
Abstract
Multidrug resistance (MDR) occurs frequently after long-term chemotherapy, resulting in refractory cancer and tumor recurrence. Therefore, combatting MDR is an important issue. Autophagy, a self-degradative system, universally arises during the treatment of sensitive and MDR cancer. Autophagy can be a double-edged sword for MDR tumors: it participates in the development of MDR and protects cancer cells from chemotherapeutics but can also kill MDR cancer cells in which apoptosis pathways are inactive. Autophagy induced by anticancer drugs could also activate apoptosis signaling pathways in MDR cells, facilitating MDR reversal. Therefore, research on the regulation of autophagy to combat MDR is expanding and is becoming increasingly important. We summarize advanced studies of autophagy in MDR tumors, including the variable role of autophagy in MDR cancer cells.
Similar content being viewed by others
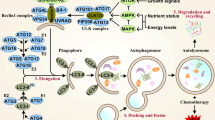

Background
Biosynthesis and degradation are two processes involved in the maintenance of metabolic homeostasis, which is the basis of all biological activities. The major protein degradation systems include the ubiquitin–proteasome pathway (targeting short-lived and misfolded proteins) and the lysosome-autophagy system (targeting long-lived macromolecular complexes and organelles) [1, 2]. Accumulating evidence indicates that autophagy plays a vital role in maintaining homeostasis in cells, and deficient autophagy seriously impacts embryonic differentiation. Autophagy is also closely related to the development of many diseases, including Alzheimer’s disease, cancer, and microorganism infection [3, 4]. Autophagy is a highly conserved cellular process in which cytoplasmic materials are degraded and recycled to maintain energy homeostasis. Autophagy can be classified as macroautophagy, microautophagy, and chaperone-mediated autophagy [5]. Given that current researches primarily focus on macroautophagy and the mechanisms are more clearly established than other types of autophagy, the term “autophagy” is commonly used to refer to macroautophagy.
Autophagy is a successive process which is initiated by the formation of an isolation membrane called phagophore. The phagophore is often seen as a thin cisterna with a clear lumen and is the structure that recruits autophagy-related proteins to induce autophagy. This lipid-based membrane then elongates and creates a complete, closed, double-membrane structure containing damaged organelles or long-lived proteins. Ultimately, autolysosomes are formed to degrade the contents, recycling amino acids, fatty acids, and nucleotides to maintain energetic homeostasis and viability [4, 6]. Autophagy occurs frequently during tumorigenesis and cancer chemotherapy. In general, constructive autophagy protects cancer cells during chemotherapy, leading to cancer drug resistance and refractory cancer [7].
Multidrug resistance (MDR) is another refractory outcome of chemotherapy and is defined as the resistance of cancer cells to multiple chemotherapeutic drugs with different structures and mechanisms of action [8]. MDR is a major cause of chemotherapy failure and responsible for increasing cancer-related mortality. Interestingly, recent mechanistic investigations have demonstrated that autophagy pathways are involved in the development of MDR [9]. Recent studies have explored approaches of using autophagy to hijack MDR cancer cells during anticancer therapy, but the mechanisms underlying the relationship between autophagy and MDR have not been fully studied.
Mechanisms of autophagy
Autophagy is a highly conserved cellular process. Approximately 30 autophagy-related genes (Atgs) in yeast and many mammalian genetic homologs have been identified [10]. Atgs are essential in responses to microenvironmental stresses such as hypoxia, heat stress, and accumulation of reactive oxygen species (ROS). Autophagy proceeds in successive stages, including initiation of phagophore assembly, autophagosomal formation, and lysosomal fusion [11, 12]. Here, we briefly discuss some primary pathways that regulate autophagy.
Phagophore assembly
The formation of autophagosomes begins with the expansion of the membrane core. The serine/threonine-protein kinase ULK1 complex containing unc-51-like autophagy activating kinase 1 (ULK1), Atg13, and focal adhesion kinase (FAK) family kinase-interacting protein 200 (FIP200) is at the most upstream position during autophagosome formation [13, 14]. Autophagosomal membrane contains a high level of phosphatidylinositol 3-phosphate (PI3P) in comparison to other types of membrane within the cell [15]. The formation of the autophagosomal membrane is regulated by class III PtdIns3K complexes containing Vacuolar protein sorting-associated protein 34 (Vps34), Vps15, Beclin1, and Atg14, which regulates the process that generates PtdIns(3)P-rich membranes [16]. Activating molecule in BECN1-regulated autophagy protein 1-deleted in liver cancer 1 (Ambra1-DLC1) released from the dynein motor complex acts as a cofactor of Beclin1 in a ULK1-dependent manner and is essential to autophagosome [17]. The autophagosomal membrane is thought to be derived from endoplasmic reticulum-Golgi [18]. The phagophore may be built up from the endoplasmic reticulum-mitochondria contact site [19]. Other compartments, such as the endosomes and the plasma membrane, also contribute to the formation of autophagosomes [20].
Autophagosome formation and maturation 3
Following autophagy initiation by the formation of phagophores, double-membrane autophagosomes (which load degradative cargos) are assembled under the control of the Atg12 conjugation system. In this system, the E1-like enzyme Atg7 and E2-like enzyme Atg10 jointly catalyze the formation of the Atg12–Atg5 complex, which is covalently conjugated [21]. Ultimately, the Atg12–Atg5–Atg16 (L1) complex is formed and directly binds membranes and constructs autophagosomes [22]. The complex is efficient for the microtubule-associated protein 1 light chain 3 (LC3) conjugation system. LC3 is first conjugated with lipid phosphatidylethanolamine (PE). During the conversion, Atg4 plays a role in lipoxidating LC3 to LC3-I and exposes the C-terminal glycine of LC3 for the subsequent conjugation of PE [23]. PE is conjugated to the C-terminal glycine of LC3-I, and this conjugation needs to be catalyzed by the E1-like enzyme Atg7 and the E2-like enzyme Atg3 [24].
