(1):437–45.
Lopez-Izquierdo A, Ponce-Balbuena D, Ferrer T, Sachse FB, Tristani-Firouzi M, Sanchez-Chapula JA. Chloroquine blocks a mutant Kir2.1 channel responsible for short QT syndrome and normalizes repolarization properties in silico. Cell Physiol Biochem. 2009;24(3–4):153–60.
Luo C, Wang K, Zhang H. Modelling the effects of chloroquine on KCNJ2-linked short QT syndrome. Oncotarget. 2017;8(63):106511–26.
Rodriguez-Menchaca AA, Navarro-Polanco RA, Ferrer-Villada T, Rupp J, Sachse FB, Tristani-Firouzi M, et al. The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc Natl Acad Sci U S A. 2008;105(4):1364–8.
Takanari H, Nalos L, Stary-Weinzinger A, de Git KC, Varkevisser R, Linder T, et al. Efficient and specific cardiac IK(1) inhibition by a new pentamidine analogue. Cardiovasc Res. 2013;99(1):203–14.
Ji Y, Veldhuis MG, Zandvoort J, Romunde FL, Houtman MJC, Duran K, et al. PA-6 inhibits inward rectifier currents carried by V93I and D172N gain-of-function KIR2.1 channels, but increases channel protein expression. J Biomed Sci. 2017 Jul 15;24(1):44. https://doi.org/10.1186/s12929-017-0352-x
Ravens U, Poulet C, Wettwer E, Knaut M. Atrial selectivity of antiarrhythmic drugs. J Physiol. 2013;591(Pt 17):4087–97.
Hancox JC, James AF, Marrion NV, Zhang H, Thomas D. Novel ion channel targets in atrial fibrillation. Expert Opin Ther Targets. 2016;20(8):947–58.
McPate MJ, Zhang H, Ideniran I, Cordeiro JM, Witchel HJ, Hancox JC. Comparative effects of the short QT N588K mutation at 37°C on hERG K+ channel current during ventricular, Purkinje fibre and atrial action potentials: an action potential clamp study. J Physiol Pharmacol. 2009;60:23–41.
Acknowledgements
The authors thank Dr. Stephen Harmer for useful discussion.
Funding
No specific funding was involved in the preparation of this review. JCH, AGS and HZ acknowledge receipt of research funding from the British Heart Foundation and SADS UK, whilst DGW acknowledges receipt of a Wellcome Trust Fellowship.
Availability of data and materials
Data sharing not applicable to this review article as no datasets were generated or analysed.
Author information
Authors and Affiliations
Contributions
JCH conceived and drafted the article. AGS helped draft and revise the article critically for important intellectual content and provided Fig. 1. HZ and DGW helped draft and revise the article critically for important intellectual content and provided Fig. 2. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Review article; not applicable.
Consent for publication
ECG in Fig. 1 is anonymized and accordingly consent for publication is not required.
Competing interests
AGS is Editor-in-Chief of the Journal of Congenital Cardiology.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hancox, J.C., Whittaker, D.G., Zhang, H. et al. Learning from studying very rare cardiac conditions: the example of short QT syndrome. J Congenit Heart Dis 3, 3 (2019). https://doi.org/10.1186/s40949-019-0024-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40949-019-0024-7