
Abstract
Background
The Palaearctic butterfly genus Pseudophilotes Beuret, 1958 (Lycaenidae: Polyommatinae), that today occurs in North Africa and in Eurasia, includes ten described species with various distribution ranges, including endemics such as the Sardinian P. barbagiae. Phylogenetic relationships among these species are largely unresolved. In the present study, we analysed 158 specimens, representing seven species out of ten described in the genus, from widely distributed sites throughout the western Palaearctic region, using nuclear markers (28S rRNA, wingless, Internal Transcribed Spacer 2 and Elongation Factor 1α) and the barcoding region of the mitochondrial cytochrome c oxidase subunit 1 gene to investigate if the current taxonomic entities match the phylogenetic pattern. Further, we attempt to infer the geographic origin of the genus Pseudophilotes and estimate the timing of its radiations, including the split of the Sardinian endemic P. barbagiae.
Results
Maximum Likelihood and Bayesian inference analyses confirmed the monophyly of the genus Pseudophilotes and clearly supported the closer affinity of P. barbagiae to the species assemblage of P. baton, P. vicrama and P. panoptes as opposed to P. abencerragus. The currently accepted species P. baton, P. vicrama and P. panoptes turned out to be weakly differentiated from each other, while P. bavius and P. fatma emerged as highly distinct and formed a well supported clade. The split between the lineage comprising bavius and fatma (sometimes treated as a distinct genus, Rubrapterus) with Salvia species as larval host plants, and the remaining Pseudophilotes that utilize Thymus and other Lamiaceae (but not Salvia), dates back to about 4.9 million years ago (Mya).
Conclusions
Our results show that the last common ancestor of the genus probably lived in the Messinian period (5.33–7.25 Mya). At species level, they support the current taxonomy of the genus, although P. panoptes, P. baton and P. vicrama display complex patterns based on phylogeographic relationships inferred from mtDNA. The Sardinian endemic P. barbagiae turned out to be a young endemic, but clearly with European instead of North African origin and evolved through allopatric isolation on the island of Sardinia only about 0.74 Mya.
Similar content being viewed by others


Background
Pseudophilotes Beuret, 1958 (Lycaenidae: Polyommatinae) is a Palaearctic genus of blue butterflies mainly distributed in Eurasia, but also in North Africa and the Levante (Fig. 1a). The phylogenetic position of the genus within the Glaucopsyche section (Polyommatinae) has been lately inferred by Ugelvig et al. [1]. They found that the genera Pseudophilotes and Scolitantides are clearly separated, although they had been lumped together by other studies [2, 3]. This separation is explained by the affiliation with different larval host plants: Pseudophilotes with Lamiaceae, and Scolitantides with Crassulaceae.

a Sampling localities and approximate geographic distribution areas of the Pseudophilotes species considered in the present study. Sampling localities for the species included in this study are showed with differently coloured circles, diamonds, and triangles respectively. Geographic distribution areas are redrawn after [2, 3, 26, 74,75,76,77] and shaded in colours referring to the sample localities. The map was prepared using Quantum GIS 2.8.2 based on a map from Natural Earth (www.naturalearthdata.com). b Median-Joining Network of Pseudophilotes COI sequences. The circle size is proportional to haplotype frequency and the different colours refer to the different species used in the study (cf. a). Numbers of mutations between haplotypes are shown at the connections, except for single or double substitutions. Connections creating loops that are less probable according to frequency and geographical criteria are indicated in dashed lines. Haplotype codes match those in Additional file 1. A1 - A3: haplogroups including P. baton, P. vicrama and P. panoptes
At the species level, the same study [1] showed that the Bavius Blue, Pseudophilotes bavius forms a sister clade to the rest of Pseudophilotes, which is also supported by its association to Salvia (Lamiaceae), whereas species in the remaining clades mainly feed on Thymus (Lamiaceae). Recent efforts to resolve the evolution of Pseudophilotes based on the COI barcoding region [4] indicated a closer relationship of the Sardinian Blue, P. barbagiae to the False Baton Blue, P. abencerragus than to the Baton Blue, P. baton. Further studies based on mtDNA [5] showed P. baton and the Eastern Baton Blue, P. vicrama to display complex patterns. Species delimitation in the genus remains thus a work-in-progress.
Species such as the Sardinian Blue, Pseudophilotes barbagiae [6], with highly restricted distributions and taxa representing unique lineages within a species flock can be suitable models to investigate the processes of differentiation and speciation and often provide the key for answers to broader-scale biogeographic questions [7]. In this respect, the study of islands where many endemic species or subspecies evolved has provided fundamental insights into the relationships between geographical patterns and evolutionary processes [8, 9]. P. barbagiae has been described as distinct from the continental and Corsican P. baton by characteristics of the male genitalia and wing markings. Its divergence has been hypothetically linked to the geographic isolation of the Sardo-Corsican plate [10], but this assumption has never been tested on the basis of molecular data. Gerace [11] hypothesized that P. barbagiae probably diverged from P. baton as a result of the separation of Corsica and Sardinia due to important climatic changes, and later P. barbagiae became isolated in mountainous areas.
Due to its geographic position and geological history, the island of Sardinia is characterized by a remarkable richness of endemic species [12] and represents one of the most prominent biodiversity hotspots in the Mediterranean basin [13]. The question how its endemic fauna was formed and when it originated has been previously investigated using molecular analyses for a variety of vertebrate [14,15,16] and invertebrate animals [9, 17,18,19,20,21]. In all these cases, phylogenetic patterns could be related to several aspects of the geological history of the Mediterranean basin [11, 12], but the specific evolutionary histories turned out to vary distinctly between taxa. About 5 Mya ago, during the Messinian period, the closure of the Strait of Gibraltar led to an almost complete desiccation of the Mediterranean Sea, and to a subsequent formation of salt deposits in all deep basins [22]. This event, called the Messinian Salinity Crisis (MSC), was often advocated as one of the main drivers for the emergence of a distinctive fauna and flora in the Mediterranean. The newly exposed land corridors permitted faunal exchange between southern Europe and the Maghreb region (Northern Africa) [23, 24], as well as between Sardinia and northern Italy and southern France.