Autolysosome degradation
The autophagosome is degraded by docking and fusing with a lysosome to construct an autolysosome. The autophagosomal membrane is conjugated with LC3-PE. During fusion with the lysosome, the outer membrane is cleaved and recycled by Atg4, while LC3-PE associated with the inner membrane is degraded by lysosomal proteases along with the cargo of the autophagosome, thus recycling amino acids, fatty acids, and nucleotides [25].
Core regulator of autophagy
The mechanistic target of rapamycin (mTOR), which regulates cell growth and survival, is the central modulator of autophagy regulation. As an environmental sensor, mTOR responds to intracellular microenvironmental changes including amino acids and glucose, as well as extracellular stresses such as agent treatments, hypoxia, and ultraviolet radiation. mTOR is active under nutrient-rich conditions and inhibits autophagy and protein degradation. By contrast, mTOR is inactive under nutrient-poor conditions: dephosphorylated ULK1 dissociates from the mTOR complex and then phosphorylates Atg13 and RB1-inducible coiled-coil (1RB1CC1/FIP200), thus triggering autophagy [26]. Inactivation of mammalian target of rapamycin complex 1 (mTORC1) by amino acid starvation can activate Atg14-containing type III phosphatidylinositol (PtdIns) 3-kinase (PIK3C3/VPS34) and induce autophagy both in vitro and in vivo [27]. The PIK3C3/VPS34 inhibitor SAR405 inhibits autophagy induced by mTOR inhibition, further indicating a crucial role of kinase regulation by mTOR in regulating autophagy [28]. In addition, mTOR can regulate autolysosome reformation by directly activating PIK3C3, leading to autolysosomal tubule sorting and lysosome regeneration [29].
Mechanisms of MDR
Mechanisms of MDR can be divided into seven categories: (1) increasing drug efflux by membrane transporters, with ATP-binding cassette (ABC) transporters as the main transporters [30]; (2) reducing drug uptake by influx transporters, such as solute carriers [31]; (3) boosting drug metabolism, including elimination by glutathione S-transferase and cytochrome P450 enzymes [32, 33]; (4) blocking apoptotic signaling pathways due to change in the expression level of B cell lymphoma (BCL) family proteins or mutations in the p53 pathway [34, 35]; (5) elevating adaptability by epigenetic regulation and miRNA regulation [36, 37]; (6) mutation in drug targets or feedback activation of other targets and signaling pathways [38]; and (7) chemoresistance induced by changes in the microenvironment, such as hypoxia response and cancer stem cell regulation [39, 40] (Fig. 1). Cellular-based resistant mechanisms are further classified into transporter-based classical MDR phenotypes and non-classical MDR phenotypes.

Complicated mechanisms of multidrug resistance (MDR) in tumor. The main mechanism of MDR is overexpressing ATP-binding cassette (ABC) transporters to increase drug efflux, resulting in a decrease in intracellular drug concentration. Other mechanisms of MDR are reducing drug uptake by influx transporters, boosting drug metabolism, blocking apoptotic signaling pathways, elevating adaptability by epigenetic regulation and microRNA regulation, mutation in drug targets or feedback activation of other targets and signaling pathways, and change of tumor microenvironment. ABCB1, ATP-binding cassette subfamily B member 1; ABCG2, ATP-binding cassette subfamily G member 2; ABCC1, ATP-binding cassette subfamily C member 1; CYP450, cytochrome P450
The ABC superfamily contains 49 different types of transporters and can be classified into seven subfamilies from ABC-A to ABC-G based on sequence similarities and structural organization [9]. Among them, P-glycoprotein (P-gp/ABCB1), multidrug-resistant protein 1 (MRP1/ABCC1), breast cancer resistant protein (BCRP/ABCG2/MXR/ABCP), and multidrug-resistant protein 10 (ABCC10/MRP7) transporters frequently drive chemosensitive cancers to MDR [41]. Human ABCB1 was the first identified ABC transporter. Overexpression of ABCB1 contributes to resistance against a wide variety of chemotherapeutic drugs. ABCC1 also leads to resistance to a wide range of anticancer drugs, and extensive evidence indicates that resistance of cancer cells to mitoxantrone, saquinavir, epipodophyllotoxins, and anthracyclines is mediated by ABCC1 [42,43,44,45]. The ABCG2 transporter is primarily expressed in breast cancer, colon cancer, gastric cancer, small cell lung cancer, and ovarian cancer [31]. ABC transporters are recognized as chief culprits in the development of MDR. Therapies continue to be developed with the goal of blocking or inactivating ABC transporters to increase the concentration of anticancer drugs within cells [46].
The relationship between autophagy and MDR
Chemotherapeutic agents that kill cancer cells primarily act by inducing apoptosis. Deficient apoptosis has been proposed to contribute to the development of MDR. Thus, alternative anticancer drugs that can directly induce apoptosis of MDR cancer cells would be valuable. However, the inactivation of apoptosis pathways adds to the complexity and difficulty of the development of such drugs. Importantly, MDR is inevitable following prolonged exposure to a new agent. Accordingly, other types of programmed cell death in MDR cells have attracted increasing attention, with autophagy emerging as a promising candidate.
