
Abstract
Background
Mitochondrial markers are widely used as a first approach in determining evolutionary relationships among vertebrate taxa at different hierarchical scales. Cytochrome b and cytochrome oxidase I are among the most common markers; they are particularly useful in phylogeography and species delineation studies. Simulation and empirical studies show that increasing the taxon sampling has a clear and strong effect on the accuracy of the inferred trees and therefore on hypothesized phylogenetic relationships (and eventually in new taxonomic rearrangements); this should be considered in the design of studies. The lizard genus Liolaemus is widely distributed in southern South America and includes more than 250 described species. The number of taxa and the distribution of Liolaemus species/populations makes them a good model for testing different hypotheses in systematics.
Methods
We studied two Liolaemus species, Liolaemus nigroviridis and L. monticola as focal species to evaluate their monophyly and the influence of adding new samples from related taxa in the resulting phylogenies. We performed phylogenetic analyses (maximum likelihood and Bayesian inference) using 141 sequences of the mitochondrial DNA Cytochrome b (cyt-b) of 11 Liolaemus species.
Results
Our study show that using intensive taxon sampling for phylogenetic reconstructions, two species (L. uniformis and L. nitidus) are placed within the clades of the two focal species (L. nigroviridis and L. monticola, respectively).
Conclusions
Our study confirms the importance of taxon sampling to infer more accurate phylogenetic relationships, particularly to reveal hidden polyphyly or paraphyly, which may have a strong impact on taxonomic proposals and/or inferring cryptic diversity.
Similar content being viewed by others


Background
Mitochondrial markers are widely and frequently used as the first approach to determining evolutionary relationships among vertebrates at different hierarchical scales [1, 2]. Cytochrome b and cytochrome oxidase I are among the most common markers; they are particularly useful in phylogeography and species delineation studies [3,4,5,6,7,8].
Although a number of methods and types of data are available, mitochondrial markers are a common tool to support descriptions of new taxa in phylogenetic analyses. This is probably due to the nature of the mitochondrial genome, i.e. as the smaller effective population size that will cause mtDNA haplotypes of a given group to coalesce four times more quickly than nuclear markers [2].
Simulation analyses show that increasing sampling of the taxon has a clear and strong effect on the accuracy of the inferred trees and therefore on hypothesized phylogenetic relationships (and eventually on new taxonomic proposals), which should be considered in the design of studies [9]. In systematics, for example, a pattern of reciprocal monophyly may be interpreted as evidence of no gene flow at the genetic locus of interest, and major factors influencing the probability of this pattern include time since isolation and effective population size. The probability of recovering a monophyletic group also depends on random processes related to the number of sequences sampled per population [2, 10]. Thus there is a probability that by sampling a few individuals a false monophyly pattern may be exhibited, therefore requiring additional samples to strengthen the hypothesis. For example, a study showed that if gene flow ceased very recently between two populations there is a higher probability of finding monophyly when few sequences are considered [11]. Also, the number of individuals required to evaluate a hypothesis adequately may vary according to the substitution rate of the marker analyzed. More individuals seem to be necessary in the analysis when the sequenced marker evolves more quickly [12]. Several empirical and simulations studies have shown that taxon sampling may be critical to achieve accurate phylogenetic results, either to retrieve the correct topology or improve bootstrap support [13,14,15,16,17,18,19,20]. Therefore, the decision of including or excluding certain taxa may result in different topologies, which in turns, may lead to new taxonomic rearrangements and/or phylogenetic relationships.
The lizard genus Liolaemus is widely distributed in southern South America and includes more than 250 described species [21]. This genus is distributed from arid Patagonian to high-altitude Andean environments, including valleys and coastal ranges. The adaptive radiation of Liolaemus has produced interesting patterns of genetic and morphological variation, resulting in a highly species-rich genus [22]. In Chile, 96 species of Liolaemus are currently recognized [23], which inhabits most environments from the arid desert in the north to the cold southern Patagonia, as well as Coast Range and Andean environments from 0 to 4500 m [24]. Thus, the number of taxa and the distribution of Liolaemus species/populations represents a good model for testing different hypotheses in systematics. Taxonomy within Liolaemus is dynamic [25], with new species reported almost every year. Most of the studies use traditional morphological analyses to diagnose the new taxa, and some include integrative approaches with other data such as molecular phylogenetic relationships with sister or close taxa. We used two Liolaemus species as focal species to evaluate their monophyly and the influence of adding new samples from closely related taxa in the resulting phylogenies.