When the connection to the Atlantic ocean was re-established, the massive influx of water brought about a drastic change in marine flora and fauna, and at the same time terminated the connections between the Tyrrhenian islands and the Eurasian or African continental mainland, respectively, which had as a consequence the differentiation of many distinctly younger island species (e.g. butterflies, cave-beetles, mayflies and lizards). Land bridges between Sardinia and Corsica and the mainland continued to be created well into the Pleistocene due to glacial/interglacial sea-level oscillations [25]. During the Würm glaciation (115,000–11,700 years ago), Sardinia could have been in contact with the mainland via the island of Elba as the sea level was up to 120 m lower than today [11, 12] and with the neighbouring Corsica.
In the present study, we analysed four nuclear genes (28S ribosomal [28S], wingless [wg], Internal Transcribed Spacer 2 [ITS2] and Elongation Factor 1α [EF1α]) and the barcoding region of the mitochondrial cytochrome c oxidase subunit 1 (COI) gene for seven out of ten Pseudophilotes species, from widely distributed sites throughout the Palaearctic region, including for the first time the Sardinian endemic P. barbagiae, as well as the African P. fatma. We aimed to (i) reconstruct the phylogeny of the genus Pseudophilotes and check if the obtained clades correspond to current taxonomic units, (ii) to trace the geographical origin of the genus since extant distributions of Pseudophilotes species cover both Eurasia and North Africa, and (iii) to shed light on the historical biogeographic processes that determined the present-day isolation of the Sardinian endemic P. barbagiae.
Methods
Taxon sampling and molecular procedures
We examined 96 Pseudophilotes individuals representing seven species out of ten described from sites across Europe and North Africa (Maghreb region) (Fig. 1a and Additional file 1). We were unable to obtain sequences from P. sinaicus, P. jordanicus and P. jacuticus. We included sequences from the Eurasian species Scolitantides orion Pallas, 1771 in our analysis, since the Pseudophilotes species complex is frequently attributed to represent just a clade within a more inclusive genus Scolitantides Hübner, (1819) [2, 26].
Specimens had either been dried or stored in 99% ethanol after collection. Subsequently, samples were stored at − 20 °C until DNA extraction. The selected mitochondrial (mt) and nuclear (nc) genetic markers (COI, 28S, wg, ITS2, EF1α) were amplified with varying success. When necessary, new specific PCR primers were used (see Additional file 2), as the specimens had been collected between the years 2000 and 2014; older samples performed often worse in Polymerase Chain Reaction (PCR) than the more recent ones.
Extraction and amplification of mt and nc DNA was conducted in two different laboratories, the Canadian Centre for DNA Barcoding (CCDB) (University of Guelph, Guelph, Canada) and the Department of Botany and Biodiversity Research (University of Vienna, Austria), according to the following procedures. In the CCDB lab, total DNA was extracted from one leg of each individual on Biomek FX liquid handling robot using a semi-automated DNA extraction protocol [27] on glass fiber plates (PALL Acroprep 96 with 3 μm GF membrane over 0.2 μm Bioinert membrane). DNA was eluted in 35-40 μl of ddH20 pre-warmed to 56 °C and stored at − 80 °C. PCR and sequencing for mtDNA were carried out following standard procedures for Lepidoptera [28, 29]. A fragment of 658 bp of the mt COI gene corresponding to the barcoding region was obtained. In the Vienna lab, total DNA was extracted from legs of butterflies homogenized with ceramic beads using a Precellys 24 homogenizer set to 5000 min-1 for 2x20s. DNA extraction was performed according to the standard protocol supplied with the Analytik Jena innuPREP DNA Micro Kit (Analytik Jena AG). Four nuclear gene fragments were amplified using the Fermentas PCR system. PCR reactions were set up as follows: 2.5 μl of 10× (NH4)2SO4 PCR buffer, 2.5 μl MgCl2 (25 mM/l), 0.5 μl dNTPs (10 mM/μl), 0.5 μl of each primer (10 pg/μl), 0.2 μl BSA, 1 μl template DNA, 0.2 μl Taq polymerase and filled to 25 μl with PCR-grade H2O. PCR reactions were purified by digestion with shrimp alkaline phosphatase and exonuclease for 15 min at 37 °C followed by 15 min at 80 °C for enzyme deactivation. Sequencing reactions were set up with 1 μl ABI BigDye 3.1 (Applied Biosystems, Carlsbad, CA, USA), 1 μl primer, 1.5 μl sequencing buffer, 1 μl template DNA and filled to 10 μl with PCR grade H2O. Primer names, references, primer sequences as well as respective annealing temperature and time are shown in Additional file 2. Sequences were obtained by using an ABI capillary sequencer following the manufacturer’s recommendations. All gene fragments were sequenced in both directions and they were edited and assembled using CodonCode Aligner 6.0.2. All sequences were submitted to GenBank (Accession Numbers in Additional file 1) and in BOLD system repository (dataset name: DS-PSEUDOPH; DOI: dx.doi.org/10.5883/DS-PSEUDOPH).
Data set compilation and tree reconstruction procedures
To reconstruct the phylogenetic relationships of Pseudophilotes, the 96 specimens sequenced in our labs were complemented by 62 publicly available specimens of Pseudophilotes and 53 outgroup species of the subfamily Polyommatinae and two species of the subfamily Lycaeninae, mainly selected from Vila et al. [30] (Additional file 1).
All mt and nc protein coding genes were aligned using CodonCode Aligner 6.0.2. The nc ribosomal 28S rRNA gene fragment and the nc ITS2 sequences were aligned with the software MAFFT v7.245, using the iterative refinement method G-INS-i [31] via the MAFFT online server (http://mafft.cbrc.jp/alignment/server/). As the nc ITS2 region consists of highly variable sections, its alignment remained partly ambiguous. We therefore used the software Aliscore v.2.0 [32] to identify ambiguously aligned or randomly similar sections (RSS) within the nc ITS2 alignment. Aliscore was applied with the following conditions: gaps were treated as ambiguous characters, a window size of six positions was selected and the maximum number of pairs was compared to check for replications. Taken together, the resulting alignments of all mt and nc target fragments consisted of 3929 bp.