Complex and controversial evidence indicating a role of autophagy in tumorigenesis has emerged in recent years. Autophagy can play a protective role against cancer by eliminating damaged organelles and recycling degradation products in normal cells. Paradoxically, excessive autophagy can devote cancer cells to “autophagic cell death” or “type II programmed cell death.” Thus, autophagy induced by metabolic and therapeutic stresses can have a pro-death or pro-survival role. Autophagy also plays dual roles in tumorigenesis, tumor progression, and resistance of cancer cells to chemotherapy [47]. Autophagy can be activated as a protective mechanism to mediate MDR during treatment. Thus, the inhibition of autophagy can re-sensitize resistant cancer cells and enhance the effect of chemotherapeutic agents. However, autophagy may also induce autophagic cell death, which differs from type I programmed cell death (apoptosis). Thus, autophagy can be used to promote the efficacy of treatment on MDR cancer if applied properly, and the role of autophagy in MDR must be clarified.
Autophagy as a cytoprotective mechanism mediating MDR
Because ABC transporters are associated with MDR, agents that modulate ABC transporters have been advocated to be developed as targets for chemotherapeutic drugs to overcome MDR. However, current ABC transporter modulators have not been as effective in the clinic as expected. Furthermore, MDR is an extremely complex phenotype. Recent studies have focused on clarifying the connection between autophagy and MDR based on clinical data. The expression level of ABCB1 was positively correlated with LC3, Beclin1, Rictor expression levels and negatively correlated with Raptor expression level in tumor samples from colorectal cancer patients surviving 5 years [48], strongly indicating that autophagy is related to the development and progression of cancer and MDR in clinical settings.
Autophagy promotes the development of MDR
Substantial evidence demonstrates that MDR develops after autophagy. The enhanced autophagy levels detected in patients with poor prognosis indicate that the presence of autophagy may catalyze the development of MDR. Yang et al. [49] demonstrated that S100 calcium-binding protein A8 (S100A8) is involved in the development of MDR by regulating autophagy in leukemia cells. Adriamycin and vincristine treatment can up-regulate the expression of S100A8, which is required for the formation of the Beclin1–Class III phosphatidylinositol 3-kinase (PI3KC3) complex [49, 50]. MDR may be mediated by high-mobility group box 1 (HMGB1), which promotes protective autophagy in response to anticancer agents. HMGB1 translocates from the nucleus to the cytoplasm and promotes the formation of Beclin1–PI3KC3 complexes by activating the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling pathway [51]. Similarly, Fan et al. [52] demonstrated that peptidylarginine deiminase IV may lead to MDR in hepatocellular carcinoma cells by inducing autophagy as a protective function.
MicroRNAs (miRNAs) can target Atgs and are considered as important modulators in MDR cancer cells. Xu et al. [53] confirmed that miR-199a-5p is down-regulated after cisplatin-based chemotherapy, resulting in cisplatin resistance via activation of autophagy. By contrast, miR-181a targets Atg5 and inhibits autophagy, thus enhancing the cytotoxicity of cisplatin in cisplatin-selective MDR SGC7901/CDDP cells in vitro and in vivo [54].
Recent studies have revealed that autophagy triggered by chemotherapeutics may facilitate resistance of cancer cells to paclitaxel [55], tamoxifen [56], epirubicin [57], or trastuzumab [58]. Sun et al. [57] found that epirubicin induced autophagy not only in MCF-7 cells but also in epirubicin-resistant MCF-7er cells. The induction of autophagy in MCF-7er cells may defend against epirubicin-mediated apoptosis, act as a pro-survival factor, and lead to deficient apoptosis [57]. Sun et al. [59] further studied two epirubicin-resistant cell lines, MCF-7er and SK-BR-3er, which overexpress ABCB1 and are simultaneously insensitive to paclitaxel and vinorelbine. Their study revealed that paclitaxel and vinorelbine induce autophagy, protecting MDR cells from apoptosis [59]. Although the expression of ABCB1 is independent of autophagy, the induction of MDR by autophagy cannot be ignored.
Autophagy inhibition facilitates the efficiency of chemotherapy in MDR cancer
As MDR-promoted autophagy is well-documented, new therapeutic strategies incorporating a combination of autophagy inhibitors have been proposed (Table 1). Inhibition of autophagy via genetic silencing of Atgs such as Beclin1, Atg5, Atg7, and Atg12 sensitizes MDR cells to therapeutic agents [73,74,75]. For example, chemoresistant cancer cell line SGC7901/VCR develops upon exposure to vincristine and exhibits increased autophagy, which is regulated by Atg12 and high-mobility group box 2 (HMGB2). Overexpression of miR-23b-3p, which targets Atg12 and HMGB2, sensitizes chemoresistant cells to multiple chemotherapeutics such as vincristine, 5-fluorouracil, and cisplatin in vitro and can restore the sensitivity of MDR cells to chemotherapy in vivo [60]. Furthermore, inhibition of autophagy by small interfering RNAs (siRNAs) targeting Atg12 and HMGB2 or by the autophagy inhibitor chloroquine (CQ) also sensitizes SGC7901/VCR cells to chemotherapeutics [60]. This finding indicates that vincristine-based MDR is related to autophagy. In response to 5-fluorouracil, cisplatin, or other chemotherapeutic drugs, some types of MDR cells exhibit protective autophagy and rapidly recover after the removal of chemotherapeutic agents [61]. Basal autophagic flux is higher in the anthracycline-resistant lines MDA-MB-231-R8 and SUM159PT-R75 than in their respective parent lines MDA-MB-231 and SUM159PT [76]. Furthermore, Atg5/Atg7 knockdown as well as pharmacologic inhibition of autophagy with CQ or hydroxychloroquine (HCQ) significantly reduce viability of both epirubicin-sensitive and epirubicin-resistant triple-negative breast cancer cells [76]. These findings suggest that autophagy responds to anthracycline-induced MDR and protects MDR cells from injury.
