
Abstract
Background
The aim of this study was to determine the in-vitro anti-ROS and in-vivo anticancer activities of Tabebuia pallida leaves (abbreviated as TPL) against Ehrlich’s ascite carcinoma (EAC) cells.
Methods
The crude ethanolic extract (CEE) (70% v/v) was fractionated successively with different solvents to get petroleum ether (PEF), chloroform (CHF), ethyl acetate (EAF) and aqueous fraction (AQF). The phytochemical studies were done by standard spectrophotometric methods. The in-vitro cytotoxic and in-vivo anticancer activity were evaluated against brine shrimp nauplii and EAC-induced tumor bearing mice, respectively.
Results
Among the extracts, the EAF showed the highest phenolic (158.17 ± 1.54 mg/g GAE) and flavonoid (5.43 ± 0.017 mg/g CAE) contents. The total antioxidant capacity of the EAF was 2.30 ± 0.080, which was higher than other extracts and the standard catechin (2.16 ± 0.038). The ferrous reducing antioxidant capacity was in the following order: AA>EAF > CEE > AQF > CHF > PEF. In DPPH and hydroxyl radical scavenging assay, the EAF showed the highest DPPH and hydroxyl radical scavenging activity with IC50 of 6 ± 0.25 μg/ml and 3.58 ± 0.28 μg/ml, respectively, when compared to the standard BHT (IC50 of 16.08 ± 0.28 μg/ml). Also, EAF showed potent inhibitory activity against lipid peroxidation with IC50 of 14.33 ± 0.14 μg/ml. A positive correlation (p < 0.01) between the total phenolics and antioxidant potentials as well as lipid peroxidation inhibition with the hydroxyl radical scavenging was observed. In addition, the EAF showed the moderate cytotoxic effect with ED50of 8.50 ± 0.70 μg/ml and significant cell growth inhibition (71.17 ± 0.44%) when compared to the standards and the cell growth inhibition was positively correlated (p < 0.01) with phenolic contents.
Conclusions
Our findings suggest that the leaves of T. pallida might be considered as a new natural source for antioxidant and anticancer compounds, which could be a potent and novel candidate for anticancer therapy.
Similar content being viewed by others


Introduction
In the last decades, our understanding about the term “oxidative stress” (OS), an imbalance between the production and scavenging of reactive oxygen species (ROS) by a living system (e.g. cells), has been widened considerably [1].OS induced by ROS such as hydroxyl, peroxyl and superoxide radicals, have the tendency to become stable through electron pairing with biological macromolecules such as proteins, lipids and DNA in healthy human cells [2, 3]. Consequently, the formation of lipid peroxidation along with DNA damage and protein dysfunction leads to etiology of different diseases including cancer, coronary heart disease, neurodegenerative disorders, diabetes, arthritis, inflammation and lung damage [4, 5]. Moreover, the damages can become more prominent due to weakened cellular defense systems. All biological systems have antioxidant defense mechanism that protects against oxidative damages to remove damaged molecules. In many cases, this natural antioxidant defense mechanism can be inefficient; hence dietary intake of antioxidant becomes imperative.
Antioxidants are secondary molecules or metabolites that act as ROS scavenger and activator of cellular antioxidative enzymes to prevent the damages induced by OS in biological system. Some synthetic antioxidants such as BHT, BHA, and tertiary butyl hydroquinone are commonly used in processed foods for preservation and elongation of shelf-life, but their unwanted toxic effects make them restricted to use [6]. Hence, naturally occurring antioxidants from plant polyphenols are getting more attention in terms of practical usage as safe and potent bioactive compounds. It has been reported that the scavenging properties of ROS are mainly due to the presence of bioactive compounds such as, flavonoids, polyphenols, carotenoids and vitamin E and C [7, 8]. Therefore, the antioxidant enriched medicinal plants, fruits and vegetables may contribute to the protection from various diseases including cancer caused by OS [9].
Cancer is a major cause of death throughout the world and the number of individuals living with cancer is increasing day by day. This is not only a problem of Bangladesh but also a global problem. Moreover, the currently available anticancer drugs have limited use due to its health-related other serious complications. It is therefore important to search a novel anticancer drug from other biological sources, which have fewer side effects. Over 60% of currently used anticancer agents are derived from natural sources, including plants, marine organisms and microorganisms [10]. Among them, plants have a long history of use in the treatment of cancer. Out of 250,000 plant species worldwide, more than 3000 species have been reported to have significant anticancer potential [11]. Over the past 30 years, approximately 45% of all anticancer drugs have been derived directly or indirectly from plant compounds, in which 12% are natural products and 33% are semi-synthetic derivatives of natural related products [12, 13]. Different types of cancer cell lines are widely used for the screening of anticancer agents from plant sources. Recently, researchers have focused on the use of Ehrlich Ascites Carcinoma (EAC) (transplantable tumour cells) cells, to investigate the anticancer agents from different plant species [14, 15]. Accumulating evidence suggests that EAC cells have been used as a model cancer cell line for the study of many plant species in cancer research. The in vitro and in vivo anticancer activity of black tea [16], honey [17], white mulberry [18], Syzygium fruticosum [3] and many others in EAC cells has been reported. These published evidences suggest that EAC is now a good model cell line to identify anticancer agents from plant species.
Tabebuia pallida (T. pallida), a member of Bignoniaceae family, is commonly known as white trumpet tree. It is one of the species of the largest genera Tabebuia, distributed in northern Mexico, South to Northern Argentina, Caribbean Islands, Cuba, Chile and Paraguay [19, 20]. To the best of our knowledge, the in vitro and in vivo biological activity of T. pallida has not been reported. Therefore, we performed a detailed literature review on Tabebuia genus and found that a diverse range of secondary metabolites has been isolated (Additional file 1: Table S1) [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56] and various species of Tabebuia have been used for the treatment of different types of diseases, including cancer. In addition, Nirmala et al. [21] reported that β-lapachone is now in clinical trial or drug development phase as plant derived anticancer agents. So, we were encouraged to investigate the anticancer activity of T. pallida in EAC cells-induced tumor bearing mice.
Recently, our group reported that the antioxidant-rich white mulberry has anti-ROS and anticancer activity [18]. Previously, we reported that the highest antioxidants were in the leaves of T. pallida compared to other parts (flowers, stem and root barks) of this plant [57]. Therefore, in this study, the leaves of T. pallida were subjected to evaluate its in vitro effect on ROS and in vivo effect on cancer in EAC-induced tumor bearing mice.
Materials and methods
Plant collection
Leaves of T. pallida were collected from Rajshahi University Campus, Rajshahi, Bangladesh in May 2013. The plant was identified by an expert taxonomist at the Department of Botany, University of Rajshahi, where a voucher specimen (Voucher No. MN-03) was deposited. The dirty materials were removed from leaves before shaded sun drying for several days. After drying, the dried leaves were grounded to coarse powder by a grinding machine. The processed plant materials kept at room temperature (RT) for future use.