Haplotype diversity and the number of variable sites of mt and nc target fragments were calculated using DnaSP 5.0 [33]. To represent the genealogical relationships among mtDNA haplotypes we calculated a Median Joining (MJ) network using NETWORK 5 [34].
Pseudophilotes relationships were assessed via Maximum Likelihood (ML), including all mt and nc target fragments (3929 bp), and using the tree reconstruction method IQ-TREE v.1.61 [35]. Prior to analysis, the data set was subdivided into eleven partitions, according to codon positions of the protein coding genes and the single ribosomal genes. To reduce over-parameterization and increase model fit, these partitions were further merged if possible and for each of the remaining partitions, the best fitting model was estimated within IQ-TREE, which resembles the partitioning algorithm of the software PartitionFinder v2.0 [36]. Node support was assessed by ultrafast bootstrap (UFBoot) analysis [37] with 1000 replications.
Multilocus coalescent species delimination
We tested the status of all Pseudophilotes species according to current taxonomy with the multilocus Bayesian Phylogenetics and Phylogeography software (BPP v4.0; [38]). Within BPP we employed the joint species delimitation and species-tree inference approach, for which the species tree inferred from the ML tree reconstruction was used as a guide tree. Prior to these analyses, we excluded all outgroups except Glaucopsyche, Philotes and Scolitantides, which resulted in a condensed data set, comprising 169 specimens and 3253 bp. BPP now implements inverse gamma priors for the parameters theta (θ) and tau (τ). This allows integrating out θ estimates analytically, which helps with moves between trees and delimitation models. We used an inverse gamma prior with mean 0.002 for θ and 0.001 for τ. The analyses were run for 200,000 generations with a burn-in of 10,000 generations and a sample frequency of five and were run three times to confirm consistency between runs.
Divergence times estimations
Divergence times of the species were further assessed using a species tree approach on the condensed data set in *BEAST 1.8.2. [39]. Each Pseudophilotes specimen in the dataset was assigned to one of the potential species delimited with a posterior probability of > 0.95 in BPP. The condensed dataset was further partitioned into the five gene fragments, which were subsequently loaded into PartitionFinder v2.0 to find the best fitting model scheme for each gene. In *BEAST, all substitution, clock and tree models were unlinked. We assigned Brower’s rate of 0.0115 substitutions/site/million years [40] to the COI partition and estimated the rates of the other partitions. A lognormal relaxed clock with uncorrelated rates was assigned to each clock model. The Tree model was set to “Speciation: Yule Process”. *BEAST analyses were run on the CIPRES Science Gateway v.3.3 [41], consisting of 150 million generations with sampling every 10,000 generations. The first 75 million generations were discarded as burn-in and convergence and mixing of the parameters were checked in Tracer 1.6 (http://tree.bio.ed.ac.uk/software/tracer/). Post-burnin samples were subsequently used to construct a maximum clade credibility tree in TreeAnnotator [42].
Tests of demographic equilibrium and population expansion in the mtDNA haplogroup
To test demographic equilibrium or events of recent expansion in the genus, different sets of mtDNA sequences were selected (Table 1) on a geographical basis and taking into account the results of the previous phylogenetic analysis. Demographic equilibrium was tested by calculating FS [43] and R2 [44] parameters, which have both been shown to provide sensitive signals of historical population expansions [44]. Arlequin 3.0 [45] and DnaSP 5.0 [33] were used to compute FS (P < 0.02) and R2 (P < 0.05) respectively, and to test their statistical significance by simulating random samples (10,000 replicates) under the null hypothesis of selective neutrality and constant population size using coalescent algorithms (both modified from Hudson [46]). Expected mismatched distributions and parameters of sudden expansion τ = 2 μ t (τ, sudden demographic expansion parameter; t, true time since population expansion; μ is the substitution rate per gene) were calculated using Arlequin 3.0 by a generalized least-squares approach [47], under models of pure demographic expansion and spatial expansion [48, 49]. The probability of the data under the given model was assessed by the goodness-of-fit test implemented in Arlequin 3.0. Parameter confidence limits were calculated in Arlequin 3.0 through a parametric bootstrap (1000 simulated random samples).
Biogeographical analyses
We applied the R software package BioGeoBEARS v.0.21 [50, 51] to infer the biogeographical history of Pseudophilotes and to test the support for its origin either in Eurasia or in North Africa. BioGeoBEARS uses the Lagrange DEC model [52, 53], as well as likelihood interpretations of the DIVA model [54] and the BayArea model [55]. For each of these biogeographical models, it further implements a parameter describing founder-event speciation (+j). All model setups were compared in a statistical framework allowing the selection of the best-fitting model. The analyses were conducted with the BEAST maximum clade credibility tree (MCC) from the *BEAST analyses, excluding the outgroup species Glaucopsyche spp. This was necessary, as the relationships within this genus are still ambiguous [1] and therefore the unclear primary distribution of Glaucopsyche (Nearctic vs. Palaearctic) would have blurred the biogeographical reconstruction of Pseudophilotes. The following areas were designated (see Fig. 2c): Eurasia (A), Iberian Peninsula (B), North Africa (C), Sardinia (D), and North America (E). The maximum number of ancestral areas was set to three in both sets of analyses.