Inducing autophagic cell death overcomes MDR
Autophagy can also play a pro-death role and trigger autophagic cell death in apoptosis-deficient MDR cells. Several studies have sought to identify a novel anticancer agent that effectively kills MDR cancer cells by inducing excessive autophagy (Table 2). Suberoylanilide hydroxamic acid (SAHA), a newly developed prototype histone deacetylase inhibitor, induces autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells and significantly suppresses tumor growth [77]. Two lipophilic tanshinones, cryptotanshinone and dihydrotanshinone, also inhibit the growth of apoptosis-resistant colon cancer cells by inducing autophagic cell death and p53-independent cytotoxicity [78]. The bisindolic alkaloid voacamine induces autophagy, which causes MDR tumor cell death [75]. The resistance of the leukemic cell line K562 to edelfosine can be reversed by edelfosine lipid nanoparticles, which induce caspase-independent and autophagic cell death [79]. These results indicate that autophagic cell death can be induced in MDR cells as an alternative cell death mechanism when the cells fail to undergo apoptosis.
Several signaling pathways contribute to autophagic cell death in MDR cancer cells (Table 1). For instance, the AMP-activated protein kinase (AMPK)-protein kinase B (Akt)-mTOR pathway is critical to the regulation of autophagic cell death. A Ganoderma microsporum immunomodulatory (GMI) protein inhibits the phosphorylation of Akt and the 70 kDa S6 protein kinase (p70S6K) in the A549 lung cancer sub-cell lines A549/D16 and A549/V16, inducing autophagy and overcoming MDR in lung cancer [80]. Similarly, the dual phosphoinositide 3-kinase (PI3K) and mTOR inhibitor NVP-BEZ235 suppresses the proliferation of cisplatin-resistant urothelial cancer cells by activating autophagic flux independent of apoptotic cell death [81]. Other cellular signaling cascades may also be involved in autophagic cell death in MDR cells. For example, a human single-chain fragment variable, HW1, kills tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-resistant cancer cells by inducing autophagic cell death, which can be inhibited by autophagy inhibitors. HW1-mediated autophagic cell death occurs primarily through the c-Jun NH2-terminal kinase pathway [89].
Autophagy facilitates cell death in apoptosis-deficient MDR cancer
In apoptosis-deficient MDR cells, an adaptive response of autophagy may aggravate MDR cancer resistance to chemotherapeutics. However, under certain conditions, autophagy can be a scavenger in apoptosis-blocked signaling pathways, sensitizing MDR tumors to apoptosis. Feroniellin A, a novel furanocoumarin derives from Feroniella lucida, elicits autophagy dependent on the mTOR/Beclin1/Atg5 pathway in etoposide-resistant, ABCB1-overexpressed A549 cells. Interestingly, inhibition of autophagy by Beclin1 siRNA could eliminate Feroniellin A-induced apoptosis. Moreover, further stimulation of autophagy by rapamycin accelerates Feroniellin A-induced apoptosis [90]. Coincidentally, autophagy induced by metformin could also assists TRAIL-mediated apoptosis in TRAIL-resistant lung adenocarcinoma. Metformin is found to down-regulate cellular FADD-like IL-1β-converting enzyme (FLICE)-inhibitory protein (c-FLIP) and markedly enhance autophagic flux, ultimately facilitates apoptosis triggered by TRAIL [91].
Autophagy mediates chemosensitization
A plenty of studies have revealed that autophagy, the chemotherapeutics’ appendant, is a patron of MDR cancer cells, consequently harass sensitive effect of MDR-reversal agents. Conversely, emerging evidence arises that the increase in autophagy upon some agents could facilitate MDR reversal (Fig. 2). For example, the nanocrystal of underivatized fullerene C60 (nano-C60) exhibits potential anticancer property to several neoplasms in vitro [92]. Nano-C60 is capable of triggering oxidative stress and then induces autophagy which is enhanced by photoactivation. In particular, autophagy induced by nano-C60 is able to sensitive drug-resistant MCF-7 cancer cells. The chemosensitizing effect of nano-C60 can be dampened after autophagy inhibition by a ROS scavenger, N-acetyl-l-cysteine. Furthermore, the chemosensitizing effect of nano-C60 depends on Atg5 and is vanished in Atg5 −/− cells and Atg5 siRNA-treated cells [93]. In addition, a derivative of nano-C60 is also capable to vulnerable doxorubicin-resistant MCF-7 cells to doxorubicin by modulating autophagy, and its activity is much greater than that of nano-C60 [94]. Similarly, cysteamine-elicited autophagy could also reverse resistance of adriamycin-resistant MCF-7/ADR cells to doxorubicin in vitro and in vivo [95]. Although the exact molecular mechanisms remain blurred, those results still strongly highlight a novel biological function of autophagy in MDR reversal strategy.