Liolaemus nigroviridis is a saxicolous species distributed in central Chile between latitudes ca. 32° to 34°S (Region of Valparaiso to O’Higgins) above 1100 m [26]. The species’ distribution is discontinuous, inhabiting both Andean and coastal mountain environments and absent in low elevations and valleys [27]. In the Coast Range it has been reported in the Cantillana, Chicauma, La Campana and El Roble mountains. Because of the allopatric distribution of its populations, three subspecies were described: L. n. nigroviridis in the Valle del Río San Francisco (Andes), L. n. minor in the Valle del Río Volcán (Andes), and L. n. campanae in the Coast Range [28, 29]. However, based on similarities in morphology and chromosomal characters, a subsequent study did not recognize the polytypic status of the species [30]. A new species was described recently in central Chile, Liolaemus uniformis, sister to L. nigroviridis [31]. The phylogenetic inference for the description of L. uniformis included L. nigroviridis from one population, the type locality Farellones (33°21′S, 70°17′W). A previous study reported at least four major lineages allowing to infer strong phylogeographic structure within L. nigroviridis [27]; therefore, an increase taxon sampling seems recommendable to determine the position of the new proposed taxon (L. uniformis) related to its sister species (L. nigroviridis). The second model, L. monticola, is a saxicolous lizard species from the mediterranean Andes [32, 33] and the Coast Range [34] in central Chile from between 600 m to 1900 m. This species displays an extensive variation and complexity in chromosome number in a latitudinal gradient [35, 36], with riverine barriers playing a major role in the divergence of populations [37, 38]. Phylogenetic inferences show that L. nitidus is sister to L. monticola [3, 39]. However, intensive sampling showed paraphyly of L. monticola populations, with two samples (one haplotype) of L. nitidus placed within the L. monticola clade, which was suggested as potential mitochondrial introgression [40].
In this study, we performed phylogenetic analyses using mitochondrial Cytochrome b (cyt-b) DNA to determine the influence of taxon sampling in finding monophyletic groups in Liolaemus, a main result for proposing new taxa. We also discuss the importance of including as much as information available from several sources and databases (i.e. Genbank), and discuss the limitations of the studies which do not take into account evolutionary population processes that may hide the history of species differentiation.
Methods
Specimens analyzed
Mitochondrial gene sequence data of cyt-b were obtained from 141 lizards of different species of Liolaemus, 32 sequences generated in this study and 109 sequences downloaded from Genbank (Fig. 1, See Additional file 1). Genbank accession numbers KY575287-KY575318. The species used were L. nigroviridis, L. uniformis, L. monticola, and L. nitidus. Other species included were L. confusus, L. curicensis, L. lemniscatus, L. tenuis, L. pseudolemniscatus, L. schroederi and L. chiliensis based on phylogenetic studies with mitochondrial and nuclear genes [31, 39, 41]. Due to a deep phylogeographic structure reported for L. nigroviridis [27], we added 14 sequences from a new locality (La Campana, 32° 57′S, 71° 07′W). We included L. uniformis to test the monophyly/paraphyly within L. nigroviridis.

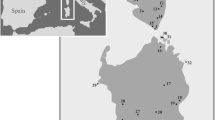
Study area. Map of 41 localities of focal species. The colors represent the four different species: L. nigroviridis (green), L. uniformis (pink), L. monticola (orange) and L. nitidus (light blue). The Maipo River is shown to represent the geographic barrier to the northern and southern clades of L. monticola reported by [38]
DNA extraction, amplification and sequencing
Total genomic DNA was extracted from liver/muscle tissues preserved in 80% ethanol using the QIA quick DNeasy Blood & Tissue kit (Qiagen Inc., Valencia, CA, USA) following the protocol provided by the manufacturer. A fragment of 775bp of the mtDNA region encoding the cyt-b gene was amplified via polymerase chain reaction (PCR) using the light strand primer GLUDG (5′-TGACTTGAARAACCAYCGTTG-3′) and the heavy strand primer CB3 (5′ GGCAAATAGGAARTATCATTC-3′) previously used [40]. The thermocycling conditions were previously documented [27]; amplification reactions were carried out in 11.5 μl reaction mixture containing 1.5μl template DNA (1–50 ng), 2μl 5X Taq buffer, 1.2 μl 25 mM MgCl2, 0.2 μl dNTPs (10 mM), 0.124 μl each primer (10 mM) and 0.1 μl Taq polymerase (5 U/μl Promega Corp.). Double-stranded PCR-amplified products were checked by electrophoresis on a 1% agarose gel in 1x Tris-Borate-EDTA (TBE) TBE running buffer and stained with Gel Red (Biotum, Inc., Hayward CA, USA). Purification and sequencing reactions were conducted by Macrogen Inc, (South Korea). All sequences were edited and aligned using the BioEdit Sequence Alignment Editor [42] and checked by eye. Missing data were coded as “?” and cyt-b sequences were translated to check for premature stop codons using DnaSP [43].
Phylogenetic analyses
To assess interspecific relationships of Liolaemus species focusing on L. nigroviridis and L. monticola and to determine changes in the topologies with different numbers of sequences, we performed Maximum-likelihood (ML) and Bayesian analyses using three approaches: 1) Eleven Liolaemus species (114 haplotypes) that included L. nigroviridis (N = 78), L. monticola (N = 36) plus their sister species and outgroups (see below). Cytochrome b sequences were downloaded from Genbank and obtained in this study (See Additional file 1). 2) L. nigroviridis as focal species that included seven populations, plus two sequences of L. uniformis (sister species of L. nigroviridis; [31], resulting in 64 cyt-b haplotypes. 3) L. monticola as focal species from 31 localities plus L. nitidus (sister species of L. monticola; [3] from 5 localities, resulting in 44 cyt-b haplotypes.