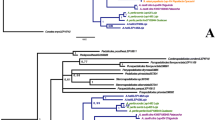
a Chronogram obtained from *BEAST species tree analyses of the COI, ITS2, wg, 28S and EF1α sequences. Nodal scale bars indicate the 95% highest posterior density interval for divergence time estimates in Mya. Black nodes: pP ≥ 0.95; white node: pP < 0.95. b Ancestral area reconstruction results of the best BioGeoBEARS model (BayArea+j). The letters preceding the species indicate the distribution of extant species. c Map (from Natural Earth: www.naturalearthdata.com) showing four biogeographical regions in different colours, as defined in this study. Biogeographical region abbreviations: Eurasian (A), Iberian Peninsula (B), North Africa (C), and Sardinia (D). The four images on the right side of the figure show representatives of the genus Pseudophilotes. Photos: V. Dincă
Results
Genetic diversity
For COI (658 bp), 158 sequences (96 sequences analysed in this study plus 62 sequences retrieved from GenBank) showed 50 different haplotypes (Fig. 1b; Additional file 1) with 64 variable sites. The total haplotype diversity (h) was 0.917 (±0.013 Standard Deviation (SD)). The thirteen sequences of P. barbagiae displayed two mtDNA haplotypes. P. abencerragus sequences displayed 19 mtDNA haplotypes (23 sequences) and h was 0.98 (±0.02 SD) (Table 1). The polyphyletic group including sequences from P. baton, P. vicrama and P. panoptes (haplogroups A1 - A3; Fig. 1b) displayed 20 mtDNA haplotypes (98 sequences) and h was 0.80 (±0.027 SD). The 37 28S sequences (832 bp) displayed 17 haplotypes and 20 variable sites. The 31 wg sequences (403 bp) displayed 19 haplotypes and 34 variable sites. The 35 EF1α sequences (1154 bp) displayed 72 variable sites. The 25 ITS2 sequences (627 bp) displayed 13 haplotypes and 36 variable sites.
Phylogenetic reconstruction and coalescent species delimitation
The ML tree reconstruction (see Additional file 3) recovered a clade consisting of Philotes sonorensis and Scolitantides orion as the closest relative to Pseudophilotes (Bootstrap support, BS = 98). Our results further support the monophyly of the genus Pseudophilotes (BS = 96) recognizing two well-supported main clades. One of them comprised P. bavius (distributed from the Balkan Peninsula and Romania eastwards to Turkey, southern Russia and Iran), and the Maghrebian endemic species P. fatma (BS = 100). In the other clade (BS = 100), P. abencerragus (distributed from Israel across the Maghreb into the Iberian Peninsula) was recovered as sister to all remaining species. This latter clade (BS = 96) was split into the Sardinian P. barbagiae (BS = 100) and a clade comprising P. vicrama, P. baton and P. panoptes (BS = 90). Although this clade shows three distinct groups, all species accepted in current taxonomy appeared polyphyletic with some members of P. panoptes scattered in the P. baton clade and members of P. baton appearing in P. vicrama. In contrast, the species delimitation analyses in BPP recovered all different taxonomical units of Pseudophilotes as species with highest posterior probabilities (pP = 1.0).
Divergence time estimations
The relationships inferred with ML were congruent with the results of the *BEAST analyses (Fig. 2a), except for the position of P. vicrama, which appeared as sister group to P. panoptes in the ML analyses (although with low support, Additional file 3) and as sister group to P. baton in the *BEAST analyses (well supported, Fig. 2a). The chronogram in Fig. 2a shows the median ages and 95% highest probability density (95% HPDs) computed from the post burn-in posterior topologies. We estimated the origin of the genus Pseudophilotes at 5.71 Mya (95% HPD: 3.24–9.13 Mya). The split between P. bavius and P. fatma was estimated at 1.65 Mya (95% HPD: 0.79–3.13 Mya). The split between P. abencerragus and the remaining species was estimated at 1.25 Mya (95% HPD: 0.59–2.24 Mya) and finally the emergence of P. barbagiae was estimated at 0.74 Mya (95% HPD: 0.32–1.32 Mya).
Geographic distribution of mtDNA haplotypes
The mtDNA haplotype network (Fig. 1b) highlighted high haplotype diversity for P. abencerragus (0.98 ± 0.02) and P. panoptes (0.85 ± 0.06; haplogroup A3) and a star-like configuration of the haplogroup A2, where the central haplotype H32, including P. vicrama haplotypes distributed from Finland to Iran, was also shared with P. baton from Corsica and southern Italy (see Additional file 1). The H23 haplotype, including almost all P. baton sequences, is instead widely distributed from Spain to southern Italy and was also shared with two P. panoptes individuals, similarly to the H22 haplotype. Besides two haplotypes (H22 and H23) shared with P. baton, the analyses highlighted that P. panoptes (A3) owns nine characteristic (i.e. not shared with any other taxon) mtDNA haplotypes. The thirteen sequences of P. barbagiae displayed two haplotypes that were connected to a central unresolved loop with P. abencerragus. Eight and nine mutational steps joined P. barbagiae with P. abencerragus from North Africa and Europe respectively, while thirteen mutational steps joined P. barbagiae with P. abencerragus from Israel. In contrast, 16 mutational steps joined P. barbagiae with the closest haplotype of the A1-A3 haplogroup (including P. panoptes, P. baton and P. vicrama), a result not reflected in the molecular analyses using both mt and nc markers, where P. barbagiae was recovered as sister to the clade including P. panoptes, P. baton and P. vicrama (Fig. 1a, Additional file 3).
Tests of demographic equilibrium
According to both the Fs and R2 parameters, calculated for six mtDNA sequence sets (Table 1), the null hypothesis of constant population size could be rejected for two phylogeographic units (set in bold in Table 1): P. abencerragus (number of mtDNA sequences (N) = 23) and haplogroup A2 (N = 40) including P. vicrama sequences and P. baton from Corsica and southern Italy (Fig. 1b). The mismatched distribution of these sequence sets was also examined according to a sudden expansion model. Goodness of fit tests indicated significant deviations from expected distributions only for P. abencerragus that showed a multimodal and ragged shape, revealing demographic equilibrium or a stable population [53]. Conversely, for the haplogroup (A2) the parameter τ = 2 μ t could be used to estimate the time (t) elapsed from population expansion. According to Brower’s mutation rate [40], the demographic expansion of the haplogroup A2 could be traced back to about 41 thousand years ago (kya) (0−207 kya).
Biogeographical analyses
The results of the ancestral area reconstructions in BioGeoBEARS are summarized in Tables 2 and 3. All models, which implemented founder-event speciation parameter (+j), were significantly better supported than their more simple counterparts. The AIC weights further showed the DEC + j model to achieve the highest relative probability (70%; see Table 2). However, the geographical origin of Pseudophilotes remained unclear, as the DEC + j model indicated several potential area combinations, none of these significant (Fig. 2b). Also the ancestral area for the P. abencerragus group (Pseudophilotes s. str.) remained unclear, with Eurasia+Iberia+Africa having the highest probability (60%; see Table 3). More details on the biogeographic modelling are given in the discussion.