Autophagy chemosensitizes MDR cancer cells to chemotherapeutics. A novel role of autophagy induced by certain autophagy inducers such as cysteamine, the nanocrystal of underivatized fullerene C60 (nano-C60) and its derivative was confirmed. Autophagy triggered by those inducers could sensitive MDR cancer cells to chemotherapeutics. PAS, pre-autophagosomal structure
Conclusions
In recent years, a large body of evidence has indicated that autophagy plays dual roles in MDR. The pro-death or pro-survival roles of autophagy are highly dependent on the tumor type and treatment characteristics. Autophagy protects MDR cancer cells from apoptosis and promotes resistance to chemotherapy treatment, and inhibition of autophagy may sensitize MDR cells to anticancer drugs. The combination of autophagy inhibitors with cytotoxic drugs is highly anticipated. Excitingly, CQ and its derivative HCQ, in combination with several anticancer drugs, have been approved to augment cytotoxicity and to sensitize refractory cancers. They are expected to be used in the battlefield of MDR tumor. On the other hand, autophagic cell death which is induced by autophagy inducers could directly bypass apoptosis and ultimately eliminate MDR cells. It represents a new battle line in the fight against MDR cancer. On the other battleground, emerging evidence demystifies that autophagy is a strong propulsor to sensitize apoptosis-resistant MDR cells to anticancer drugs and reverse MDR. It shows a novel biological function of autophagy in MDR cancer cells and will enable the development of promising strategies to overcome MDR. Although the exact mechanisms of the interaction between autophagy and MDR reversal remain obscure, it provides us a vast research space to elucidate the mysteries.
References
Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15(8):713–20. doi:10.1038/ncb2815.
Deschenes-Simard X, Lessard F, Gaumont-Leclerc MF, Bardeesy N, Ferbeyre G. Cellular senescence and protein degradation breaking down cancer. Cell Cycle. 2014;13(12):1840–58. doi:10.4161/cc.29335.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018.
Sridhar S, Botbol Y, Macian F, Cuervo AM. Autophagy and disease: always two sides to a problem. J Pathol. 2012;226(2):255–73. doi:10.1002/path.3025.
Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989;246(4928):382–5.
Hundeshagen P, Hamacher-Brady A, Eils R, Brady NR. Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of autophagy. BMC Biol. 2011;9:38. doi:10.1186/1741-7007-9-38.
Buchser WJ, Laskow TC, Pavlik PJ, Lin HM, Lotze MT. Cell-mediated autophagy promotes cancer cell survival. Cancer Res. 2012;72(12):2970–9. doi:10.1158/0008-5472.CAN-11-3396.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–26. doi:10.1038/nrc3599.
Kumar P, Zhang DM, Degenhardt K, Chen ZS. Autophagy and transporter-based multi-drug resistance. Cells. 2012;1(3):558–75. doi:10.3390/cells1030558.
Klionsky DJ. Look people, “Atg” is an abbreviation for “autophagy-related.” That’s it. Autophagy. 2012;8(9):1281–2. doi:10.4161/auto.21812.
Ganley IG, Wong PM, Gammoh N, Jiang XJ. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42(6):731–43. doi:10.1016/j.molcel.2011.04.024.
Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20(21):5971–81. doi:10.1093/emboj/20.21.5971.
Chan EYW, Kir S, Tooze SA. SiRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282(35):25464–74. doi:10.1074/jbc.M703663200.
Hara T, Takamura A, Kishi C, Iemura SI, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181(3):497–510. doi:10.1083/jcb.200712064.
Obara K, Noda T, Niimi K, Ohsumi Y. Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes Cells. 2008;13(6):537–47. doi:10.1111/j.1365-2443.2008.01188.x.
Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11(4):385–96. doi:10.1038/ncb1846.
Fimia GM, Di Bartolomeo S, Piacentini M, Cecconi F. Unleashing the Ambra1-Beclin 1 complex from dynein chains Ulk1 sets Ambra1 free to induce autophagy. Autophagy. 2011;7(1):115–7. doi:10.4161/auto.7.1.14071.
Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife. 2013;2:00947. doi:10.7554/eLife.00947.
Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495(7441):389–93. doi:10.1038/nature11910.
Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell. 2013;154(6):1285–99. doi:10.1016/j.cell.2013.08.044.
Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature. 1998;395(6700):395–8. doi:10.1038/26506.
Fujioka Y, Noda NN, Nakatogawa H, Ohsumi Y, Inagaki F. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J Biol Chem. 2010;285(2):1508–15. doi:10.1074/jbc.M109.053520.
Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, Kraft C, et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012;31(22):4304–17. doi:10.1038/emboj.2012.278.
Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408(6811):488–92.
Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13(5):495–504. doi:10.1016/j.cmet.2011.04.004.
Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11. doi:10.1128/Mcb.06159-11.
Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy. 2013;9(12):1983–95. doi:10.4161/auto.26058.
Pasquier B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy. 2015;11(4):725. doi:10.1080/15548627.2015.1033601.
Yu L, McPhee CK, Zheng LX, Mardones GA, Rong YG, Peng JY, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–6. doi:10.1038/nature09076.
Chun SY, Kwon YS, Nam KS, Kim S. Lapatinib enhances the cytotoxic effects of doxorubicin in MCF-7 tumorspheres by inhibiting the drug efflux function of ABC transporters. Biomed Pharmacother. 2015;72:37–43. doi:10.1016/j.biopha.2015.03.009.
Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KLR, Chu XY, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–36. doi:10.1038/nrd3028.