Preparation of the extract
The extraction procedure was performed according to Islam et al. [3]. The dried powdered leaves (about 500 g) were soaked in an amber colored extraction bottle with 70% ethanol (1 L × 3 times) and preserved for 7 days at room temperature with occasional shaking and stirring. First cotton filtration was performed to extract and then Whatman No.1 filter paper was used. Afterwards they were concentrated with a rotary evaporator (BibbySterlin Ltd., UK) under reduced pressure at 45 °C to afford 46 g crude extract. Slurry prepared with water was taken in a separating funnel to perform partitioning with petroleum ether, chloroform, ethyl acetate and finally water. After vigorous shaking, the funnel was allowed to stand for a few minutes to obtain petroleum ether fraction (PEF, 13.51 g), chloroform fraction (CHF, 9.98 g), ethyl acetate fraction (EAF, 7.79 g) and aqueous fraction (AQF, 8.50 g), respectively. The process was repeated three times. Finally, the different fractionated parts were evaporated using rotary evaporator at 40 °C.
Chemicals
2,2-diphenyl-1-picrylhydrazyl (DPPH), potassium ferricyanide, catechin (CA), ferrous ammonium sulphate, butylatedhydroxytoluene (BHT), gallic acid (GA), ascorbic acid (AA), aluminum trichloride (AlCl3), trichloro acetic acid (TCA), sodium phosphate, sodium nitrate, ammonium molybdate, 2-deoxyribose, sodium hydroxide, EDTA and FeCl3 were purchased from Sigma Chemical Co. (St. Louis, MO, USA); potassium acetate, phosphate buffer, thiobarbituric acid (TBA), HCl, H2SO4, H2O2were purchased from Sigma-Aldrich, vinblastine sulphate (VBS) from Cipla India, Folin-Ciocalteu phenol reagent and sodium carbonate were obtained from Merck (Darmstadt, Germany).
Determination of total phenolics
Total phenolic contents of the extracts were determined by the modified Folin-Ciocalteu method described by Wolfe et al. [58]. An aliquot of the extract was mixed with 2 ml Folin-Ciocalteu reagent (previously diluted with water 1:10 v/v) and 2 ml (75 g/l) of sodium carbonate. The tubes were vortexed for 15 s and allowed to stand for 20 min at 25 °C for color development. Absorbance was then measured at 760 nm UV- spectrophotometer (Shimadzu, USA). Samples of the extract were evaluated at a final concentration of 0.1 mg/ml. Total phenolic contents were expressed in terms of gallic acid equivalent, GAE (standard curve equation: y = 0.095x + 0.097, R2 = 0.998), mg of GA/g of dry extract. Gallic acid stock solution (5 mg/1 ml methanol) was diluted in six concentrations (2.5-40 μg/ml) to obtain standard curve, where the range of absorbance values was 0.362–3.905 (Additional file 2: Figure S1). Moreover, there were four fractions of sample with the concentrations ranged from 12.5 to 150 μg/ml and the range of absorbance values was 0.103 to 3.854.The experiment was repeated three times at each concentration.
Determination of total flavonoids
Total flavonoids were estimated using aluminum chloride colorimetric assay described by Zhishen et al. [59]. To 0.5 ml of samples/standard, 150 μl of 5% sodium nitrate and 2.5 ml of distilled water were added. After 5 min, 0.3 ml of 10% AlCl3 was added. At 6 min, 1 ml of 0.001 M NaOH and 0.55 ml distilled water was added to the mixture and left at RT for 15 min. Absorbance of the mixtures was measured at 510 nm. Samples of extract were evaluated at a final concentration of 0.1 mg/ml. Total flavonoid contents were expressed in terms of catechin equivalent, CAE (standard curve equation: y = 0.000x + 0.001, R2 = 0.998), mg of CA/g of dry extract. There were six concentrations ranged from 10 to 320 μg/ml of CA with the absorbance values ranged from 0.0046 to 0.092, respectively. The experiment was repeated three times at each concentration.
Determination of total antioxidant capacity
Total antioxidant capacity (TAC) of the extracts was determined by phosphomolybdate method using catechin as a standard reported by Prieto et al. [60]. The assay is based on the reduction of Mo (VI) to Mo (V) by samples and formation of green colored phosphate/Mo(V) complex at acidic pH. 0.5 ml of samples/standard at different concentrations (12.5–150 μg/ml) was mixed with 3 ml of reaction mixture containing 0.6 M sulphuric acid, 28 mM sodium phosphate and 1% ammonium molybdate into the test tubes. The test tubes were incubated at 95 °C for 10 min to complete the reaction. The absorbance was measured at 695 nm using a spectrophotometer against blank after cooling at RT. CA was used as standard. The absorbance values of the samples were 0.193–2.307 at the concentrations ranged from 12.5–150 μg/ml, respectively. A typical blank solution contained 3 ml of reaction mixture and the appropriate volume of the same solvent used for the samples/standard was incubated at 95 °C for 10 min and the absorbance was measured at 695 nm. Increased absorbance of the reaction mixture indicates increased antioxidant capacity.
Ferrous reducing antioxidant capacity assay
The ferrous reducing antioxidant capacity of extracts was evaluated by the method of Oyaizu [61]. The Fe2+ can be monitored by measuring the formation of Perl’s Prussian blue at 700 nm. 0.25 ml samples/standard solution at different concentrations (12.5–150 μg/ml), 0.625 ml of potassium buffer (0.2 M) and 0.625 ml of 1% potassium ferricyanide, [K3Fe (CN)6] solution were added into the test tubes. The reaction mixture was incubated for 20 min at 50 °C to complete the reaction. Then 0.625 ml of 10% TCA solution was added into the test tubes. The total mixture was centrifuged at 3000 rpm for 10 min. After which, 1.8 ml supernatant was withdrawn from the test tubes and was mixed with 1.8 ml of distilled water and 0.36 ml of 0.1% ferric chloride (FeCl3)solution. The absorbance of the solution was measured at 700 nm using a spectrophotometer against blank. The range of the absorbance values of the samples was 0.033–2.943 at the concentrations ranged from 12.5–150 μg/ml, respectively. A typical blank solution contained the same solution mixture without plant extracts/standard was incubated under the same conditions and the absorbance of the blank solution was measured at 700 nm. Increased absorbance of the reaction mixture indicates increased reducing capacity. The experiment was repeated three times at each concentration.
DPPH radical scavenging assay
Free radical scavenging ability of the extracts was tested by DPPH radical scavenging assay as described by Blois [62] and Desmarchelier et al. [63]. The hydrogen atom donating ability of the plant extracts was determined by the decolorization of methanol solution of 2,2-diphenyl-1-picrylhydrazyl (DPPH). DPPH produces violet/purple color in methanol solution and fades to shades of yellow color in the presence of antioxidants. A solution of 0.1 mM DPPH in methanol was prepared and 2.4 ml of this solution was mixed with 1.6 ml of extracts in methanol at different concentration (12.5–150 μg/ml). The reaction mixture was vortexed thoroughly and left in the dark at RT for 30 min. The absorbance of the mixture was measured spectrophotometrically at 517 nm. BHT was used as reference. Percentage DPPH radical scavenging activity was calculated by the following equation,
Where, A0 is the absorbance of the control, and A1 is the absorbance of the extracts/standard. Then % of inhibition was plotted against concentration, and from the graph IC50 was calculated. The range of the absorbance values of all the samples was 0.535–0.023 at the concentrations ranged from 12.5 to150 μg/ml, respectively. The experiment was repeated three times at each concentration.