Discussion
The taxonomical status of the genus Pseudophilotes and its species
Mitochondrial and nuclear phylogenetic analyses (Figs. 1b, 2a, Additional file 3) confirmed the findings of Ugelvig et al. [1] that Pseudophilotes and Scolitantides are closely related, but clearly separated from each other. It therefore seems justified to maintain Pseudophilotes and Scolitantides as distinct genera (e.g. [56]), despite the fact that other studies have joined them [2, 3]. Our analyses further corroborate the dichotomy between the clade of the species P. bavius and P. fatma, (equivalent to what has been proposed as the genus Rubrapterus Korshunov, 1987), and Pseudophilotes s. str. This evolutionary split between the Rubrapterus clade and the remaining Pseudophilotes follows larval food plant preferences. P. bavius and fatma feed on Salvia spp. [57, 58], whereas Lamiaceae genera other than Salvia (mostly Thymus spp., but also Cleonia, Calamintha, and rarely Cuscuta as a parasite on thymes) are preferred by all other species [3, 59]. Within P. bavius, several subspecies have been described in Europe (e.g. hungarica Diószeghy, 1913; macedonicus Schulte, 1958; casimiri Hemming, 1932; see for example Tshikolovets [3]), but our dataset did not reveal any clear spatial genetic differentiation below the species level. P. fatma has sometimes been treated as a subspecies of P. bavius [60, 61], but given morphological [62] and genetic differences it is probably best regarded as a distinct species.
Among the Thymus-feeders, the southern-mediterranean species P. abencerragus is clearly distinct from all other species. Within P. abencerragus, the sequences obtained from North African and Israeli specimens appear to be derived from those of Iberian individuals, reflecting previously proposed subspecies (North Africa: colonarium Turati, 1927; Israel: nabateus Graves, 1925 and amelia Hemming, 1927). However the support of these relationships is weak, prohibiting final statements on their taxonomic status. The Sardinian endemic P. barbagiae belongs to the baton-group but is clearly separated from the clade comprising P. baton, P. panoptes and P. vicrama. Although species delimitation analyses support the status of all of the latter (Fig. 2a), our tree reconstruction analyses (Additional file 3) and the mtDNA haplotype network (Fig. 1b) indicate a more complex pattern representing a challenging taxonomic case that requires further study to clarify species boundaries and their precise geographic distribution, as well as possible hybrid zones.
Biogeographical history of the genus Pseudophilotes
Our results are consistent with other studies on Polyommatinae [63] indicating that the most recent common ancestor of the genus Pseudophilotes probably lived in the Messinian period (5.33–7.25 Mya). This ancestor had already colonized Lamiaceae as larval hosts, a synapomorphic character differentiating Pseudophilotes from Scolitantides and Philotes; affiliation with Lamiaceae is a rather rare trait in the Polyommatinae worldwide [64, 65].
During the Messinian, the closure of the Strait of Gibraltar led to an almost complete desiccation of the Mediterranean Sea, permitting massive faunal exchange between southern Europe and the Maghreb region [23, 24]. Within Pseudophilotes s. str., the ancestor of P. abencerragus split into the extant P. abencerragus that occurs in southwestern Europe and from northwestern Africa eastwards through to southern Israel, and the ancestor of P. baton, P. panoptes, P. vicrama and P. barbagiae. Although climate modelling analyses [66] showed differences in climate requirements that possibly suggest different evolutionary histories for the more western P. baton and its eastern vicariant P. vicrama, the two species are parapatric in Central Europe (Fig. 1a) and all P. baton analysed from populations in Corsica share their mtDNA haplotype (H32) with P. vicrama. The large geographical distance of the Corsican populations from the extant contact zone in eastern central Europe prompts some degree of historical introgression between these two species. This hypothesis is supported both from the distribution of the H32 haplotype (Fig. 1a; Additional file 1) and the genetic traces of demographic expansion in P. vicrama (haplogroup A2: Fig. 1b) pointing to the Late Pleistocene (about 41 kya), when sea-level oscillations created land bridges between Corsica and mainland [25] permitting faunal exchange.
Isolation of the Sardinian P. barbagiae
The Sardinian Blue butterfly, Pseudophilotes barbagiae [6] is restricted to mountainous habitats on the slopes of the Gennargentu massif in Central Sardinia. Its larvae develop exclusively on Thymus herba-barona, which mostly grows between 800 and 2000 m [67,68,69]. In Sardinia, this plant is restricted to a few slopes in the Barbagiae and Gennargentu Mountains, and on Mount Limbara, although in other Mediterranean regions it has a much wider distribution. Interestingly, the plant also occurs in Corsica, while the butterfly does not. In the International Union for Conservation of Nature (IUCN) Red List of Threatened Species [70], P. barbagiae is assessed as Data Deficient, as little is known about this butterfly. The thyme species it feeds on has not yet been assessed by the IUCN, however according to the European Red List of Vascular Plants [71] this plant species is not threatened. Our analyses of combined mt and nc markers show that P. barbagiae appears more closely related to the clade including P. baton, P. vicrama and P. panoptes than to P. abencerragus. On the other hand, the haplotype network (Fig. 1b) depicts a somewhat different picture. Here, P. barbagiae results more closely related to P. abencerragus than to P. baton. Such discrepancies between analyses based solely on the COI region and others based on combined data, including nuclear markers, are quite common, since the barcoding region by itself is not sufficient for drawing phylogenetic conclusions and mitochondrial inheritance often follows different pathways that do not necessarily reflect the evolutionary history of species.
Our hypothesis is that during the Pleistocene, when Sardinia was connected with Corsica and the mainland due to glacial/interglacial sea-level oscillations [25], gene flow occurred among baton-group European populations and Sardinia. Afterwards, gene flow among these became interrupted and from this founder population P. barbagiae evolved into an endemic species about 0.74 Mya. Interestingly, the mtDNA of P. baton from Corsica is of the vicrama-type (H32) shared with individuals of P. vicrama distributed from Finland to Iran (Fig. 1b; Additional file 1). An expansion from Africa, as it has been hypothesized for two other Sardinian endemic butterflies (Maniola nurag: Kreuzinger et al. [72] and Papilio hospiton: Pittaway et al. [73]), does not seem a plausible scenario, despite the smaller geographical distance between Sardinia and the African continent.