Phylogenetic reconstructions were rooted with the outgroup criterion, using L. chiliensis and L. schroederi for all Lioalemus species included in this study (first approach), L. lemniscatus for analyses with L. nigroviridis as focal species (second approach), and L. confusus and L. curicensis for L. monticola as focal species [31, 39, 41] (third approach). Outgroups used for second and third approaches were based in the phylogenetic relationships resulting from the first approach (see results). Maximum-likelihood (ML) analyses were performed in the PhyML 3.0 server (http://www.atgc-montpellier.fr/phyml/) using the best fit model of sequence evolution according to the Bayesian Inference Criterion (other criteria such as Akaike Information Criterion resulted in the same model of sequence evolution) identified by jModelTest [44] for each data set. We evaluated node support using 1000 bootstrap pseudoreplicates [45]. The selected best models of sequence evolution were: For the first approach, Hasegawa-Kishino-Yano (HKY; [46] + invariable sites (0.55) + Gamma shape parameter (1.4866). For the second approach HKY + invariable sites (0.614), and for the third approach, HKY + invariable sites (0.533) + Gamma shape parameter (1.003).
Bayesian analyses were performed using MrBAYES v.3.1.2b [47], using as prior parameters values obtained from jModeltest [44]. Computational resources available in CIPRES Science Gateway were used to run all Bayesian analyses [48]. We ran Metropolis-coupled Markov Chain Monte Carlo simulations (MCMCMC) with four incrementally heated chains. From random starting trees, four independent runs (two replicas of two simultaneous, independent runs each) of 1 x108 generations each were performed, with the resulting trees sampled every 10000 generations. Mixing and convergence were assessed using Tracer v1.6 [49] and the first 1000 trees of the sample were removed. The last 9000 trees were used to compute a 50% majority rule consensus tree. The percentage of samples that recover any particular clade in this tree represents that clade’s posterior probability; we considered BPP (Bayesian Posterior Probability) = 95% as evidence for significant support [50]. Trees were visualized using the Fig Tree v1.4.2 program [51].
Results
Major relationships of the L. nigroviridis and L. monticola (focal species of this study) were very similar in the phylogenetic inferences (Figs. 2, 3 and 4), thus we refer to the ML topology for a discussion of the results. Figure 2 shows the results including all eleven Liolaemus species (approach 1). The ML topology shows two major groups, one supported by maximum likelihood bootstrap (MLB = 92) and by Bayesian posterior probabilities (BPP = 0.99; clade A), and a second clade not supported by maximum likelihood bootstrap (MLB = 47) but supported by Bayesian posterior probabilities (BPP = 0.96; clade B). The difference in the support for the clade B may result from a variety of causes, including the different nature of both analyses and overestimation of posterior probabilities [52,53,54,55]. The two sequences of L. uniformis were placed in a supported clade (MLB = 100, BPP = 1.0) within the L. nigroviridis group, related (but unsupported) to the supported clade that included samples from La Campana, El Roble, and Chicauma (Fig. 2). The second group (clade B) included L. monticola, L. nitidus, L. confusus, L. curicensis and L. lemniscatus (Fig. 2). Within this clade, L. monticola, L. nitidus and L. confusus plus L. curicensis were recovered in a supported clade (MLB = 87; BPP = 0.99). The focal species L. monticola was recovered within an unsupported clade together with L. nitidus. However, all sequences from the northern distribution of L. monticola (northern race, see [38] were placed in a supported clade (MLB = 99, BPP = 1.0; clade B1) with sequences of L. nitidus (from Chicauma, El Yeso, Dunas de Ritoque and Cantillana localities; See Additional file 1). Sequences of L. monticola of the southern race were recovered within a supported clade together with L. nitidus from Cantillana (MLB = 93, BPP = 0.99; clade B2). One L. nitidus sequence (L. nitidus EU22083) was placed closer to all L. monticola from the northern race (Fig. 2). Figure 3 shows the relationships of L. nigroviridis as focal species (approach 2). The ML topology does not differ from Fig. 2, but differs in the position and support of the L. nigroviridis sequences from Cantillana. In Fig. 3, 13 out of 14 sequences from Cantillana were placed in a supported clade (MLB = 89; BPP = 0.99), but in Fig. 2 all sequences were recovered in a supported (MLB = 99, BPP = 1.0) clade. Similar to Fig. 2, sequences from L. uniformis were placed in supported clade (MLB = 99, BPP = 1.0) with an unresolved position compared to all L. nigroviridis clades. Figure 4 shows the relationships with L. monticola as focal species. L. confusus and L. curicensis were used as outgroups. The topology recovered was similar to that depicted in Fig. 2, but the clade including all L. monticola plus L. nitidus sequences was supported (MLB = 96, BPP = 1.0). The sequence of L. nitidus (EU22083) was recovered unresolved with L. monticola northern race sequences but in an unsupported clade.