Hydroxyl radical scavenging activity
Hydroxyl radical scavenging activity of the extracts was determined by the method of Halliwell and Gutteridge [64]. Hydroxyl radical was generated by the Fe3+-ascorbate-EDTA-H2O2 system (Fenton reaction). The assay is based on the quantification of the 2-deoxy-D-ribose degradation product, which forms a pink chromogen upon heating with TBA at low pH. The reaction mixture contained 0.8 ml of phosphate buffer solution (50 mmol L− 1, pH 7.4), 0.2 ml of extracts/standard at different concentration (12.5–150 μg/ml), 0.2 ml of EDTA (1.04 mmol L− 1), 0.2 ml of FeCl3 (1 mmol L− 1), and 0.2 ml of 2-deoxy-D-ribose (28 mmol L− 1) was taken in the test tubes. The mixtures were kept in a water bath at 37 °C and the reaction was started by adding 0.2 ml of AA (2 mmol L− 1) and 0.2 ml of H2O2 (10 mmol L− 1). After incubation at 37 °C for 1 h, 1.5 ml of TBA (10 g L− 1) was added to the reaction mixture followed by 1.5 ml of HCl (25%). The mixture was heated at 100 °C for 15 min and then cooled down with water. The absorbance of solution was measured at 532 nm with a spectrophotometer. The hydroxyl radical scavenging capacity was evaluated with the inhibition percentage of 2-deoxy-D-ribose oxidation on hydroxyl radicals. The percentage of hydroxyl radical scavenging (%HRSA) activity was calculated according to the following formula:
Where, A0 is the absorbance of the control without a sample. A1 is the absorbance after adding the sample and 2-deoxy-D-ribose. A2 is the absorbance of the sample without 2-deoxy-D-ribose. At the concentrations ranged from 12.5 to150 μg/ml, the range of the absorbance values of the samples was0.068–0.478, respectively. The experiment was repeated three times at each concentration.
Lipid peroxidation inhibition assay
The lipid peroxidation inhibition assay was determined according to the method described by Haenen and Bast [65] with a slight modification. Excised rat liver was homogenized in buffer and then centrifuged to obtain liposome. To make 10% liver homogenate, excised Wister rat liver (weight of ~ 150 g) was minced using glass Teflon homogenizer in icecold phosphate buffered saline (50 mm, pH 7.4). The homogenate was centrifuged at 12000 rpm for 15 min at 4°c. The supernatant was used as liposome for in vitro lipid peroxidation assay. The process of homogenization and filtration was carried out in ice cold condition. Firstly, 0.5 ml of supernatant, 1 ml of 0.15 M KCl and 0.3 ml of extractives or standard at different concentrations were mixed. Peroxidation was initiated by the addition of 300 μL of 0.5 mm FeCl3. The mixture was incubated at 37°c for 30 min and the reaction was stopped by adding 2 ml of ice-cold TBA-TCA-HCl-BHT solution. The TBA-TCA-HCl solution was prepared by dissolving 1.68 g TCA and 41.60 mg TBA in 10 ml of 0.125 M HCl. One ml BHT solution (1.5 mg/ml ethanol) was added to 10 ml TBA-TCA-HCl solution. The reaction mixture was heated for 60 min at 90 °C and then cooled on ice and centrifuged at 3000 rpm for 5 min. The supernatants were removed, and the intensity of the pink colored complex was measured using a spectrophotometer at 532 nm. The degree of lipid peroxidation was assayed by estimating the TBARS (TBA-reactive substances) content. Control experiment was performed in the presence of distilled water without the extract. The percentage of lipid peroxidation inhibition in the samples was calculated using the following formula:
Where, A0 is the absorbance of the control (300 μl of distilled water), and A1 is the absorbance of extracts/standard. Then % of inhibition was plotted against concentration, and from the graph IC50 was calculated. The experiment was performed in triplicate and repeated three times at each concentration. The range of the absorbance values of all samples were 0.048–0.200 at the concentrations ranged from 12.5 to150 μg/ml, respectively.
Determination of cytotoxic and anticancer activities
Brine shrimp lethality bioassay
Brine shrimp lethality bioassay is a general bioassay which is indicative of cytotoxic activity, pharmacological actions and pesticidal effects. Extracts with ED50 ≤ 30 μg/ml are considered to be cytotoxic [66]. All fractionated extracts (12.5-150 μg/ml) and VBS (0.16-160 μg/ml) were diluted with hatching medium and were subjected to standard procedure for the brine shrimp lethality bioassay in a 24 well plate (n = 10 live shrimps/well). The live healthy nauplii with constant motion were counted after 24 h. The percentage of viability of the nauplii was calculated at each concentration by the following formula:
Where, Nt = Number of viable nauplii after 24 h of incubation, N0 = Number of total nauplii transferred i.e. 10.
Experimental animal
Swiss albino male mice, aged 4 weeks and weighing between 25 and 30 g, were purchased from ICDDRB (International Centre for Diarrheal Disease Research in Bangladesh), Dhaka, Bangladesh. The animals were housed in propylene cages in a controlled environment (temperature 25 ± 2 °C and 12 h dark and light cycle) and received feed formulated by ICDDRB and water ad libitum. The animals were acclimatized to laboratory conditions for 10 days prior to initiation of experiments. To keep the hydration rate constant, food and water supply were stopped 12 h before the experiments.
Cell lines
EAC cells were obtained by the courtesy of Indian Institute of Chemical Biology (IICB), Kolkata, India. Mice were injected with EAC cells by successive transplantation of 1 × 105 cells/mouse in peritoneal cavity by needle aspiration with a volume of 0.2 ml in PBS.
Cell growth inhibition
In vivo tumor cell growth inhibition was carried out by the method previously described by Sur and Ganguly [67]. For this study, 6 groups of mice (6 in each group) were used. For therapeutic evaluation, 1 × 104 Ehrlich ascites carcinoma (EAC) cells /mouse were inoculated into each group of mice on the first day. Treatment was started after 24 h of EAC inoculation and continued for 5 days. Group 1 to 4 received the test compounds (EAF, AQF, CHF and PET) at the doses of 50 mg/kg, respectively per day per mouse. In each case, the volume of the test solutions injected intraperitoneally (i.p.) was 0.1 ml/day per mouse. Group 5 received standard bleomycin (0.3 mg/kg, i.p) and was considered as positive control. Finally, the group 6 mice were treated with the vehicle (normal saline) and were considered as untreated control. The mice were sacrificed on the 6th day after transplantation and tumor cells were collected by repeated i.p.wash with 0.9% saline. Viable tumor cells per mouse of the treated group were compared with those of control.
The cell growth inhibition was calculated using the following formula:
Where, Tw = Mean of number of tumor cells of the treated group of mice and Cw = Mean of number of tumor cells of the control group of mice.
Statistical analysis
All tests were carried out in triplicates. Data were presented as mean ± SD. Free EViews 4.1 Software and Microsoft Excel 2007 (Roselle, IL, USA) were used for the statistical and graphical evaluations. The results with a value ofP < 0 .05 were considered significant.