Conclusions
This study confirmed the monophyly of the genus Pseudophilotes and shed further light on the validity of its component species, as described by current taxonomy. We found evidence for a deeper split between the Salvia feeders (P. bavius and P. fatma) and the Thymus feeders (Pseudophilotes s. str.). However, the precise species boundaries and possible signals of introgression and hybridization within the baton-group further merit closer scrutiny. Finally, P. barbagiae turned out to be a young endemic, but clearly with European instead of North African origin.
Abbreviations
- 28S:
-
28S ribosomal gene
- BPP v4.0:
-
Bayesian Phylogenetics and Phylogeography software
- BS:
-
Bootstrap support
- CCDB:
-
Canadian Centre for DNA Barcoding
- COI:
-
Cytochrome c oxidase subunit 1 gene
- EF1α:
-
Elongation Factor 1α gene
- h :
-
Haplotype diversity
- H:
-
Number of unique mtDNA haplotypes
- HPD:
-
Highest probability density
- ITS2:
-
Internal Transcribed Spacer 2 gene
- IUCN:
-
International Union for Conservation of Nature
- kya:
-
Thousand years ago
- MCC:
-
Maximum clade credibility tree
- MJ:
-
Median Joining
- ML:
-
Maximum Likelihood
- MSC:
-
Messinian Salinity Crisis
- mt:
-
Mitochondrial
- Mya:
-
Million years ago
- N:
-
Number of mtDNA sequences
- nc:
-
Nuclear
- PCR:
-
Polymerase Chain Reaction
- pP:
-
Posterior probabilities
- RSS:
-
Randomly similar sections
- SD:
-
Standard Deviation
- wg:
-
Wingless gene
References
Ugelvig LV, Vila R, Pierce NE, Nash DR. A phylogenetic revision of the Glaucopsyche section (Lepidoptera: Lycaenidae), with special focus on the Phengaris–Maculinea clade. Mol Phylogenet Evol. 2011;61:237–43.
Kudrna O, Harpke A, Lux K, Pennerstorfer J, Schweiger O, Settele J, Wiemers M. Distribution atlas of butterflies in Europe. Halle: Gesellschaft für Schmetterlingsschutz eV; 2011. p. 576.
Tshikolovets VV. Butterflies of Europe & the Mediterranean area. Pardubice: Tshikolovets publications; 2011.
Todisco V, Fiedler K, Puliga E, Dinca V, Voda R, Grill A. The origin of the Sardinian Blue, Pseudophilotes barbagiae (Lepidoptera, Lycaenidae): out-of-Europe or out-of-African? Genome. 2015;58(Suppl 5):288.
Dincă V, Montagud S, Talavera G, Hernández-Roldán J, Munguira ML, García-Barros E, Hebert PDN, Vila R. DNA barcode reference library for Iberian butterflies enables a continental-scale preview of potential cryptic diversity. Sci Rep. 2015;5:12395.
De Prins W, Van der Poorten D. Overzicht van het genus Pseudophilotes in Europa en Noord Afrika met beschrijving van een soort uit Sardinie, nieuw voor de wetenschap. Phegea. 1982;10(Suppl 2):61–76.
Castellanos-Morales G, Gámez N, Castillo-Gámez RA, Eguiarte LE. Peripatric speciation of an endemic species driven by Pleistocene climate change: the case of the Mexican prairie dog (Cynomys mexicanus). Mol Phylogenet Evol. 2016;94:171–81.
Ketmaier V, Argano R, Caccone A. Phylogeography and molecular rates of subterranean aquatic stenasellid isopods with a peri-Tyrrhenian distribution. Mol Ecol. 2003;12:547–55.
Sbordoni V, Allegrucci G, Todisco V. Il genere Dolichopoda in Sardegna: filogenesi molecolare e ipotesi sulla evoluzione del popolamento. Studi Trentini Sci Nat Acta Biol. 2004;81:103–11. ISSN 0392-0542
Grill A, Raijmann L, Van Ginkel W, Gkioka E, Menken S. Genetic differentiation and natural hybridization between the Sardinian endemic Maniola nurag and the European Maniola jurtina. J Evol Biol. 2007;20:1255–70.
Gerace O. Un modello spazio-temporale per il popolamento dell’areale Sardo-Corso, dell’arcipelago toscano e della zona costiera della Toscana meridionale relativo ai Lepidoptera Rhopalocera (Farfalle diurne). Atti Soc Toscana Sci Nat, Mem, Serie B. 2013;120:65–82.
Grill A, Casula P, Lecis R, Menken SBJ. Endemism in Sardinia. In: Weiss S, Ferrand N, editors. Phylogeography of southern European refugia. Dordrecht: Springer; 2007. p. 273–96.
Zachos EF, Habel JC, editors. Biodiversity hotspots: distribution and protection of conservation priority areas. Berlin-Heidelberg: Springer- Verlag; 2011.
Caccone A, Milinkovitch MC, Sbordoni V, Powell JR. Mitochondrial DNA rates and biogeography in European newts (genus Euproctus). Syst Biol. 1997;46:126–44.
Oliviero M, Bologna M, Monciotti A, Amnesi F, Mariottini P. Molecular phylogenetics of the Italian Podarcis lizards (Reptilia, Lacertidae). Ital J Zool. 1998;65:315–24.
Guicking D, Joger U, Wink M. Molecular phylogeography of the viperine snake Natrix maura (Serpentes: Colubridae): evidence for strong intraspecific differentiation. Org Divers Evol. 2008;8:130–45.
Marchi A, Addis G, Hermosa VE, Crnjar R. Genetic divergence and evolution of Polyommatus coridon gennargenti (Lepidoptera: Lycaenidae) in Sardinia. Heredity. 1996;77:16–22.
Caccone A, Sbordoni V. Mitochondrial biogeography of cave life: a study using mitochondrial DNA from Bathysciine beetles. Evolution. 2001;55:122–30.
