
Abstract
Background
Saccharopolyspora erythraea (S. erythraea) is a Gram-positive erythromycin–producing filamentous bacterium. The lack of comprehensive S. erythraea genome-scale metabolic models (GEMs) hinders the efficiency of metabolic engineering as well as fermentation process optimization.
Results
In this study, the GEMs model of S. erythraea iZZ1342 was reconstructed according to the latest genome annotations, omics databases, and literatures. Compared with the previous S. erythraea model—GSMR, the new model iZZ1342 presented great improvements both on scope and coverage in the number of reactions, metabolites, and annotated genes. In detail, the number of unique reactions in iZZ1342 was increased from 1482 to 1684, the number of metabolites was increased from 1546 to 1614, and the number of unique genes was increased from 1272 to 1342. We also added 1441 gene-protein-reaction associations in iZZ1342 which lacks in the previous model to overcome the limitation in the application of strain designing. Compared with the transcriptomics data obtained from the published literature, 86.3% ORFs and 92.9% reactions in iZZ1342 can be verified. The results of the sensitivity analysis showed the similar trend in the E. coli GEMs. The prediction of growth on available 27 kinds of carbon sources and 33 kinds of nitrogen sources showed the accuracy rate was 77.8 and 87.9%, respectively. Compared with the physiological data obtained from chemostat cultivation, the simulation results showed good consistency. The correlation coefficient between the 13C-labeled experiment data and the flux simulation result was 0.97. All the above results showed that the iZZ1342 model has good performance. Furthermore, four genes are in the range of successful knockout by comparing these targets with the results which have been earlier published.
Conclusion
The new model iZZ1342 improved significantly in model size and prediction performance, which will lay a good foundation to study the systematic metabolic engineering of S. erythraea system in vivo.

Similar content being viewed by others

Background
Saccharopolyspora erythraea, formerly called Streptomyces erythraeus (Weber et al. 1985), is a kind of Gram-positive filamentous bacterium which produces the medically useful antibiotic erythromycin A (Oliynyk et al. 2007). Erythromycin is an important broad-spectrum 14-membered macrolide antibiotic which has been widely used in the treatment of many diseases caused by pathogenic Gram-positive bacteria (Mironov et al. 2004). Nowadays, industrial production of erythromycin is mainly through submerged culture system. Similar to the production of antibiotics through secondary metabolism of other actinomyces (Bibb 2005; Medema et al. 2010; Wentzel et al. 2012), the synthesis process of erythromycin is also complex and is largely influenced by the composition of the media and culture condition (Martin and Bushell 1996; Mcdermott et al. 1993; Rostamza et al. 2008).
Systems biology is a kind of discipline which combines experimental and computational methods (Feist and Al 2009). This method is useful to make a comprehensive analysis and prediction of complex intracellular biological systems. Using different omics techniques has made it possible to analyze the abundant data intracellularly, which includes the correlation between the various components within the cell. The significance of genomics is self-evident in today’s most widely used omics analysis (Ellis and Goodacre 2012). The development of mathematical models improves the ability to analyze and integrate these omics data (Gehlenborg et al. 2010; Hollywood et al. 2006; Stefanovic et al. 2017). Abstract genome data and intracellular metabolic pathways are transformed into mathematical models, which make systems biology developed tremendously (Ellis and Goodacre 2012). Most importantly, the Genome-Scale Metabolic Models (GEMs) are becoming one of the most significant tools for analyzing different metabolites and metabolic pathways in metabolic engineering (Kim et al. 2012). With the development of modern genome sequencing, it is possible to integrate the reconstruction of metabolic pathways into GEMs.
The genome-scale metabolic reconstructions (GSMRs) of S. erythraea were built in 2012 (Licona-Cassani et al. 2012). In this model, the metabolic reactions of S. erythraea in intracellular were sorted out by the author and the medium suitable for erythromycin growth was optimized by using this model. In order to promote the quality of the GEMs of S. erythraea, there are at least four aspects that need to be improved according to GEMs of other model strains (O’Brien et al. 2013; Tomàs-Gamisans et al. 2016). Firstly, the balance of mass and electrical charge should be checked; secondly, the ineffective reactions should be deleted and the lacking metabolic pathways need to be filled; thirdly, the gene-protein-reactions (GPRs) relationship should be constructed so that we can use our model to design strain by gene target prediction. Finally, the accuracy and predictive power of the model should be validated by comparing the biomass growth parameters, metabolic fluxes, and other physiological parameters with the simulation results.