Maximum likelihood tree of eleven species of the genus Liolaemus. Maximum likelihood tree of eleven species of the genus Liolaemus based on the mitochondrial gene cytochrome b. Numbers on the branches indicate bootstrap values for maximum likelihood and posterior probabilities for Bayesian inference. Branches of the focal species are shown in colors: L. nigroviridis (green) and L. monticola (orange): L. uniformis is shown in pink and L. nitidus in light blue. Information on haplotypes is given in Additional file 1: Table S1

Maximum likelihood tree for the focal species L. nigroviridis. Maximum likelihood tree based on cytochrome b gene for the focal species L. nigroviridis. Numbers on the branches indicate bootstrap values for maximum likelihood and posterior probabilities for Bayesian inference. Colors of the branches represent L. nigroviridis (in green) and L. uniformis (in pink) haplotypes

Maximum likelihood tree for the focal species L. monticola. Maximum likelihood tree based on cytochrome b gene for the focal species L. monticola. Numbers on the branches indicate bootstrap values for maximum likelihood and posterior probabilities for Bayesian inference. Colors of the branches represent L. monticola (in orange) and L. nitidus (in light blue) haplotypes
Discussion
Phylogenetic analyses are widely used in systematics to support species status complimentarily with other types of data (e.g. isozyme electrophoresis [56], traditional morphology-based methods [57], chromosomes [34, 58], and some physiological and ecological traits [59, 60]), especially when monophyly is recovered. A number of studies have emphasized the role of using intense taxon sampling of species and the number of individuals to elucidate spurious monophyletic groups [2, 9, 10, 12], and the degree of polymorphism in diagnosing new or hidden taxa [61]. Reciprocal monophyly may not be achieved in taxa with different evolutionary trajectories [62, 63] leading to inconclusive support for species designation. The genus Liolaemus provides several examples of this situation [5, 12, 64,65,66,67], with a number of taxa still waiting to be studied or validated. Here we used a higher number of samples that included sister species for phylogenetic reconstructions, with two species (L. uniformis and L. nitidus) placed within the clades of the two focal species, L. nigroviridis and L. monticola, respectively.
Topologies for L. nigroviridis with ML and Bayesian inference recovered all haplotypes in a supported clade, with strong phylogeographic structure congruent with previous results [27]. Almost all sampled populations were recovered in a supported clade, with L. uniformis placed within the L. nigroviridis clade. Liolaemus uniformis was described from one locality (Laguna Chepical, 32°15′S, 70°30′W), and the analysis of the phylogeny included sequences from 37 Liolaemus species, with 11 sequences of L. nigroviridis from the locality of Farellones [31]. These taxa were inferred as sister species. Morphological descriptions and analyses were also performed including other species from the L. nigroviridis group to support the new proposed taxon. L. nigroviridis and L. uniformis were reported to be sympatric, with significant differences in a few morphological characters [31]. The phylogenetic placement of L. uniformis within the L. nigroviridis clade may be explained by several processes including mitochondrial introgression, coalescent stochasticity, imperfect taxonomy, inadequate phylogenetic information, paralogy, hybridization, the taxon sampling effect and incomplete lineage sorting [68,69,70,71,72]. Discriminating among these processes may be difficult, particularly in groups with complex evolutionary histories and/or widely distributed species inhabiting diverse landscapes. Liolaemus nigroviridis inhabits mountain areas of central Chile, and several populations are currently in allopatry, inhabiting both in Coast Range and Andean environments [27, 29]. Although it is not the goal of this study to determine the main processes that explain a potential paraphyly of L. nigroviridis, our results show that increasing the sampling from additional localities of closer (e.g. sister) taxa may result in absence of monophyly. According to our results and a previous study [27], the molecular divergence of L. nigroviridis populations and the placement of L. uniformis suggests cryptic divergence within L. nigroviridis. However, our study is limited to one mitochondrial locus, and the robustness of phylogenetic inferences (i.e. for delimiting species) increases when more independent loci are considered [2, 61, 73]. Therefore, to evaluate the processes that may explain this pattern additional molecular analyses coupled with morphology and ecology are required [74,75,76,77].
Our topologies also show polyphyly of L. monticola (Figs. 2 and 4). Haplotypes of L. monticola were placed in two divergent clades (northern and southern races; [37, 38], with several haplotypes of L. nitidus within L. monticola clades. Eight haplotypes of L. nitidus were sister (and supported; clade B1) to a clade that included all L. monticola haplotypes that belong to the northern race plus one (supported) additional L. nitidus haplotype (H96; Fig. 2). Two haplotypes of L. nitidus were closer to haplotypes of L. monticola that belong to the southern race (clade B2). Several studies using molecular phylogenetic analyses have inferred L. monticola and L. nitidus as sister species [3, 31, 39, 60, 78], a result that might contribute to the complexity of determining the main process (either deterministic or stochastic) that explains our results. Incomplete lineage sorting and mitochondrial introgression have been discussed as probable causes to explain paraphyletic patterns in Liolaemus [4, 5, 40, 64, 66]. We found that almost all haplotypes of L. nitidus and L. monticola (except L. nitidus H34; Fig. 4) were placed in divergent and supported clades, which would not support the incomplete lineage sorting hypothesis. L. nitidus is widely distributed in Central Chile, including Administrative Regions IV to VIII and from 0 to 3050 m [60], and this distribution partially overlaps that of L. monticola. In our study two out of five localities (El Yeso from the Andes and Cantillana from Coast Range) included samples of both species; these populations are in contact and may potentially exchange mitochondrial genes. Therefore, alternative processes such as historical mitochondrial introgression should be further evaluated.