Results
Total phenolic and flavonoid contents
The total polyphenol contents in the CEE and its four fractions: EAF, AQF, CHF and PEF expressed as GAE and CAE were shown in Table 1. The EAF showed the highest phenolic (158.17 ± 1.54 mg GA/g) and flavonoid (5.43 ± 0.017 mg CA/g) content among the extracts. Strong correlation (p < 0.01) of total phenolic content of the extracts with free radical scavenging efficiency and % lipid peroxidation inhibition were observed (Table 2).
Determination of TAC and ferrous reducing antioxidant capacity
The TAC of CEE and its four fractions of leaves of T. pallida were shown in Fig. 1a. The total antioxidant activity of EAF was higher (p < 0.01) than all other extracts even in comparison to standard CA followed by EAF > CA > CEE > CHF > AQF > PEF. At the concentration of150μg/ml, the absorbance of CEE, EAF, AQF, CHF, PEF and standard CA were in the range of 0.901 ± 0.01–2.30 ± 0.08. Increasing the extracts concentration increased the total antioxidant activity.

Determination of (a) total antioxidant capacity and (b) ferrous reducing antioxidant capacity of CEE and its various fractions (EAF, AQF, CHF and PEF). Data expressed as mean ± SD (n = 3, p < .01) for all tested dosages
The ferrous reducing antioxidant capacity of CEE and its four fractions was shown in Fig. 1b. It was found that the reducing power increased with concentration of each sample. At 150 μg/ml, the absorbance of CEE, EAF, AQF, CHF, PEF and standard AA were in the range of 0.608 ± 0.005–3.22 ± 0.06. EAF exhibited very strong radical scavenging (p < 0.01) activity in comparison to other fractions but less than the standard ascorbic acid. The descending order of strength of ferrous ion reducing activity of the extracts were AA> EAF > CEE > AQF > CHF > PEF. A higher absorbance indicates a higher reducing capacity. The reducing activity was increased with the increasing concentration of the extracts.
DPPH radical scavenging activity
The free radical scavenging activity of CEE, EAF, AQF, CHF and PEF at a concentration of 50 μg/ml was 89.19 ± 0.60, 96.18 ± 0.16, 81.78 ± 1.25, 74.16 ± 0.66 and 26.81 ± 0.92%, respectively; while at the same concentration the activity of BHT was 95.74 ± 0.25% (Fig. 2a). The IC50 of CEE, EAF, AQF, CHF, PEF and standard BHT were 9.33 ± 0.38, 6 ± 0.25, 10 ± 0.01, 26.58 ± 0.52, 94.5 ± 1.39 and16.08 ± 0.28 μg/ml, respectively. Lower the IC50indicates higher the radical scavenging activity. The free radical scavenging activity of different extracts and BHT were in the following order: EAF > AQF > CEE > BHT > CHF > PEF.

Determination of (a) DPPH radical scavenging activity and (b) OH radical scavenging activity of CEE and its various fractions (EAF, AQF, CHF and PEF). Data expressed as mean ± SD (n = 3, p < .01) for all tested dosages
Hydroxyl radical scavenging activity
The hydroxyl radical scavenging activity of CEE of T. pallida leaves was dose dependent. Among the extracts, EAF had higher activity than that of the other extracts. At a concentration of 100 μg /ml, the scavenging activity of CEE, EAF, AQF, CHF and PEF were found to be 62 ± 0.57, 88.24 ± 0.76, 81.60 ± 0.59, 84.14 ± 0.14 and 78.53 ± 0.56%, respectively; while at the same concentration, the activity of standard AA was 80.27 ± 0.46% (Fig. 2b). The IC50 of CEE, EAF, AQF, CHF, PEF and AA were 47.25 ± 2.28, 3.58 ± 0.28, 4 ± 0.00, 4.66 ± 0.14, 7.05 ± 0.18 and 10.25 ± 0.25 μg/ml, respectively. The result demonstrates that the radical scavenging activity of EAF was higher than not only other extracts but also standard drug AA.
Lipid peroxidation inhibition assay
The lipid peroxides scavenging activity of CEE of T. pallida leaves was investigated and compared with standard CA. The scavenging activity of CEE, EAF, AQF, CHF and PEF at a concentration of 100 μg/ml, were 71.57 ± 0.46, 75.87 ± 0.83, 72.02 ± 0.29, 65.67 ± 0.52 and 58.89 ± 0.36% respectively; whereas the activity of CA was 85.53 ± 1.01% (Fig. 3a). The IC50 of CEE, EAF, AQF, CHF and PEF were 20.5 ± 1.08, 14.33 ± 0.14, 22.08 ± 0.76, 25.25 ± 0.66 and 67.25 ± 1.95 μg/ml, respectively; on the other hand, the IC50 of CA was 11.46 ± 0.38 μg/ml (Fig. 3b). The EAF had the highest inhibitory activity among the extracts and almost equal inhibitory activity to that of CA. Significant correlations (p < 0.01) were observed between % lipid peroxidation inhibition and % hydroxyl radical scavenging (Table 3).

Determination of (a) lipid peroxidation inhibition of CEE and its various fractions (EAF, AQF, CHF and PEF) and (b) IC50 of CEE and its various fractions (EAF, AQF, CHF and PEF) determined from lipid peroxidation inhibition assay. Data expressed as mean ± SD (n = 3, p < .01) for all tested dosages
Brine shrimp lethality bioassay
The effect of CEE, EAF, AQF, CHF and PEF of T. pallida was investigated at different concentrations (12.5–150 μg/ml) on the brine shrimp nauplii (Fig. 4b). The ED50 (effective dose of the extracts at which 50% nauplii are viable) of the test samples was calculated using the concentration versus % of nauplii viability curve of the samples (Fig. 4c). The lower ED50 means higher toxicity. Among the extracts, EAF showed most potent activity (p < 0.01) with ED50of 8.50 ± 0.70 μg/ml. The ED50values of other extracts such as CEE, AQF, CHF and PEF were 10.50 ± 0.93, 9.75 ± 0.48, 11.25 ± 0.21 and 11 ± 0.67 μg/ml, respectively. On the contrary, control VBS (Fig. 4a) showed viability of nauplii when the concentration was lowered gradually from 10 μg/ml (zero viability) to 0.16 μg/ml (100% viability). The ED50 of VBS was found to be 1.05 ± 0.02 μg/ml. Our results demonstrated that all the extracts of T. pallida leaves had mild cytotoxic activity. The cytotoxic activities of different extracts were in the following order:
EAF>AQF>CEE>PEF>CHF

Determination of (a) cytotoxic effect of VBS(Standard) on the viability of brine shrimp nauplii (b) cytotoxic effect of leaves of T. pallidaon the viability of brine shrimp nauplii (c) Determination of ED50 (μg/ml) values of T. pallida leaves and VBS on brine shrimp viability. Data expressed as mean ± SD (n = 3, p < .01) for all tested dosages
Tumor cell growth inhibition
The significant tumor cell growth inhibition was observed after treatment with the extracts at the dose of 50.0 mg/kg (i.p) on day six of tumor inoculation. The EAF showed maximum cell growth inhibition (71.17 ± 0.44%, p < 0.01) when compared with other extractives such as AQF (63.60 ± 0.81%), CHF (58.88 ± 0.35%) and PEF (51.94 ± 0.56%). On the other hand, standard bleomycin (positive control) at a dose of 0.3 mg/kg i.p inhibited the tumor cell growth by 83.46 ± 0.75% (Table 4). This result implies that various fractions of leaves especially EAF had considerable anticancer activity compared with the standard bleomycin and the plant might therefore be considered as an effective source of active chemopreventive agents.