In this study, a GEM model based on constraints of flux balance analysis (FBA) was reconstructed and in silico analysis was conducted to compare the physiological data and metabolic states of S. erythraea among different cultivation environments. We integrated the latest omics information into our GEM model (Oliynyk et al. 2007; Peano et al. 2012), then predicted the essential genes, the secretion of product, and the growth condition in different media with our model accurately. Overall, the reconstructed model could better describe the metabolic characteristics, and for this reason, we create a better platform to study the systems metabolic engineering of S. erythraea in vivo.
Methods
Microorganism
The S. erythraea strains NRRL23338 and E3 were used in this study.
Media and culture conditions
The composition of the chemically defined medium used for the pre-batch culture of the microorganism contains (per liter of deionized water): 30 g glucose, 7 g K2HPO4, 3 g KH2PO4, 5.5 g (NH4)2SO4, 0.25 g MgSO4·7H2O, 25 mg FeSO4·7H2O, 0.53 mg CuCl2, 0.55 mg CoCl2, 13.8 mg CaCl2·7H2O, 10.4 mg ZnCl2, 6.2 mg MnCl2, 0.3 mg Na2MoO4 (Bushell et al. 1997). The medium composition for the carbon limitation chemostat culture was the same as for the pre-batch cultivation except the glucose concentration was changed to 15 g/L (Ghojavand et al. 2011; Mcdermott et al. 1993).
The culture conditions for seed culture, pre-batch culture, and chemostat culture were determined by McDermott et al. (1993). Pre-batch culture and chemostat culture were carried out in a 5-L bioreactor (National Engineering Research Center for Biotechnology, Shanghai, China) with a working volume of 3 L. The dissolved oxygen (DO) was maintained above 40% by adjusting the aeration and the agitation to ensure fully aerobic conditions. The OUR, CER, and RQ were measured online using a process mass spectrometer (MAX300-LG, Extrel, America). Temperature, pH, and pressure were set as 34 °C, 7.0 (with adding 1 M NaOH), and 0.05 MPa, respectively. The specific growth rate (μ) of chemostat culture was controlled using the dilution ratio (D).
Analyses
The cell concentration was monitored by measuring the OD600. The dry cell weight (DCW) was measured as described by Carreras et al. (2002). The fermentation broth supernatant was used for measuring the concentration of residual glucose and organic acids. Residual glucose concentration was analyzed using a glucose kit (Sinopharm Chemical Reagent Co., Ltd, China) as per manufacturer’s protocol. Organic acids concentration was analyzed by high-performance liquid chromatography (HPLC) as described by Albert and Martens (1997).
Procedures for model reconstruction
The GEMs of S. erythraea was built on the basis of the whole genome annotation of S. erythraea and other information from the databases (KEGG, UniProtKB, BioCyc, Enzyme) and literatures (Caspi et al. 2012; Licona-Cassani et al. 2012; Oliynyk et al. 2007), which was followed by the standard three step process (Thiele and Palsson 2010). Firstly, considering the lack of metabolites list and GPR relationship of the model created in 2012, this research reassembled all metabolic reactions and metabolites referring to annotated genes from KEGG and PubChem (Kanehisa et al. 2006; Kim et al. 2016). The reactions in this paper were all manually refined and checked to ensure that the structure of each metabolite is consistent, the charge and mass in every reaction is balanced.