Filomeni G, Turella P, Dupuis ML, Forini O, Ciriolo MR, Cianfriglia M, et al. 6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol, a specific glutathione S-transferase inhibitor, overcomes the multidrug resistance (MDR)-associated protein 1-mediated MDR in small cell lung cancer. Mol Cancer Ther. 2008;7(2):371–9. doi:10.1158/1535-7163.Mct-07-0487.
Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene. 2006;25(11):1679–91. doi:10.1038/sj.onc.1209377.
Wilson CS, Medeiros LJ, Lai R, Butch AW, McCourty A, Kelly K, et al. DNA topoisomerase II alpha in multiple myeloma: a marker of cell proliferation and not drug resistance. Mod Pathol. 2001;14(9):886–91. doi:10.1038/modpathol.3880407.
Bedi A, Barber JP, Bedi GC, El-Deiry WS, Sidransky D, Vala MS, et al. BCR-ABL-mediated inhibition of apoptosis with delay of G2/M transition after DNA damage: a mechanism of resistance to multiple anticancer agents. Blood. 1995;86(3):1148–58.
Shoemaker RH. Genetic and epigenetic factors in anticancer drug resistance. J Natl Cancer Inst. 2000;92(1):4–5. doi:10.1093/jnci/92.1.4.
Li H, Yang BB. Friend or foe: the role of microRNA in chemotherapy resistance. Acta Pharmacol Sin. 2013;34(7):870–9. doi:10.1038/aps.2013.35.
Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11(8):473–81. doi:10.1038/nrclinonc.2014.104.
Milane L, Duan ZF, Amiji M. Role of hypoxia and glycolysis in the development of multi-drug resistance in human tumor cells and the establishment of an orthotopic multi-drug resistant tumor model in nude mice using hypoxic pre-conditioning. Cancer Cell Int. 2011;11:3. doi:10.1186/1475-2867-11-3.
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–84. doi:10.1038/nrc1590.
Kathawala RJ, Wang YJ, Ashby CR Jr, et al. Recent advances regarding the role of ABC subfamily C member 10 (ABCC10) in the efflux of antitumor drugs. Chin J Cancer. 2014;33(5):223–30. doi:10.5732/cjc.013.10122.
Morrow CS, Peklak-Scott C, Bishwokarma B, et al. Multidrug resistance protein 1 (MRP1, ABCC1) mediates resistance to mitoxantrone via glutathione-dependent drug efflux. Mol Pharmacol. 2006;69(4):1499–505. doi:10.1124/mol.105.017988.
König SK, Herzog M, Theile D, et al. Impact of drug transporters on cellular resistance towards saquinavir and darunavir. J Antimicrob Chemother. 2010;65(11):2319–28. doi:10.1093/jac/dkq324.
Anreddy N, Gupta P, Kathawala RJ, et al. Tyrosine kinase inhibitors as reversal agents for ABC transporter mediated drug resistance. Molecules. 2014;19(9):13848–77. doi:10.3390/molecules190913848.
Wang YJ, Zhang YK, Kathawala RJ, et al. Repositioning of tyrosine kinase inhibitors as antagonists of ATP-binding cassette transporters in anticancer drug resistance. Cancers. 2014;6(4):1925. doi:10.3390/cancers6041925.
Zhang GN, Ashby CR, Zhang YK, et al. The reversal of antineoplastic drug resistance in cancer cells by β-elemene. Chin J Cancer. 2015;34(3):1. doi:10.1186/s40880-015-0048-0.
Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961–7. doi:10.1038/nrc2254.
Shuhua W, Chenbo S, Yangyang L, Xiangqian G, Shuang H, Tangyue L, et al. Autophagy-related genes Raptor, Rictor, and Beclin1 expression and relationship with multidrug resistance in colorectal carcinoma. Hum Pathol. 2015;46(11):1752–9. doi:10.1016/j.humpath.2015.07.016.
Yang M, Zeng P, Kang R, et al. S100A8 contributes to drug resistance by promoting autophagy in leukemia cells. PLoS ONE. 2014;9(5):e97242. doi:10.1371/journal.pone.0097242.
Yang LC, Yang MH, Zhang H, Wang Z, Yu Y, Xie M, et al. S100A8-targeting siRNA enhances arsenic trioxide-induced myeloid leukemia cell death by down-regulating autophagy. Int J Mol Med. 2012;29(1):65–72. doi:10.3892/ijmm.2011.806.
Pan BZ, Chen DQ, Huang JY, Wang R, Feng B, Song HZ, et al. HMGB1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol Cancer. 2014;13:165. doi:10.1186/1476-4598-13-165.
Fan TT, Zhang CS, Zong M, Zhao QD, Yang X, Hao C, et al. Peptidylarginine deiminase IV promotes the development of chemoresistance through inducing autophagy in hepatocellular carcinoma. Cell Biosci. 2014;4:49. doi:10.1186/2045-3701-4-49.
Xu N, Zhang JJ, Shen CH, Luo Y, Xia L, Xue F, et al. Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem Biophys Res Commun. 2012;423(4):826–31. doi:10.1016/j.bbrc.2012.06.048.
Jiao XY, Zhao L, Ma MT, Bai XF, He M, Yan YY, et al. MiR-181a enhances drug sensitivity in mitoxantone-resistant breast cancer cells by targeting breast cancer resistance protein (BCRP/ABCG2). Breast Cancer Res Treat. 2013;139(3):717–30. doi:10.1007/s10549-013-2607-x.