Strikingly, the eight haplotypes of L. nitidus from the locality of Cantillana were placed in two divergent clades: six haplotypes (haplotypes 35, 38, 41, 42, 43, 44; Fig. 4) were closer to the clade that included the L. monticola northern race, and two other haplotypes (Haplotypes 36, 37; Fig. 4) were closer to the clade of the L. monticola southern race. Despite our sampling bias, the high molecular divergence in L. nitidus may indicate a substantial history of genetic isolation among populations [68]. Quaternary fluctuations may have played a major role in the current population structure of lizards in Chile [6, 38, 79], connecting populations that may have been allopatric in the past [80].
The divergence found in L. nitidus may be a result of ancestral allopatric populations that converged in Cantillana as a result of historical climatic changes. However, the latter hypothesis does not explain the overall phylogenetic pattern found in L. monticola and L. nitidus.
Imperfect taxonomy has also been suggested as a hypothesis to explain paraphyletic patterns. However, these species have strong differences in morphology (size, shape, scales, and color; [32], chromosome number [34, 58], and certain ecological and physiological traits [60, 81]. Molecular analyses have revealed several examples of cryptic diversity that have impacted the taxonomy of some species (e.g. [82, 83]); our topologies show that clade divergences may hide greater diversity than expected, which should be further evaluated within each of the focal species (i.e. adding more independent loci).
Conclusion
Based on a mitochondrial marker, we showed that using intensive taxon sampling for phylogenetic reconstructions, two lizard species (L. uniformis and L. nitidus) are placed within the clades of two different species (L. nigroviridis and L. monticola, respectively). Our study confirms the importance of taxon sampling to infer more accurate phylogenetic relationships, particularly to reveal hidden polyphyly or paraphyly, which may have a strong impact in taxonomic proposals and inference of cryptic diversity.
Abbreviations
- BPP:
-
Bayesian posterior probabilities
- HKY:
-
Hasegawa-Kishino-Yano
- MCMCMC:
-
Metropolis-coupled markov chain monte carlo
- ML:
-
Maximum likelihood
- MLB:
-
Maximum likelihood bootstrap
References
Townsend T, Larson A. Molecular phylogenetics and mitochondrial genomic evolution in the Chamaeleonidae (Reptilia, Squamata). Mol Phylogenet Evol. 2002;23:22–36.
Wiens JJ, Penkrot TA. Delimiting species using DNA and morphological variation and discordant species limits in spiny lizards (Sceloporus). Syst Biol. 2002;51:69–91.
Schulte JA, Robert MJ, Espinoza RE, Larson A. Phylogenetic relationships in the iguanid lizard genus Liolaemus: multiple origins of viviparous reproduction and evidence for recurring Andean vicariance and dispersal. Biol J Linn Soc. 2000;69:75–102.
Avila LJ, Morando M, Sites JW. Congeneric phylogeography: Hypothesizing species limits and evolutionary processes in Patagonian lizards of the Liolaemus boulengeri group (Squamata: Liolaemini). Biol J Linn Soc. 2006;89:241–75.
Morando M, Avila LJ, Turner CR, Sites JW. Molecular evidence for a species complex in the patagonian lizard Liolaemus bibronii and phylogeography of the closely related Liolaemus gracilis (Squamata: Liolaemini). Mol Phylogenet Evol. 2007;43:952–73.
Victoriano PF, Ortiz JC, Benavides E, Adams BJ, Sites JW. Comparative phylogeography of codistributed species of Chilean Liolaemus (Squamata: Tropiduridae) from the central-southern Andean range. Mol Ecol. 2008;17:2397–416.
Kornilios P, Thanou E, Lymberakis P, Sindaco R, Liuzzi C, Giokas S. Mitochondrial phylogeography, intraspecific diversity and phenotypic convergence in the four-lined snake (Reptilia, Squamata). Zool Scr. 2014;43:149–60.
Tonione MA, Fisher RN, Zhu C, Moritz C. Deep divergence and structure in the Tropical Oceanic Pacific: A multilocus phylogeography of a widespread gekkonid lizard (Squamata: Gekkonidae: Gehyra oceanica). J Biogeogr. 2016;43:268–78.
Zwickl DJ, Hillis DM. Increased taxon sampling greatly reduces phylogenetic error. Syst Biol. 2002;51:588–98.
Hedin M, Wood DA. Genealogical exclusivity in geographically proximate populations of Hypochilus thorelli Marx (Araneae, Hypochilidae) on the Cumberland Plateau of North America. Mol Ecol. 2002;11:1975–88.
Hey J. Bridging phylogenetics and population genetics with gene tree models. In: Schierwater B, Streit B, Wagner GP, DeSalle R, editors. Molecular Ecology and Evolution: Approaches and Applications. Birkhäuser Basel. 1994. p. 435–49.