Subsequently, in order to expand the S. erythraea GEMs, we found the latest gene annotation information from three major databases (KEGG, UniProtKB, IMG) to add the number of effective reactions and metabolites (Markowitz et al. 2012). At the same time, we used the GapFind algorithm to check the connectivity of all the pathway in this model and if there are identified missing links somewhere (Kumar et al. 2007), then we will use two steps to fill these gaps: first, by referring to the literature add new reactions from other organisms’ metabolic pathway; second, if the synthetic pathway of this metabolite cannot be found, then introduce transport reactions to allow for metabolite exchange. The format of each new reaction was referred to the standard protocol to ensure the quality of the final model to achieve the standardization requirements (Caspi et al. 2012).
Lastly, because there is lack of the gene-protein-reactions (GPRs) relationships in the model created in 2012, we established the GPRs in our model GEM-iZZ1342. We found out all the genes in the NCBI database, and then correlated the genes with enzymes and reactions according to the function of this gene in the KEGG database. According to the GPRs, the number of ineffective reactions was greatly reduced, and the relationship between the reactions and genes was confirmed clearly. Finally, we re-added the non-gene-associated reactions to the model, including metabolites transport reactions, exchange reactions, and other reactions which are lack of gene annotations. The detailed information about the databases used in this study can be found in Additional file 1.
Sensitivity analysis
In order to investigate the sensitivity of iZZ1342, the qs and the qO2 were set to 0–1.5 mmol glucose/gDCW h and 0–1.0 mmol O2/gDCW h, respectively. In all of the six elements (protein composition, RNA composition, DNA composition, cofactor composition, GAM, NGAM), we changed only one element each time. The range of variations for each simulation is the protein (22.8–68.4%), the RNA (4.9–14.7%), the DNA (2.2–6.6%), the cofactor (1.5–4.5%), the GAM (16–48 mmol ATP/gDCW h), and the NGAM (1.25–3.75 mmol ATP/gDCW h). Finally, we calculated the specific growth rate (μ) and specific oxygen uptake rate (qO2) to reflect the result on changing every element of our model. All simulations were performed using the available software Matlab (Mathworks, Inc).
Biomass composition
An equation describing the conversion of every cellular component into biomass can be derived from previous published literature publication on Streptomyces coelicolor (Borodina et al. 2005). The biomass is composed of the following macromolecules: protein, DNA, RNA, lipid, carbohydrates, and cofactors. The detailed biomass components (Additional file 2) of S. erythraea were referred from Donachie and Begg 1970 and Borodina et al. 2005.
In silico computation using flux balance analysis
Metabolic fluxes of the S. erythraea were defined by using flux balance analysis (FBA), constrains of which are imposed by the stoichiometry matrix in the metabolic network (Bordbar et al. 2014; Orth et al. 2010). The matrix of stoichiometry imposes flux balance constraints on the system, ensuring that the total amount of metabolites produced must be equal to the total amount of consumed at the steady state; this is the so-called pseudo-steady state. The net sum of all fluxes which contains production and consumption for each internal metabolite is set to zero. In FBA, a special objective function, written as a linear combination of fluxes, can be used to calculate the optimal solution. And we all know that according to the linear optimization theory, the optimal solution is at a corner in the feasible flux space. Using the matrix representation, this problem can be stated as follows:
where S is the stoichiometric matrix indicating the stoichiometric coefficient of metabolic reactions in the network and v is the vector of all metabolic fluxes. vmin and vmax represent the minimum and maximum constraints on the fluxes, and which are also used to define the constraints for maximal enzymatic rate and irreversibility of reaction. cT is a vector representing the linear combination of metabolic fluxes. In our research, the biomass production rate is used as the objective function which is targeted to be maximized. We adopted this method to estimate the metabolic fluxes under the assumption that our strain is under exponential phase at which cells grow at the maximum speed. In all of the simulations, glucose was chosen as the sole carbon source and other external metabolites in the transport reactions are set to freely transport through the cell membrane: H2O, CO2, NH4+, PO4, and SO4. All calculations were performed using the available software Matlab (Mathworks, Inc).