Discussion
From therapeutical and pharmaceutical point of view, plants are still considered as a great source of novel medicinal principles for new drug discovery. So, the possibility of getting pharmacologically active compounds is high from traditional and native medicinal plants. Thus, phytochemical investigation of the plant species is of eminent importance. Therefore, the assessment of the antioxidant, cytotoxicity, and anticancer potential of traditional medicinal plants by using safe and easy assay method may serve as an efficient tool for the chemical identification of active molecules which may be used as antioxidant as well as anticancer compounds; or lead compounds which could be chemically modified to effective chemotherapeutic agents such as antioxidant or anticancer drugs. These drugs could be used to manage different diseases as well as slow down cancer growth or even cure cancer and consequently contributing positively to safety of human life as well as the economy of the country and the world at large.
Total phenolic and flavonoid contents
Polyphenols are the foremost antioxidants found in plant kingdom, which have anticancer property [68]. It is believed that antioxidant properties of the polyphenolic compounds are mainly for their redox capacity, which neutralizes the free radicals [69]. In addition, flavonoids which are also present abundantly as secondary metabolites in plant showed potential antioxidant, antimutagenic, and anti-malignant effects [70]. Besides, the chemo-preventive role of flavonoids is due to their signal transduction in cell proliferation followed by angiogenesis [71]. In our study, the results showed that the leaves extracts especially EAF contains significant amount of phenolic and flavonoid compounds (Table 1). Thus, our findings indicate that these polyphenolic compounds are the vital components of this plant, and it can be assumed that the presence of such constituents may play an important role for pharmacological effects like anticancer activity.
Total antioxidant capacity
The total antioxidant potentials of leaves extracts were determined by their ability to reduce the reduction of Mo (VI) to Mo (V) followed by the formation of a green complex [phosphate/Mo (V)] at acidic pH. To show antioxidant activity the abundance of bioactive compounds in the extract is more significant. As extracts with higher concentrations shows higher antioxidant activity. In our experiment, the higher concentration of extracts showed higher antioxidant activity with a reducing ability range between 0.19 ± 0.005 to 2.30 ± 0.008 μm of green phosphate/Mo (V) (Fig. 1a). The antioxidant activity of EAF was significantly higher (p < 0.01) than the standard antioxidant. Furthermore, our results were consistence with the data published previously [72] and revealed that the antioxidant capacity is due to the presence of polyphenol contents in the extracts.
Ferrous reducing antioxidant capacity
In order to evaluate the antioxidant activity of plant extract, reducing power determination is widely used which exert antioxidant action by breaking the free radicals. In this assay, the polyphenolic enriched-antioxidant sample causes the reduction of the Fe3+ /ferricyanide complex to the Fe2+/ ferrous form where the reducing power is measured at 700 nm after the formation of blue complex of Perl’s Prussian [73]. In our study, the iron reducing capacity of CEE, EAF, AQF, CHF and PEF was evaluated from their reducing ability where they showed the activity with a range of 0.033 ± 0.002–2.94 ± 0.036 μm of Fe (II)/g (Fig. 1b) and the reducing activity was increased with concentration dependent manner (Fig. 1b). Thus, it can be revealed that the presence of polyphenols is attributed to the antioxidant activity and reducing power capacity.
DPPH radical scavenging activity
DPPH free radical scavenging is a well-established experiment for screening the antioxidant activity of plant extracts. In this assay, violet color DPPH solution is reduced to the yellow color of diphenylpicryl hydrazine after adding the extracts in a concentration dependent manner. This method now a day is very popular to predict the antioxidant activities due to its accuracy and short timing for assay. All the fractions of T. pallida leaves extract exhibited better DPPH radical scavenging activity with the increase of concentration (Fig. 2a). The IC50 of EAF was significantly lower (p < 0.01) than that of other fractions and the standard BHT. The inhibitory activity of the extracts was found to be as following order EAF > AQF > CEE > BHT > CHF > PEF. Previously, it has been reported that hydrogen donating ability of phenolics, flavonoids and tocopherols is the main factor to reduce the DPPH radicals [74, 75]. Therefore, this study revealed that T. pallida leaves act as free radical scavengers by reducing DPPH radicals that might be through hydrogen donation.
Hydroxyl radical scavenging activity
Breakdown of DNA basically involves in the mutagenic capacity of free radicals where hydroxyl radicals directly interact with DNA and finally trigger the cancer formation [76]. Usually, hydroxyl radicals can be generated by biochemical reaction in the presence of superoxide dismutase and divalent metal ions, such as iron and copper. Our results demonstrated that all the fractions of leaves extract had higher hydroxyl radical scavenging activity compared with standard antioxidant BHT. Among the fractions and standard antioxidant, EAF showed the highest scavenging activity (Fig. 2b) and may serve as anticancer agent by inhibiting the interaction of hydroxyl radical with DNA.
Lipid peroxidation inhibition assay
Lipid peroxidation refers to the chain reaction where ROS induce membrane damage by peroxiding lipid moieties [77] that lead to functional abnormalities of cells. Lipid peroxidation has been reported to be elevated in the cancer [78]. In vitro lipid peroxidation was induced in rat liver by generating OH radical through Fenton’s reaction in presence of ferric ion and potassium chloride. In respect to antioxidant activity, inhibition of lipid peroxidation is considered the most important index. In this study, the CEE of leaves and its four fractions especially EAF exhibited potential inhibition of lipid peroxidation (Fig. 3a). The lipid peroxidation inhibition was significantly correlated with hydroxyl radical scavenging (Table 3). This result indicates that the leaves extracts can inhibit lipid peroxidation differentially by varying capacity of free radical quenching potential and may be used to treat several diseases caused by free radicals.
Cytotoxic effect on brine shrimp
The cytotoxic effect may be due to the presence of diverse phytochemicals such as saponins, triterpenes, tannins and polyphenolic compounds in the extract and this phytochemical compound exist in plants exhibited anti-tumorigenic effects via multiple anticancer pathways [68]. It has been reported that polyphenolic compounds have a correlation between cytotoxic activity and antioxidant property [79]. Among the tested fractions of T. pallid leaves especially EAF showed (Fig. 4a and b) the highest cytotoxic properties which was significantly correlated (p < 0.01) with polyphenol compounds and antioxidant properties that comply with the data published elsewhere [80]. This result suggests that all the extracts showed cytotoxicity due to the presence of bioactive compounds which might have anti-tumor effect.