Morando M, Avila LJ, Sites JW. Sampling strategies for delimiting species: genes, individuals, and populations in the Liolaemus elongatus-kriegi complex (Squamata: Liolaemidae) in Andean-Patagonian South America. Syst Biol. 2003;52:159–85.
Agnarsson I, May-Collado LJ. The phylogeny of Cetartiodactyla: The importance of dense taxon sampling, missing data, and the remarkable promise of cytochrome b to provide reliable species-level phylogenies. Mol Phylogenet Evol. 2008;48:964–85.
Albert EM, San Mauro D, García-París M, Rüber L, Zardoya R. Effect of taxon sampling on recovering the phylogeny of squamate reptiles based on complete mitochondrial genome and nuclear gene sequence data. Gene. 2009;441:12–21.
Johnson RF. Breaking family ties: Taxon sampling and molecular phylogeny of chromodorid nudibranchs (Mollusca, Gastropoda). Zool Scr. 2011;40:137–57.
Frost DR, Etheridge R. A phylogenetic analysis and taxonomy of iguanian lizards (Reptilia: Squamata). Univ Kansas Museum Nat Hist Misc Publ. 1989;81:1–65.
Hillis DM. Inferring complex phylogenies. Nature. 1996;383:130–1.
Harris DJ, Marshall JC, Crandall KA. Squamate relationships based on C-mos nuclear DNA sequences: increased taxon sampling improves bootstrap support. Amphibia-Reptilia. 2001;22:235–42.
Pollock DD, Zwickl DJ, McGuire JA, Hillis DM. Increased taxon sampling is advantageous for phylogenetic inference. Syst Biol. 2002;51:664–71.
Streicher JW, Schulte JA, Wiens JJ. How should genes and taxa be sampled for phylogenomic analyses with missing data? An empirical study in iguanian lizards. Syst Biol. 2016;65:128–45.
Abdala CS, Quinteros AS. Los últimos 30 años de estudios de la familia de lagartijas más diversa de Argentina. Actualización taxonómica y sistemática de Liolaemidae. Cuad Herpetol. 2014;28(2):55–82.
Fontanella FM, Feltrin N, Avila LJ, Sites JW, Morando M. Early stages of divergence: Phylogeography, climate modeling, and morphological differentiation in the South American lizard Liolaemus petrophilus (Squamata: Liolaemidae). Ecol Evol. 2012;2:792–808.
Ruiz De Gamboa M. Lista actualizada de los reptiles de Chile. Boletín Chil Herpetol. 2016;12:7–12.
Pincheira-Donoso D, Scolaro JA, Sura P. A monographic catalogue on the systematics and phylogeny of the South American iguanian lizard family Liolaemidae (Squamata, Iguania). Zootaxa. 2008;1800:1–85.
Vidal MA, Iturra-Cid M, Ortiz JC. Clasificación de anfibios y reptiles. In: Vidal M, Labra A, editors. Herpetología de Chile. Santiago de Chile: Science Verlag Chile; 2008. p. 79–106.
Torres-Pérez F, Gomez-Lobo FD, Garin C. Liolaemus nigroviridis (black-green tree iguana). Herpetological Review. 2005;36(1):80.
Cianferoni F, Yáñez RP, Eduardo RE, Garin CF, Torres-Pérez F. Deep divergences within Liolaemus nigroviridis (squamata, liolaemidae) lineages associated with sky islands in central Chile. Zootaxa. 2013;3619(1):59–69.
Hellmich W. Die Eidechsen der Ausbeute Schröder (Gattung Liolaemus, Iguan.) (Beiträge zur Kenntnis der Herpetofauna Chiles XIII). Veröffentlichungen der Zool Staatsammlung. 1950;1:129–94.
Müller L, Hellmich W. Beitrage zur kenntnis der herpetofauna Chiles. II Neue Liolaemus arten und rassen aus den hoch-Anden Chiles. Zool Anz. 1932;97:307–29.
Valencia J, Veloso A, Sallaberry M. Análisis biométrico y cromosómico en poblaciones de Biometric and chromosomic analysis of populations of Liolaemus nigroviridis Müller and Hellmich (Squamata-iguanidae). Arch Biol Med Exp. 1979;12:209–20.
Troncoso-Palacios J, Elorza AA, Puas GI, Alfaro-Pardo E. A new species of Liolaemus related to L. nigroviridis from the Andean highlands of central Chile (Iguania, Liolaemidae). Zookeys. 2016;555:91–114.
Donoso-Barros R. Reptiles de Chile. Santiago: Editorial Universitaria; 1966.
Lamborot M, Alvarez E, Campos I, Espinoza A. Karyotypic characterization of three Chilean subspecies of Liolaemus monticola. J Hered. 1981;72:328–34.
Lamborot M, Alvarez-Sarret E. Karyotypic variation within and between populations of Liolaemus monticola (Tropiduridae) separated by the Maipo River in the Coastal Range of Central Chile. Herpetologica. 1993;49:435–49.
Lamborot M. Chromosomal evolution and speciation in some Chilean lizards. Evol Biol. 1993;7:133–51.
Lamborot M, Ossa CG, Vásquez M. Population cytogenetics of the “Northern Mod 1” chromosomal race of Liolaemus monticola Müller & Helmich (Iguanidae) from Central Chile. Gayana (Concepción). 2012;76:10–21.