Ajabnoor GMA, Crook T, Coley HM. Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in MCF-7 breast cancer cells. Cell Death Dis. 2012;3:e260. doi:10.1038/cddis.2011.139.
Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, et al. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat. 2008;112(3):389–403. doi:10.1007/s10549-007-9873-4.
Sun WL, Chen J, Wang YP, Zheng H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy. 2011;7(9):1035–44. doi:10.4161/auto.7.9.16521.
Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS ONE. 2009;4(7):e6251. doi:10.1371/journal.pone.0006251.
Sun WL, Lan D, Gan TQ, Cai ZW. Autophagy facilitates multidrug resistance development through inhibition of apoptosis in breast cancer cells. Neoplasma. 2015;62(2):199–208. doi:10.4149/neo_2015_025.
An Y, Zhang Z, Shang Y, Jiang X, Dong J, Yu P, et al. miR-23b-3p regulates the chemoresistance of gastric cancer cells by targeting ATG12 and HMGB2. Cell Death Dis. 2015;6:e1766. doi:10.1038/cddis.2015.123.
O’Donovan TR, O’Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy. 2011;7(5):509–24. doi:10.4161/auto.7.5.15066.
Zhang LH, Yang AJ, Wang M, Liu W, Wang CY, Xie XF, et al. Enhanced autophagy reveals vulnerability of P-gp mediated epirubicin resistance in triple negative breast cancer cells. Apoptosis. 2016;21(4):473–88. doi:10.1007/s10495-016-1214-9.
Shin SW, Kim SY, Park JW. Autophagy inhibition enhances ursolic acid-induced apoptosis in PC3 cells. BBA-Mol Cell Res. 2012;1823(2):451–7. doi:10.1016/j.bbamcr.2011.10.014.
Zhang N, Qi YF, Wadham C, Wang LJ, Warren A, Di W, et al. FTY720 induces necrotic cell death and autophagy in ovarian cancer cells: a protective role of autophagy. Autophagy. 2010;6(8):1157–67. doi:10.4161/auto.6.8.13614.
Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3(147):81. doi:10.1126/scisignal.2001017.
Ahn JH, Lee M. Suppression of autophagy sensitizes multidrug resistant cells towards Src tyrosine kinase specific inhibitor PP2. Cancer Lett. 2011;310(2):188–97. doi:10.1016/j.canlet.2011.06.034.
Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang CY, Chung L, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110(1):313–22. doi:10.1182/blood-2006-10-050260.
Ma XH, Piao SF, Dey S, Mcafee Q, Karakousis G, Villanueva J, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Investig. 2014;124(3):1406–17. doi:10.1172/Jci70454.
Ren JH, He WS, Nong L, Zhu QY, Hu K, Zhang RG, et al. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother Radiopharm. 2010;25(1):75–80. doi:10.1089/cbr.2009.0701.
Shi C, Zhang Z, Shi J, Wang F, Luan Y. Co-delivery of docetaxel and chloroquine via PEO–PPO–PCL/TPGS micelles for overcoming multidrug resistance. Int J Pharm. 2015;495(2):932–9. doi:10.1016/j.ijpharm.2015.10.009.
Gao M, Xu Y, Qiu L. Sensitization of multidrug-resistant malignant cells by liposomes co-encapsulating doxorubicin and chloroquine through autophagic inhibition. J Liposome Res. 2016;2:1–10. doi:10.1080/08982104.2016.1185731.
Liang B, Liu X, Liu Y, Kong D, Liu X, Zhong R, et al. Inhibition of autophagy sensitizes MDR-phenotype ovarian cancer SKVCR cells to chemotherapy. Biomed Pharmacother. 2016;82:98–105. doi:10.1016/j.biopha.2016.04.054.
Eum KH, Lee M. Targeting the autophagy pathway using ectopic expression of Beclin 1 in combination with rapamycin in drug-resistant v-Ha-ras-transformed NIH3T3 cells. Mol Cells. 2011;31(3):231–8. doi:10.1007/s10059-011-0034-6.
Ge J, Chen ZH, Huang J, Chen JX, Yuan WJ, Deng ZH, et al. Upregulation of autophagy-related gene-5 (ATG-5) is associated with chemoresistance in human gastric cancer. PLoS ONE. 2014;9(10):e110293. doi:10.1371/journal.pone.0110293.
Meschini S, Condello M, Calcabrini A, Marra M, Formisano G, Lista P, et al. The plant alkaloid voacamine induces apoptosis-independent autophagic cell death on both sensitive and multidrug resistant human osteosarcoma cells. Autophagy. 2008;4(8):1020–33.
Chittaranjan S, Bortnik S, Dragowska WH, Xu J, Abeysundara N, Leung A, et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin Cancer Res. 2014;20(12):3159–73. doi:10.1158/1078-0432.Ccr-13-2060.
Lee YJ, Won AJ, Lee J, Jung JH, Yoon S, Lee BM, et al. Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells. Int J Med Sci. 2012;9(10):881–93. doi:10.7150/ijms.5011.
Hu T, Wang L, Zhang L, Lu L, Shen J, Chan RLY, et al. Sensitivity of apoptosis-resistant colon cancer cells to tanshinones is mediated by autophagic cell death and p53-independent cytotoxicity. Phytomedicine. 2015;22(5):536–44. doi:10.1016/j.phymed.2015.03.010.
Aznar MA, Lasa-Saracibar B, Blanco-Prieto MJ. Edelfosine lipid nanoparticles overcome multidrug resistance in K-562 leukemia cells by a caspase-independent mechanism. Mol Pharm. 2014;11(8):2650–8. doi:10.1021/mp5000696.