Effect of antioxidant on EAC-induced tumor cells
In vivo, oxygen is the most common form of free radicals. When an oxygen molecule (O2) becomes electrically charged or radicalized, it damages the DNA that lead to various diseases, including cancer. On the other hand, antioxidants are often neutralizing the electrical charge and prevent the free radical to damage the cell and thereby prevent various diseases, including cancer. Literature review on different chemical, cell culture, and animal studies suggest that antioxidants may prevent or slowdown the development of cancer [80]. In this study, the anticancer activity of T. pallida leaves on EAC-induced cancer in mice was in moderate level when compared with the standard bleomycin (Table 4) that makes it as a good candidate for isolating anticancer agent.
Conclusion
The present study indicates that T. pallida leaves possess significant polyphenolic contents, which exhibited strong ROS scavenging activities. The leaves also showed strong cytotoxic effect and moderate anticancer activity. Tabebuia genus is enriched with a diverse class of chemical compounds (Additional file 1: Table S1) based on literature survey and in our study, the resulting bioactivities are basically responsible for mixture of phytochemicals possess in the extracts or may be due to the synergistic activity of various constituents retain in this plant species. Further investigation is being carried out to identify and characterize the inherent phenolic compounds as well as bioactive compounds of the plant which are responsible for the antioxidant, cytotoxic effect and anticancer activities from the ethyl acetate fraction of T. pallida leaves.
Abbreviations
- AA:
-
Ascorbic acid
- BHA:
-
Butylated hydroxyl anisole
- BHT:
-
Butylatedhydroxytoluene
- CA:
-
Catechin
- CAE:
-
Catechin equivalent
- CEE:
-
Crude ethanolic extract
- DPPH:
-
2,2-diphenyl-1-picrylhydrazyl
- DRSA:
-
DPPH radical scavenging assay
- EDTA:
-
Ethylenediaminetetraacetic acid
- FRAC:
-
Ferrous reducing antioxidant capacity
- GA:
-
Gallic acid
- GAE:
-
Gallic acid equivalent
- HRSA:
-
Hydroxyl radical scavenging activity
- LPI:
-
Lipid peroxidation inhibition assay
- OS:
-
Oxidative stress
- ROS:
-
Reactive oxygen species
- TAC:
-
Total antioxidant capacity
- TBA:
-
Thiobarbituric acid
- TCA:
-
Trichloro acetic acid
- TPL:
-
Tabebuiapallida leaves
- VBS:
-
Vinblastine sulphate
References
Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9–19.
Khan MA, Rahman AA, Islam S, Khandokhar P, Shahnaj P, Islam MB, et al. A comparative study on the antioxidant activity of the methanolic extracts from different parts of Morus alba L. (Moraceae). BMC Res Notes. 2013;6:24.
Islam S, Nasrin S, Khan MA, ASMS H, Islam F, Khandokhar P, et al. Evaluation of antioxidant and anticancer properties of the seed extracts of Syzygium fruticosum Roxb. growing in Rajshahi, Bangladesh. BMC Complement. Altern Med. 2013;13:142.
Maxwell SR. Prospects for the use of antioxidant therapies. Drugs. 1995;49:345–61.
Kalauni SK, Awale S, Tezuka Y, Banskota AH, Linn TZ, Kadota S. Methyl migrated cassane-type furan-oditerpenes of Caesalpinia crista from Myanmar. Chem Pharm Bull. 2005;53:1300–4.
Moussaid M, Elamrani AA, Berahal C, Moussaid H, Bourhime N, Benaissa M. Evaluation of the antioxidant potential of some Morocco medicinal plants. Global J Pharmacol. 2011;5:153–8.
Miliauskas G, Venskutonis PR, Beek TAV. Screening of radical scavenging activity of some medicinal and aromatic plant extracts. Food Chem. 2004;85:231–7.
Mohsen SM, Ammar AS. Total phenolic contents and antioxidant activity of corn tassel extracts. Food Chem. 2009;112:595–8.
Willett WC. Balancing life-style and genomics research for disease prevention. Science. 2002;296:695–8.
Cragg GM, Newman DJ. Plants as a source of anti-cancer agents. J Ethnopharmacol. 2005;100:72–9.
Mans DRA, Rocha AB, Schwartsmann G. Anti-Cancer drug discovery and development in Brazil: targeted plant collection as a rational strategy to acquire candidate anti-cancer compounds. Oncologist. 2000;5:185–98.
Solowey E, Lichtenstein M, Sallon S, Paavilainen H, Solowey E, Lorberboum-Galski H. Evaluating medicinal plants for anticancer activity. Sci World J. 2014;2014:12.
Greenwell M, Rahman PKSM. Medicinal plants: their use in anticancer treatment. Int J Pharm Sci Res. 2015;6:4103–12.
Gupta M, Mazumder UK, Sambath KR, Sivakumar T, Mohan MLV. Antitumor activity and antioxidant status of caesalpinia bonducella against Ehrlich ascites carcinoma in swiss albino mice. J Pharmacol Sci. 2004;94:177–84.
Bromberg N, Dreyfuss JL, Regatieri CV, Marcelly V, et al. Growth inhibition and pro-apoptotic activity of violacein in Ehrlich ascites tumor. Chem Biol Interac. 2010;186:43–52.
Bhattacharyya A, Choudhuri T, Pal S, Chattopadhyay S, Datta GK, Gaurisankar S, et al. Apoptogenic effects of black tea on Ehrlich’s ascites carcinoma cell. Carcinogenesis. 2003;24:75–80.
Hegazi A, Tahtawy RHM, Allah FA, Abdou AM. Antitumor and antioxidant activity of honey in mice bearing Ehrlich ascites carcinoma. Academic J Cancer Res. 2014;7:208–14.
Alam AHMK, Hossain ASMS, Khan MA, Kabir SR, Reza MA, Rahman MM, et al. The antioxidative fraction of white mulberry induces apoptosis through regulation of p53and NFκB in EAC cells. PLoS One. 2016;11:e0167536.
Jiménez-González FJ, Veloza LA, Sepúlveda-Arias JC. Anti-infectious activity in plants of the genus Tabebuia. Univ Sci. 2013;18:257–67.
Kavya SK, Vijusha M, Rajani M, Hemamalini K, Sundari EGR. Screening of behavioural, muscle co-ordination & anxiolytic activities of methanolic extract of Tabebuia rosea (Bertol). Asian J Pharm Clin Res. 2013;6:187–90.
Nirmala MJ, Samundeeswari A, Sankar PD. Natural plant resources in anti-cancer therapy-a review. Res Plant Biol. 2011;1:1–14.
Queiroz ML, Valadares MC, Torello CO, Ramos AL, Oliveira AB, Rocha FD, et al. Comparative studies of the effects of Tabebuia avellanedae bark extract and β-lapachone on the hematopoietic response of tumour-bearing mice. J Ethnopharmacol. 2008;117:228–35.
Hussain H, Krohn K, Ahmad VU, Miana GA, Green IR. Lapachol: an overview. ARKIVOC. 2007;2:145–71.
Pereira EM, Machado Tde B, Leal IC, Jesus DM, Damaso CR, Pinto AV, et al. Tabebuia avellanedae naphthoquinones: activity against methicillin-resistant staphylococcal strains, cytotoxic activity and in vivo dermal irritability analysis. Annals Clin Microb Antimicrob. 2006;5:5.