Lamborot M, Eaton LC. Concordance of morphological variation and chromosomal races in Liolaemus monticola (Tropiduridae) separated by riverine barriers in the Andes. Zeitschrift fur Zool Syst und Evol. 1992;30:189–200.
Torres-Pérez F, Lamborot M, Boric-Bargetto D, Hernández CE, Ortiz JC, Palma RE. Phylogeography of a mountain lizard species: An ancient fragmentation process mediated by riverine barriers in the Liolaemus monticola complex (Sauria: Liolaemidae). J Zoolog Syst Evol Res. 2007;45(1):72–81.
Pyron RA, Burbrink FT, Wiens JJ. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol Biol. 2013;13:93.
Torres-Pérez F, Méndez MA, Benavides E, Moreno RA, Lamborot M, Palma RE, Ortiz JC. Systematics and evolutionary relationships of the mountain lizard Liolaemus monticola (Liolaemidae): how morphological and molecular evidence contribute to reveal hidden species diversity. Biol J Linn Soc. 2009;96:635–50.
Troncoso-Palacios J, Schulte JA, Marambio-Alfaro Y, Hiriart D. Phenotypic variation, phylogenetic position and new distributional records for the poorly known Liolaemus silvai Ortiz, 1989 (Iguania: Iguanidae: Liolaemini). South Am J Herpetol. 2015;10:71–81.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Darriba D, Taboada G, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772.
Felsenstein J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J Mol Evol. 1981;17:368–76.
Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–74.
Ronquist F, Teslenko M, Van Der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard, MA, Huelsenbeck JP. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Gateway Computing Environments Workshop (GCE). 2010. p. 1–8.
Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer v1.6. 2014.
Huelsenbeck JP, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5.
Rambaut A. FigTree Tree Figure Drawing Tool (version 1.4.2). 2006-2014. http://tree.bio.ed.ac.uk/software/figtree/. Accessed 9 Jan 2017.
Douady CJ, Dosay M, Shivji MS, Stanhope MJ. Molecular phylogenetic evidence refuting the hypothesis of Batoidea (rays and skates) as derived sharks. Mol Phylogenet Evol. 2003;26:215–21.
Alfaro ME, Zoller S, Lutzoni F. Bayes or Bootstrap? A simulation study comparing the performance of Bayesian Markov Chain Monte Carlo sampling and bootstrapping in assessing phylogenetic confidence. Mol Biol Evol. 2003;20:255–66.
Cummings MP, Handley SA, Myers DS, Reed DL, Rokas A, Winka K. Comparing Bootstrap and Posterior Probability values in the four-taxon case. Syst Biol. 2003;52:477–87.
Simmons MP, Pickett KM, Miya M. How meaningful are Bayesian support values? Mol Biol Evol. 2004;21:188–99.
Buth DG, Murphy RW. The use of isozyme characters in systematic studies. Biochem Syst Ecol. 1999;27:117–29.
Hillis DM, Wiens JJ. Molecular versus morphological systematics: conflicts, artifacts, and misconceptions. In: Wiens JJ, editor. Phylogenetic analysis of morphological data. Washington: Smithsonian Institution Press; 2000. p. 1–19.
Lamborot M, Alvarez-Sarret E. Karyotypic characterization of some Liolaemus lizards in Chile (Iguanidae). Genome. 1989;32:393–403.
Escobar C, Labra A, Niemeyer HM. Chemical composition of precloacal secretions of Liolaemus lizards. J Chem Ecol. 2001;27:1677–90.
Pincheira-Donoso D, Nuñez H. Las especies chilenas del género Liolaemus Wiegmann, 1834 (Iguania: Tropiduridae: Liolaeminae): Taxonomía, Sistemática y Evolución. Publicación Ocas del Mus Nac Hist Nat. 2005;59:7–486.
Wiens JJ, Servedio MR. Species delimitation in systematics: inferring diagnostic differences between species. Proc R Soc Lond Ser B Biol Sci. 2000;267:631–6.
Knowles L, Maddison W. Statistical phylogeography. Mol Ecol. 2002;11:2623–35.
Knowles L, Carstens BC. Delimiting species without monophyletic gene trees. Syst Biol. 2007;56:887–95.
Olave M, Martinez LE, Avila LJ, Sites JW, Morando M. Evidence of hybridization in the Argentinean lizards Liolaemus gracilis and Liolaemus bibronii (Iguania: Liolaemini): An integrative approach based on genes and morphology. Mol Phylogenet Evol. 2011;61:381–91.
Olave M, Avila LJ, Sites JW, Morando M. Model-based approach to test hard polytomies in the Eulaemus clade of the most diverse South American lizard genus Liolaemus (Liolaemini, Squamata). Zool J Linn Soc. 2015;174:169–84.
Morando M, Avila LJ, Baker J, Sites JW. Phylogeny and phylogeography of the Liolaemus darwinii complex (Squamata: Liolaemidae): evidence for introgression and incomplete lineage sorting. Evolution. 2004;58:842–61.