Model prediction of the cell growth on different carbon and nitrogen sources
To comprehensively evaluate the prediction ability of iZZ1342, physiological data were obtained from two parts: previous publications and experiments through cultivating S. erythraea on different carbon and nitrogen sources performed in our laboratory. When predicted the utilization of carbon source, NH4+ was set as the only nitrogen source, in the same time sulfate and phosphate were maintained as the only phosphorus source and sulfur source, respectively. Then we set the flux of other exchange reactions referring to carbon source to zero except the aimed carbon source. To predict the utilization of nitrogen, similarly, we set the glucose as the only carbon source. In the simulation, the target substrate was viewed as growth supporting if the predicted growth rate was obviously above zero.
Model prediction of essential genes
To predict the essential genes, non-essential genes, and partially essential genes, Single-Gene Deletion function based on the Cobra Toolbox v2.0 was carried out (Schellenberger et al. 2011). Based on the size of the specific growth rate calculated when a certain gene is knocked out, the genes were divided into three groups: the essential genes (the predicted specific growth rate is equal to 0 or infinitely approaches to 0), non-essential genes (the predicted specific growth rate is equal to the maximum value), and partially essential genes (the predicted specific growth rate was between 0 and the maximum value). Both minimal and optimized chemically defined medium were used to predict essential genes, non-essential genes, and partially essential genes. The minimal chemically defined medium was made up of glucose, oxygen, ammonia, sulfur, and phosphorus, while the optimized synthetic medium formula was optimized by Licona-Cassani et al. (2012).
Results and discussion
Reconstruction of the S. erythraea GEMs iZZ1342
The genome-scale metabolic model (GEMs) of S. erythraea was reconstructed on a three step procedure (see “Methods”). During the reconstruction, the specific process which contains all the materials and the procedures was required to be manually curated in the model as shown in Fig. 1.

The reconstruction process of the genome-scale metabolic model of S. erythraea iZZ1342
Compared with the updated GSMR of S. erythraea published in 2012, the reconstructed GEMs iZZ1342 have shown obvious improvements. First, the ORFs (open reading frames) are increased from 1272 to 1342, the total number of reactions is decreased from 3985 to 1684 after removing the ineffective reactions and adding the GPRs associations. Furthermore, we also sorted out the metabolites list which contains the information of all the metabolites in the reaction. We also manually checked and balanced the mass and electrical charge of the elements and reactions according to the process in “Methods.” Finally, we conducted the gap find analysis procedure and found all the orphan reactions. Then solved these gaps by adding the connecting reactions. The detailed information of iZZ1342 can be found in Additional files 3 and 4.
We compared the GEMs parameters in all aspects of S. erythraea and other actinomyces (Alam et al. 2010, 2011; Kjeldsen and Nielsen 2009). The comparison result is provided in Table 1. As shown in Table 1, the number of total reactions and metabolites (Additional file 5) in iZZ1342 are larger than that in S. erythraea NRRL23338-GSMR, which indicates our model was improved on the scale. The improvements are also reflected on the assigned genes, the coverage of the annotated genes as well as the number of the reactions assigned by the genes. We also compared our GEM with other actinomyces, and the results show that our GEM has a larger scale under the conditions of almost the same genome size.
Model verification by transcriptomic analyses
We verified the new ORFs in the updated GEMs of S. erythraea with the latest transcriptomic analysis data (Carata et al. 2009; Li et al. 2013; Peano et al. 2012). The reason for this is that we can identify genes with low-expression as many as possible compared with the microarray data (Wang et al. 2009). By extracting the information of RNA sequencing, the information of gene sequence and enzymes of different pathways among sampling period can be gained. Then, we can get the information of genes and reactions by analyzing the gene sequence and enzymes. Due to the fact that we created the GPRs associations in this study, we could distinguish the single-gene-associated reactions and multi-gene-associated reactions. The transcriptomic analysis data result showed that about 7186 genes could be determined during the cultivation condition (Additional file 6). The iZZ1342 contains 1342 genes and the expression of most genes (86.3%) could be found according to the results of transcriptomic analysis data during the sampling condition (Fig. 2a). Among all the reactions (except the exchange reactions), the expression of genes which cannot be measured was at 4.6% (71 reactions) of these reactions (Fig. 2b). When removing the exchange reactions and other reactions which are without any annotated genes, 702 and 739 reactions are annotated with single and multiple genes, respectively. Furthermore, the transcriptome analysis result shows that about 85.4% of the single-gene-associated reactions and 89.2% of the multi-gene-associated reactions were verified (Fig. 2c, d), indicating that the most reactions in the model iZZ1342 were reasonable and reliable, which shows the rationality of our model to a great extent.