Nakano K, Maruyama K, Murakami K, Takaishi Y, Tomimatsu T. Iridoids from Tabebuia avellanedae. Phytochemistry. 1993;32:371–3.
Machado TB, Pinto AV, Pinto MC, Leal IC, Silva MG, Amaral AC, et al. In vitro activity of Brazilian medicinal plants, naturally occurring naphthoquinones and their analogues, against methicillin-resistant Staphylococcus aureus. Int J Antimicrob Agents. 2003;2:279–84.
Yamashita M, Kaneko M, Tokuda H, Nishimura K, Kumeda Y, Iida A. Synthesis and evaluation of bioactive naphthoquinones from the Brazilian medicinal plant, Tabebuia avellanedae. Bioorg Med Chem. 2009;17:6286–91.
Portillo A, Vila R, Freixa B, Adzet T, Canigueral S. Antifungal activity of Paraguayan plants used in traditional medicine. J Ethnopharmacol. 2001;76:93–8.
Hofling JF, Anibal PC, Obando-Pereda GA, Peixoto IA, Furletti VF, Foglio MA, et al. Antimicrobial potential of some plant extracts against Candida species. Brazi J Biol. 2010;70:1065–8.
Park BS, Lee HK, Lee SE, Piao XL, Takeoka GR, Wong RY, et al. Antibacterial activity of Tabebuia impetiginosa Martius ex DC (Taheebo) against Helicobacter pylori. J Ethnopharmacol. 2006;105:255–62.
Byeon SE, Chung JY, Lee YG, Kim BH, Kim KH, Cho JY, et al. In vitro and in vivo anti-inflammatory effects of taheebo, a water extract from the inner bark of Tabebuia avellanedae. J Ethnopharmacol. 2008;119:145–52.
Twardowschy A, Freitas CS, Baggio CH, Mayer B, Santos AC, Pizzolatti MG, et al. Antiulcerogenic activity of bark extract of Tabebuia avellanedae, Lorentz ex Griseb. J Ethnopharmacol. 2008;118:455–9.
Freitas AE, Budni J, Lobato KR, Binfaré RW, Machado DG, Jacinto J, et al. Antidepressant-like action of the ethanolic extract from Tabebuia avellanedaein mice: evidence for the involvement of the monoaminergic system. Prog Neuropsychopharmacol Biol Psych. 2010;34:335–43.
Woo HJ, Choi YH. Growth inhibition of A549 human lung carcinoma cells by b-lapachone through induction of apoptosis and inhibition of telomerase activity. Int J Oncology. 2005;26:1017–23.
Yamashita M, Kaneko M, Tokuda H, Nishimura K, Kumeda Y, Iida A. Synthesis and evaluation of bioactive naphthoquinones from the Brazilian medicinal plant, Tabebuiaavellanedae. Bioorg Med Chem. 2009;17:6286–91.
Hegnauer R, Kooiman P. The taxonomic significance of iridoids of tubifloraesen suwettstein. Planta Med. 1978;33:1–33.
Silva TM, Silva TG, Martins RM, Maia GL, Cabral AG, Camara CA, et al. Molluscicidal activities of six species of Bignoniaceae from North-Eastern Brazil, as measured against Biomphalaria glabrata under laboratory conditions. Ann Trop Med Parasitol. 2007;101:359–65.
Abreu MB, Temraz A, Vassallo A, Braca A, Tommasi A. Phenolic glycosides from Tabebuia argentea and Catalpa bignonioides. Phytochem Lett. 2014;2014:85–8.
Gómez-Estrada H, Gaitán-Ibarra R, Díaz-Castillo F, Pérez HA, Medina JD. In vitro antimalarial activity of fractions and constituents isolated from Tabebuia billbergii. Rev Cub Plant Medicin. 2012;17:172–80.
de ST C, Anderson JE, Alfonso D, McLaughlin JL. Bioactive furonaphtoquinones from Tabebuia barbata (Bignoniaceae). Acta Cient Venez. 1997;48:42–6.
Melo e Silva F, de Paula JE, Espindola LS. Evaluation of the antifungal potential of Brazilian Cerrado medicinal plants. Mycoses. 2009;52:511–7.
Bianco A, Passacantilli P, Polidori G, Nicoletti M, Alves de Lima R. Isolation of 6-epimonomelittoside from Tecoma heptaphylla and its conversion into monomelittoside. Phytochemistry. 1983;22:1189–91.
Bianco A, Passacantilli P, Nicoletti M, Alves de Lima R. Iridoids in equatorial and tropical flora - III. Isolation and partial synthesis of 6-epiaucubin, a new glucosidiciridoid. Tetrahedron. 1982;38:359–62.
Bianco A, Passacantilli P, Nicoletti M, Alves de Lima R. Iridoids in equatorial and tropical Flora. Part 4. Isolation of amareloside. Planta Med. 1982;46:33–7.
Bianco A, Passacantilli P, Polidori G, Nicoletti M, Alves de Lima R. Iridoids in equatorial and tropical Flora. VII. Two new natural esters of 6-epimonomelittoside. Gazz Chim Ital. 1983;113:465–7.
Cwikla C, Schmidt K, Matthias A, Bone KM, Lehmann R, Tiralongo E. Investigations into the antibacterial activities of phytotherapeutics against Helicobacter pylori and Campylobacter jejuni. Phytother Res. 2010;24:649–56.
Oliveira DG, Prince KA, Higuchi CT, Santos ACB, Lopes LMX, Simões MJS, et al. Antimycobacterial activity of some Brazilian indigenous medicinal drinks. Rev Ciênc Farm Básica Apl. 2007;28:165–9.
Cordeiro CHG, Do Sacramento LVS, Corrêa MA, Pizzolitto AC, Bauab TM. Herbal extractsin an experimental mouthwash: Pharmacognostics analysis and antibacterial activity. Rev Bras Cienc Farm. 2006;42:395–404.
Brandão GC, Kroon EG, dos Santos JR, Stehmann JR, Lombardi JA, de Oliveira AB. Antiviral activities of plants occurring in the state of Minas Gerais, Brazil. Part 2. Screening Bignoniaceae species. Braz J Pharmacogn. 2010;20:742–50.
Oliveira ADB, Raslan DS, Geovane G, Oliveira D, Guilherme J, Maia S. Lignans and naphthoquinones from Tabebuia incana. Phytochemistry. 1993;34:1409–12.
Morais SKR, Silva SG, Portela CN, Nunomura SM, Quignard ELJ, Pohlit AM. Bioactive dihydroxyfuranonaphthoquinones from the bark of Tabebuia incana a.H. Gentry (Bignoniaceae) and HPLC analysis of commercial paud’arco and certified T. incana bark infusions. Acta Amazon. 2007;37:99–102.
Oliveira AB, Raslan DS, Miraglia MCM, Mesquita AAL, Zani CL, Ferreira DT, et al. Chemical structures and biological activities of naphthoquinones from Brazilian Bignoniaceae. Quim Nova. 1990;13:302–7.
Franco Ospina LA, Castro Guerrero JP, OcampoBuendía YC, Pájaro Bolívar IB, Díaz CF. Antiinflammatory, antioxidant and antibacterial activity of two species of Tabebuia genus. Rev Cubana Plant Med. 2013;18:34–46.