Camargo A, Morando M, Avila LJ, Sites JW. Species delimitation with abc and other coalescent-based methods: a test of accuracy with simulations and an empirical example with lizards of the Liolaemus darwinii complex (Squamata: Liolaemidae). Evolution. 2012;66(9):2834–49.
Funk DJ, Omland KE. Frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu Rev Ecol Evol Syst. 2003;34:397–423.
Kingman JFC. The coalescent. Stoch Process their Appl. 1982;13:235–48.
Neigel JE, Avise JC. Application of a random walk model to geographic distributions of animal mitochondrial DNA variation. Genetics. 1993;135:1209–20.
Takahata N, Slatkin M. Genealogy of neutral genes in two partially isolated populations. Theor Popul Biol. 1990;38:331–50.
Templeton AR. Nested clade analyses of phylogeographic data: testing hypotheses about gene flow and population history. Mol Ecol. 1998;7:381–97.
Wiens JJ. Species delimitation: new approaches for discovering diversity. Syst Biol. 2007;56:875–8.
Padial JM, Miralles A, De la Riva IJ, Vences M. The integrative future of taxonomy. Front Zool. 2010;7:16.
Aguilar C, Wood PL, Cusi JC, Guzmán A, Huari F, Lundberg M, Mortensen E, Ramírez C, Robles D, Suárez J, Ticona A, Vargas VJ, Venegas PJ, Sites Jr JW. Integrative taxonomy and preliminary assessment of species limits in the Liolaemus walkeri complex (Squamata, Liolaemidae) with descriptions of three new species from Peru. Zookeys. 2013;364:47–91.
Aguilar C, Wood PL, Belk MC, Duff MH, Sites JW. Different roads lead to Rome: Integrative taxonomic approaches lead to the discovery of two new lizard lineages in the Liolaemus montanus group (Squamata: Liolaemidae). Biol J Linn Soc. 2016. doi:10.1111/bij.12890.
Minoli I, Morando M, Avila LJ. Integrative taxonomy in the Liolaemus fitzingerii complex (Squamata: Liolaemini) based on morphological analyses and niche modeling. Zootaxa. 2014;3856:501–28.
Pincheira-Donoso D, Harvey LP, Ruta M. What defines an adaptive radiation? Macroevolutionary diversification dynamics of an exceptionally species-rich continental lizard radiation. BMC Evol Biol. 2015;15:153.
Vidal MA, Moreno PI, Poulin E. Genetic diversity and insular colonization of Liolaemus pictus (Squamata, Liolaeminae) in north-western Patagonia. Austral Ecol. 2012;37:67–77.
Fuentes ER, Jaksic FM. Lizards and rodents: an explanation for their relative species diversity in Chile. Arch Biol Med Exp (Santiago). 1979;12:179–90.
Labra A, Escobar CA, Niemeyer HM. Chemical discrimination in Liolaemus lizards: comparison of behavioral and chemical data. Chemical signals in vertebrates. 2001;9:439–44.
Murphy JC, Jowers MJ. Treerunners, cryptic lizards of the Plica plica group (Squamata, Sauria, Tropiduridae) of northern South America. Zookeys. 2013;355:49–77.
Lobo F, Espinoza RE. Two New Cryptic Species of Liolaemus (Iguania: Tropiduridae) from Northwestern Argentina: Resolution of the purported reproductive bimodality of Liolaemus alticolor. Copeia. 1999;1:122–40.
Acknowledgements
We thank R. Stevenson, T. Ramirez and G. Quintana for fieldwork support. We also thank the Servicio Agricola y Ganadero (SAG) for trapping permits.
Funding
Financial support was provided by FONDECYT 1140929. DBB was supported by the postdoctoral project 2015-2016 VRIEA PUCV.
Availability of data and materials
Please contact author for data request.
Authors’ contributions
Conceived and designed the experiments: FTP, DBB, REP. Performed the experiments: DBB, ERV, CE. Analyzed the data: FTP, DBB, ERV, CE, REP. Contributed reagents/materials/analysis tools: FTP, REP. Wrote the paper: FTP, DBB, ERV. Critically evaluated the manuscript: FTP, DBB, ERV, CE, REP. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publications
Not applicable.
Ethics approval
Captures were made under the following permits granted by the Chilean Agriculture and Livestock Service (SAG): 306(2015)
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
List of species and museum or scientific collection codes used in the phylogeny, locality, haplotype number and map locality. SSUC: Colección flora y fauna, Profesor Patricio Sánchez Reyes; CFFPUCV: Colección flora y fauna de la Pontificia Universidad Católica de Valparaíso. MNHNCL: Museo Nacional de Historia Natural de Chile; MZUC: Museo de Zoología de la Universidad de Concepción; CUCH: Colección de la Universidad de Chile. (PDF 86 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Torres-Pérez, F., Boric-Bargetto, D., Rodríguez-Valenzuela, E. et al. Molecular phylogenetic analyses reveal the importance of taxon sampling in cryptic diversity: Liolaemus nigroviridis and L. monticola (Liolaeminae) as focal species. Rev. Chil. de Hist. Nat. 90, 5 (2017). https://doi.org/10.1186/s40693-017-0068-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40693-017-0068-z