Chiu LY, Hu ME, Yang TY, Hsin IL, Ko JL, Tsai KJ, et al. Immunomodulatory protein from Ganoderma microsporum induces pro-death autophagy through Akt-mTOR-p70S6K pathway inhibition in multidrug resistant lung cancer cells. PLoS ONE. 2015;10(5):e0125774. doi:10.1371/journal.pone.0125774.
Li JR, Cheng CL, Yang CR, Ou YC, Wu MJ, Ko JL. Dual inhibitor of phosphoinositide 3-kinase/mammalian target of rapamycin NVP-BEZ235 effectively inhibits cisplatin-resistant urothelial cancer cell growth through autophagic flux. Toxicol Lett. 2013;220(3):267–76. doi:10.1016/j.toxlet.2013.04.021.
Sirichanchuen B, Pengsuparp T, Chanvorachote P. Long-term cisplatin exposure impairs autophagy and causes cisplatin resistance in human lung cancer cells. Mol Cell Biochem. 2012;364(1–2):11–8. doi:10.1007/s11010-011-1199-1.
Kim KW, Hwang M, Moretti L, Jaboin JJ, Cha YI, Lu B. Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy. 2008;4(5):659–68.
Wang ZY, Wang N, Liu PX, Chen QJ, Situ HL, Xie T, et al. MicroRNA-25 regulates chemoresistance-associated autophagy in breast cancer cells, a process modulated by the natural autophagy inducer isoliquiritigenin. Oncotarget. 2014;5(16):7013–26.
Kong DJ, Ma SM, Liang B, Yi HQ, Zhao YL, Xin R, et al. The different regulatory effects of p53 status on multidrug resistance are determined by autophagy in ovarian cancer cells. Biomed Pharmacother. 2012;66(4):271–8. doi:10.1016/j.biopha.2011.12.002.
Law BYK, Mok SWF, Chan WK, Xu SW, Wu AG, Yao XJ, et al. Hernandezine, a novel AMPK activator induces autophagic cell death in drug-resistant cancers. Oncotarget. 2016;7(7):8090–104.
Li X, Feng J, Zhang R, Wang J, Su T, Tian Z, et al. Quaternized chitosan/alginate-Fe3O4 magnetic nanoparticles enhance the chemosensitization of multidrug-resistant gastric carcinoma by regulating cell autophagy activity in mice. J Biomed Nanotechnol. 2016;12(5):948–61.
Khurana A, Roy D, Kalogera E, Mondal S, Wen X, He X, et al. Quinacrine promotes autophagic cell death and chemosensitivity in ovarian cancer and attenuates tumor growth. Oncotarget. 2015;6(34):36354–69. doi:10.18632/oncotarget.5632.
Park KJ, Lee SH, Kim TI, Lee HW, Lee CH, Kim EH, et al. A human scFv antibody against TRAIL receptor 2 induces autophagic cell death in both TRAIL-sensitive and TRAIL-resistant cancer cells. Cancer Res. 2007;67(15):7327–34. doi:10.1158/0008-5472.Can-06-4766.
Kaewpiboon C, Surapinit S, Malilas W, Moon J, Phuwapraisirisan P, Tip-Pyang S, et al. Feroniellin A-induced autophagy causes apoptosis in multidrug-resistant human A549 lung cancer cells. Int J Oncol. 2014;44(4):1233–42. doi:10.3892/ijo.2014.2297.
Nazim UM, Moon JH, Lee JH, Lee YJ, Seol JW, Eo SK, et al. Activation of autophagy flux by metformin downregulates cellular FLICE-like inhibitory protein and enhances TRAIL-induced apoptosis. Oncotarget. 2016;7(17):23468–81. doi:10.18632/oncotarget.8048.
Isakovic A, Markovic Z, Todorovic-Markovic B, Nikolic N, Vranjes-Djuric S, Mirkovic M, et al. Distinct cytotoxic mechanisms of pristine versus hydroxylated fullerene. Toxicol Sci. 2006;91(1):173–83. doi:10.1093/toxsci/kfj127.
Zhang Q, Yang W, Man N, Zheng F, Shen Y, Sun K, et al. Autophagy-mediated chemosensitization in cancer cells by fullerene C60 nanocrystal. Autophagy. 2009;5(8):1107–17.
Wei P, Zhang L, Lu Y, Man N, Wen L. C60(Nd) nanoparticles enhance chemotherapeutic susceptibility of cancer cells by modulation of autophagy. Nanotechnology. 2010;21(49):495101. doi:10.1088/0957-4484/21/49/495101.
Wan XM, Zheng F, Zhang L, Miao YY, Man N, Wen LP. Autophagy-mediated chemosensitization by cysteamine in cancer cells. Int J Cancer. 2011;129(5):1087–95. doi:10.1002/ijc.25771.
Authors’ contributions
All authors contributed substantially to the writing of this review. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by the Science and Technology Program of China (2012ZX09103101-053), the Natural Science Foundation of Guangdong Province (S2013050014183 and 2013CXZDA006), and the Program for New Century Excellent Talents in University (D. M. Zhang), and the project was supported by Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (D. M. Zhang).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, YJ., Lei, YH., Yao, N. et al. Autophagy and multidrug resistance in cancer. Chin J Cancer 36, 52 (2017). https://doi.org/10.1186/s40880-017-0219-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40880-017-0219-2