Hemamalini K, Soujanya GL, Bhargav A, Vasireddy U. In vivo anticancer activity of Tabebuia rosea (Bertol) dc. Leaves on dalton’s ascetic lymphoma in mice. Int J Pharm Sci Res. 2012;3:4496–502.
Otero R, Núñez V, Jiménez SL, Fonnegra R, Osorio RG, García ME, et al. Snakebites and ethnobotany in the northwest region of Colombia: part II: neutralization of lethal and enzymatic effects of Bothropsatrox venom. J Ethnopharmacol. 2000;71:505–11.
Gonzalez-Coloma A, Reina M, Saenz C, Lacret R, Ruiz-Mesia L, Aran VJ, et al. Antileishmanial, antitrypanosomal, and cytotoxic screening of ethnopharmacologically selected Peruvian plants. Parasitol Res. 2012;110:1381–92.
Rahman MM, Islam MB, Biswas M, Alam AHMK. In vitro antioxidant and free radical scavenging activity of different parts of Tabebuia pallida growing in Bangladesh. BMC Res Notes. 2015;8:621.
Wolfe K, Wu X, Liu RH. Antioxidant activity of apple peels. J Agric Food Chem. 2003;51:609–14.
Zhishen J, Mengcheng T, Jianming W. The determination of flavonoid contents in mulberry and their scavenging effects on superoxide radicals. Food Chem. 1999;64:555–9.
Prieto P, Pineda M, Aguilar M. Spectrophotometric quantitation of antioxidant capacity through the formation of a phosphomolybdenu complex: specific application to the determination of vitamin E. Anal Biochem. 1999;269:337–41.
Oyaizu M. Studies on products of browning reactions: antioxidant activities of products of browning reaction prepared from glucose amine. Jpn J Nutr. 1986;44:307–15.
Blois MS. Antioxdant determinations by the use of a stable free radical. Nature. 1958;181:1199–200.
Desmarchelier C, Bermudez MJN, Coussio J, Ciccia G, Boveris A. Antioxidant and prooxidant activities in aqueous extract of argentine plants. Int J Pharm. 1997;35:116–20.
Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Clarendon Press. 1989;3:617–783.
Haenen GR, Bast A. Protection against lipid peroxidation by a microsomal glutathione-dependent labile factor. FEBS Lett. 1983;159:24–8.
Meyer BN, Ferrigni NR, Putnam JE, Jacobsen LB, Nichols DE, Mclaughlin JL. Brine shrimp: a convenient general bioassay for active plant constituents. Planta Med. 1982;45:31–4.
Sur P, Ganguly DK. Tea plant root extract (TRE) as an antineoplastic agent. Planta Med. 1994;60:106–9.
Lamoral-Theys D, Pottier L, Dufrasne F, Neve J, Dubois J, Kornienko A, et al. Natural polyphenols that display anticancer properties through inhibition of kinase activity. Curr Med Chem. 2010;17:812–25.
Zheng W, Wang SY. Antioxidant activity and phenolic compounds in selected herbs. J Agr Food Chem. 2001;49:5165–70.
Fotsis T, Pepper MS, Aktas E, Breit S, Rasku S, Adlercreutz H, et al. Flavonoid, dietary-derived inhibitors of cell proliferation and in vitro angiogenesis. Cancer Res. 1997;57:2916–21.
Wagner H, Geyer B, Kiso Y, Hikino H, Rao GS. Coumestans as the main active principles of liver drugs Eclipta alba and Wedelica calendulaceae. Planta Med. 1986;5:370–4.
Jayaprakasha GK, Girennavar B, Patil BS. Radical scavenging activities of Rio Red grapefruits and Sour orange fruit extracts in different in vitro model systems. Bioresour Technol. 2008;99:4484–94.
Singh N, Rajini PS. Free radical scavenging activity of an aqueous extract of potato peel. Food Chem. 2004;85:611–6.
Duan X, Wu G, Jiang Y. Evaluation of the antioxidant properties of litchi fruit phenolics in relation to pericarp browning prevention. Molecules. 2007;12:759–71.
Li H, Wang X, Li Y, Li P, Wang H. Polyphenolic compounds and antioxidant properties of selected China wines. Food Chem. 2009;112:454–60.
Waris G, Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog. 2006;5:14.
Scully C. Oral cancer: new insight into pathogenesis. Dent Update. 1993;20:95–100.
Klauning JE, Xu Y, Isenberg JS, Bachowski S, Kolaja KL, Jiang J, et al. The role of oxidative stress in chemical carcinogenesis. Environ Health Perspect. 1998;106:289–95.
Mitsuhashi S, Saito A, Nakajima N, Shima H, Ubukata M. Pyrogallol structure in polyphenols is involved in apoptosis-induction on HEK293T and K562 cells. Molecules. 2008;13:2998–3006.
Blot WJ, Li JY, Taylor PR, Guo W, Dawsey S, Wang GQ, et al. Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J Nat Cancer Inst. 1993;85:1483–91.
Acknowledgments
The authors acknowledge the Bangladesh Council of Scientific and Industrial Research (BCSIR) and Department of Pharmacy, University of Rajshahi for their all logistic support. We also thank Dr. AHM Mahabubur Rahman, Associate Professor, Department of Botany, University of Rajshahi for the identification of the plant. We thank Ridita Mizan, Lecturer, Department of English, Rajshahi University, Bangladesh for correcting grammatical errors.
Funding
This work was carried out with partial funding by the DSR of Imam Abdulrahman Bin Faisal University project no 2018–090 irmc.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Declarations
All authors read the manuscript and approved it for submission. No part of the manuscript has been published before, nor is any part of it under consideration for publication at another journal.
Author information
Authors and Affiliations
Contributions
MMR: Executed the experimental works according to study design. ASMSH: Analysis and acquisition of data. MGM: Performed anticancer study. MAK: Carried out the lipid peroxidation inhibition assay. RA: Performed in vitro antioxidant work. AM: Checked the initial draft of the manuscript. MGS: Checked the final manuscript. AHMKA: Supervised and designed the study as well as drafted and corrected all versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Protocol used in this study for the use of mice as animal model for cancer research was approved by the Rajshahi University Animal Ethical committee (27/08/RUBCMB). This research work was approved by Ethical Review Committee of Research Cell of Rajshahi Medical College, Bangladesh (ref. RMC/ER/2010–2013/01) under the guidelines of National Research Ethics Committee (NREC) of Bangladesh, which follow the protocols of National Health Act No. 61 of 2003, Government of Canada Interagency Advisory Panel on Research Ethics (PRE) and the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
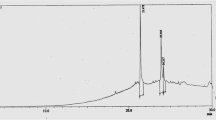
Table S1. A list of secondary metabolites of Tabebuia genus. Current bioactivity and secondary metabolites profile of Tabebuia genus. (DOC 76 kb)
Additional file 2:
Standard curve for Gallic acid. (TIF 64 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rahman, M.M., Hossain, A.S.M.S., Mostofa, M.G. et al. Evaluation of anti-ROS and anticancer properties of Tabebuia pallida L. Leaves. Clin Phytosci 5, 17 (2019). https://doi.org/10.1186/s40816-019-0111-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40816-019-0111-5