Martinsen L, Venanzetti F, Bachmann L. Phylogeography and mitochondrial DNA divergence in Dolichopoda cave crickets (Orthoptera, Rhahidophoridae). Hereditas. 2009;146:33–45.
Grill A, Gkiokia E, Alvarez N. Evolutionary history and patterns of differentiation among European Maniola butterflies (Lepidoptera: Satyrinae). Eur J Entomol. 2006;103:613–8.
Vuataz L, Rutschmann S, Monaghan MT, Sartori M. Molecular phylogeny and timing of diversification in alpine Rhithrogena (Ephemeroptera: Heptageniidae). BMC Evol Biol. 2016;16:194.
Duggen S, Hoernle K, Van Den Bogaard P, Rüpke L, Morgan JP. Deep roots of the Messinian salinity crisis. Nature. 2003;422(Suppl 6932):602–6.
Blondel J, Aronson J. Biology and wildlife of the Mediterranean region. Oxford: Oxford University Press; 1999.
Dobson R, Wright A. Faunal relationships and zoogeographical affinities of mammals in north-west Africa. J Biogeogr. 2000;27(Suppl 2):417–24.
Arias C, Azzararoli A, Bigazzi G, Bonadonna FP. Magnetostratigraphy and Pliocene–Pleistocene boundary in Italy. Quat Res. 1980;13:65–74.
Gorbunov PY. The butterflies of Russia: classification, genitalia, keys for identification (Lepidoptera: Hesperioidea and Papilionidea). Ekaterinburg: Thesis Publishers; 2001. p. 320.
Ivanova NV, deWaard JR, Hebert PDN. An inexpensive, automation friendly protocol for recovering high-quality DNA. Mol Ecol Notes. 2006;6:998–1002.
Hajibabaei M, deWaard JR, Ivanova NV, Ratnasingham S, Dooh RT, Kirk SL, Mackie PM, Hebert PDN. Critical factors for assembling a high volume of DNA barcodes. Philos Trans R Soc B. 2005;360:1959–67.
deWaard JR, Ivanova NV, Hajibabaei M, Hebert PDN. Assembling DNA barcodes: analytical protocols. In: Martin C, editor. Methods in molecular biology: environmental genetics. Totowa: Humana Press; 2008. p. 275–93.
Vila R, Bell CD, Macniven R, Goldman-Huertas B, Ree RH, Marshall CR, Balint Z, Johnson K, Benyamini D, Pierce NE. Phylogeny and palaeoecology of Polyommatus blue butterflies show Beringia was a climate-regulated gateway to the New World. Proc R Soc B. 2011;278(Suppl 1719):2737–44.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(Suppl 4):772–80.
Misof B, Misof K. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst Biol. 2009;58:21–34.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(Suppl 1):268–74.
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2017;34(Suppl 3):772–3.
Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30(Suppl 5):1188–95.
Yang Z, Rannala B. Unguided species delimitation using DNA sequence data from multiple loci. Mol Biol Evol. 2014;31:3125–35.
Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010;27(Suppl 3):570–80.
Brower AVZ. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato, inferred from patterns of mitochondrial DNA evolution. Proc Natl Acad Sci U S A. 1994;91:6491–5.
Miller MA, Pfeiffer WT, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In: GCE 2010, IEEE; 2010. p. 1–8.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Ramos-Onsins S, Rozas J. Statistical properties of new neutrality tests against population growth. Mol Biol Evol. 2002;19:2092–100.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinformatics Online. 2005;1:47–50.
Hudson RR. Gene genealogies and the coalescent process. Oxford surveys in evolutionary biology. In: Futuyma DJ, Antonovics JD, editors. , vol. 7. Oxford: Oxford University Press; 1990. p. 1–44.
Schneider S, Excoffier L. Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: application to human mitochondrial DNA. Genetics. 1999;152:1079–89.
Ray N, Currat M, Excoffier L. Intra-deme molecular diversity in spatially expanding populations. Mol Biol Evol. 2003;20:76–86.
Excoffier L. Patterns of DNA sequence diversity and genetic structure after a range expansion: lessons from the infinite-island model. Mol Ecol. 2004;13:853–64.
Matzke NJ. Probabilistic historical biogeography: new models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing. Front Biogeogr. 2013;5(Suppl 4):242–8.
Matzke NJ. Probabilistic historical biogeography: New models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing. Berkeley: University of California; 2013. p. 1–240. Dissertation
Ree RH. Detecting the historical signature of key innovations using stochastic models of character evolution and cladogenesis. Evolution. 2005;59:257–65.
Ree RH, Smith SA. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst Biol. 2008;57:4–14.
Ronquist F. Dispersal-Vicariance analysis: a new approach to the quantification of historical biogeography. Syst Biol. 1997;46:195–203.
Landis M, Matzke NJ, Moore BR, Huelsenbeck JP. Bayesian analysis of biogeography when the number of areas is large. Syst Biol. 2013;62(Suppl 6):789–804.
Hesselbarth G, Van Oorschot H, Wagener S. Die Tagfalter der Türkei, vol. 1. Bocholt: Selbstverlag Sigbert Wagener; 1995. p. 754.
Német E, Czekes Z, Markó B, Rákosy L. Host plant preference in the protected myrmecophilous Transylvanian blue (Pseudophilotes bavius hungarica) butterfly (Lepidoptera: Lycaenidae) and its relationship with potential ant partners. J Insect Conserv. 2016;20(Suppl 5):765–72.
Tennent J. The butterflies of Morocco, Algeria and Tunisia. Wallingford: Gem Publishers; 1996. p. xx + 217.
Muñoz Sariot MG. Biología y ecología de los Licénidos Españoles. Granada: Ediciones Autor; 2011. p. 383.
Higgins LG. The classification of European butterflies. London: Collins; 1975.
Tolman T, Lewington R. Collins butterfly guide. London: Harper-Collins Publishers; 2008.
Tarrier MR, Delacre J. Les papillons de jour du Maroc. Biotope: Mèze; 2008.
Talavera G, Lukhtanov VA, Pierce NE, Vila R. Establishing criteria for higher-level classification using molecular data: the systematics of Polyommatus blue butterflies (Lepidoptera, Lycaenidae). Cladistics. 2013;29:166–92.
Fiedler K. Systematic, evolutionary, and ecological implications of myrmecophily within the Lycaenidae (Insecta: Lepidoptera: Papilionoidea). Bonner zool Monogr. 1991;31:1–210.
Fiedler K. Lycaenid butterflies and plants: is myrmecophily associated with particular hostplant preferences? Ethol Ecol Evol. 1995;7(Suppl 2):107–32.
Settele J, Kudrna O, Harpke A, Kühn I, van Swaay C, Verovnik R, Warren M, Wiemers M, Hanspach J, Hickler T, Kühn E, van Halder I, Veling K, Vliegenthart A, Wynhoff I, Schweiger O. Climatic risk atlas of European butterflies. BioRisk. 2008;1:1–712.
Pignatti S. Flora d’Italia, vol. 3. Bologna: Edizioni Agricole; 1982.
Molins A, Bacchetta G, Rosato M, Rosselló JA, Mayo M. Molecular phylogeography of Thymus herba-barona (Lamiaceae): insight into the evolutionary history of the flora of the western Mediterranean islands. Taxon. 2011;60(Suppl 5):1295–305.
Leigheb G, Cameron-Curry V. Observations on the biology and distribution of Pseudophilotes barbagiae (Lycaenidae: Polyommatini). Nota Lepid. 1998;21(Suppl 1):66–73.
van Swaay C, Wynhoff I, Wiemers M, Katbeh-Bader A, Power A, Benyamini D, Tzirkalli E, Balletto E, Monteiro E, Karaçetin E, Franeta F, Pe’er G, Welch H, Thompson K, Pamperis L, Dapporto L, Šašić M, López Munguira M, Micevski N, Dupont P, Garcia-Pereira P, Moulai R, Caruana R, Verovnik R, Bonelli S, Beshkov S. Pseudophilotes barbagiae. The IUCN Red List of Threatened Species 2015:e.T173207A64553436. https://doi.org/10.2305/IUCN.UK.2015-2.RLTS.T173207A64553436.en. Accessed 28 Sept 2017.
Bilz M, Kell SP, Maxted N, Lansdown RV. European Red List of Vascular Plants. Luxembourg: Publications Office of the European Union; 2011.
Kreuzinger A, Fiedler K, Letsch H, Grill A. Tracing the radiation of Maniola (Nymphalidae) butterflies: new insights from phylogeography hint at one single incompletely differentiated species complex. Ecol Evol. 2015;5(Suppl 1):46–58.
Pittaway AR, Larsen TB, Clarke CA, Smith CR, Crnjar R, Clarke FMM. Papilio saharae Oberthür, 1879, specifically distinct from Papilio machaon Linnaeus, 1758 (Lepidoptera: Papilionidae). Entomologist’s Gazette. 1994;45:223–49.
Lukhtanov V, Lukhtanov A. Die Tagfalter Nordwestasiens (Lepidoptera, Diurna), vol. 3. Marktleuthen: Herbipoliana; 1994. p. 440.
Nazari V. Butterflies of Iran. Tehran: Dayereye-Sabz Publications; 2003.
Tshikolovets VV, Bidzilya OV, Golovushkin MI. The butterflies of Transbaikal Siberia. Brno-Kyiv: V.V. Tshikolovets Publications; 2002. p. 220.
Tshikolovets VV. The Butterflies of Tajikistan. Kyiv–Brno: Publ. by the author; 2003. p. 1–500. col. pl. I-LXXVII.
Acknowledgements
We are extremely grateful to V. Nazari, P. Huemer, A. Hausmann, M. Mutanen, A. Sourakov and B. Dardenne (DNA barcoding Lepidoptera of Upper-Normandy region’ project) for providing sequences and two anonymous reviewers for their helpful comments. Finally, Canadian Centre for DNA Biotecnology (CCDB) is acknowledged for technical support.
Funding
FWF grant V168-B17 to A. Grill, a Marie Curie IOF within the 7th European Community Framework Programme to V. Dincă (project no. 625997), the European Union’s Seventh Framework programme for research and innovation under the Marie Skłodowska-Curie grant agreement No 609402–2020 researchers: Train to Move (T2M) to R. Vodă, the Russian Science Foundation to the Zoological Institute of the Russian Academy of Sciences (N 14–14-00541) to V. Lukhtanov. Genetic analyses were further supported through funds of the Faculty of Life Sciences, University of Vienna, Austria.
Availability of data and materials
The datasets generated during the current study are available in the BOLD system repository: Dataset name: DS-PSEUDOPH; DOI: dx.doi.org/10.5883/DS-PSEUDOPH.
Specimens collection statement
We declare that all samples analysed in this study were collected complying with institutional, national, and international guidelines.
Author information
Authors and Affiliations
Contributions
VT, AG and KF designed the study. AG, RV, VD and VL collected samples. VT, BG, RV, VD and VL sequenced the specimens. VT, HL and VD analysed the data. VT, AG, HL and KF wrote the manuscript. All authors edited the manuscript and approved its final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional files
Additional file 1:
Dataset analysed in this study. Taxon = Pseudophilotes and outgroup species analysed in this study. Sample numbers (N), collection locality, population code, mtDNA haplotype code and GenBank code. (XLSX 24 kb)
Additional file 2:
Primer names, references, primer sequences as well as respective annealing temperatures used to amplify nuclear and mitochondrial markers in the present study. (XLSX 16 kb)
Additional file 3:
Maximum-likelihood tree. Maximum-likelihood phylogeny inferred from the combined genes dataset of Pseudophilotes and outgroups, with bootstrap support (%) indicated for the seven species analysed. (PDF 515 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Todisco, V., Grill, A., Fiedler, K. et al. Molecular phylogeny of the Palaearctic butterfly genus Pseudophilotes (Lepidoptera: Lycaenidae) with focus on the Sardinian endemic P. barbagiae. BMC Zool 3, 4 (2018). https://doi.org/10.1186/s40850-018-0032-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40850-018-0032-7