Verification of the GEM of iZZ1342 by transcriptomics data. a Pie chart of the expressed and unexpressed genes involved in iZZ1342. b Pie chart of the verified reactions and other reactions in total 1441 reactions (except the exchange and transport reactions). c Pie chart of the verified single-gene reactions and other reactions in total single-gene reactions. d Pie chart of the verified multi-gene reactions and other reactions in total multi-gene reactions
Sensitivity analysis of iZZ1342
To check the sensitivity of the simulation results generated from FBA with iZZ1342, we varied the content of all the four largest macromolecules in the cell (protein, RNA, DNA, and cofactor) and the two energy parameters (GAM, NGAM), respectively (Feist et al. 2007). The specific growth rate (μ) and the specific oxygen uptake rate (qO2) were investigated under aerobic and glucose-limited conditions, shown in Fig. 3. When composition of the protein, RNA, DNA, and cofactor was changed, the specific oxygen uptake rate can hardly be affected by the change. However, the specific growth rate was slightly decreased when the composition of protein and RNA was changed. On the contrary, when the energy parameter was changed, μ and qO2 were affected seriously and easily. As GAM and NGAM increased, μ was greatly decreased. In the meantime, qO2 was tremendously increased as shown in Fig. 3e, f. The results of the sensitivity analysis indicate that the model iZZ1342 is very sensitive to the energy parameters rather than the cell composition parameters, which is correspondingly consistent with the results gained from E. coli GEMs (Feist et al. 2007).

Sensitivity analysis of different model parameters by the model iZZ1342. The effects of each parameter when changing the specific growth rate (A1–F1) and the specific oxygen uptake rate (A2–F2) with iZZ1342. The simulations were performed in the glucose-limited condition by varying the protein content (22.8–68.4%). a The RNA content (4.9–14.7%), b the DNA content (2.2–6.6%), c the cofactor content (1.5–4.5%), d the GAM content (16–48 mmol ATP/gDCW h), e the NGAM content (1.25–3.75 mmol ATP/gDCW h), f red represents the simulated results of the high value of the input parameter and black represents the lower value
Model prediction by measuring availability of different carbon and nitrogen sources
To predict the physiological state of S. erythraea growing under different conditions, we collected the reported phenotype experimental data (El-Enshasy et al. 2008; Zou et al. 2009). For other carbon sources and nitrogen sources which are quite crucial but could not find the reference, we complemented experiments to verify the validity. Collectively, 27 kinds of carbon sources and 33 kinds of nitrogen sources were validated. FBA was used to analyze the growth situation on every carbon or nitrogen source. According to the physiological data from the publications and our laboratory, S. erythraea could grow on 23 carbon sources and 27 nitrogen sources. The in silico growth capabilities of S. erythraea on 17 carbon sources and 25 nitrogen sources could be predicted using iZZ1342 and the accuracy rates were 77.8 and 87.9%, respectively. Although the accuracy is already high, the remaining discrepancy will limit its impact on the partial metabolic function of the network, and we believe this will provide new improvement space for a next round of upgrading. The growth-relating results can be found in Tables 2 and 3.
Model validation using physiological growth parameters
To validate the GEMs iZZ1342, we compared the phenotype predictions with the experimental data obtained from the chemostat cultures with minimal chemically defined medium (Mcdermott et al. 1993). Firstly, the S. erythraea NRRL23338 was grown in carbon-limited medium at five dilution rates (0.01, 0.02, 0.03, 0.04, and 0.05/h), then measured the uptake and secretion rates of glucose, O2, CO2, and dry cell weight (DCW). Finally, we calculated the specific growth rate (μ), qO2, qCO2, and qs. In all cultures, we ensured that the recovery is over 91% of the substrate carbon in biomass, CO2, and organic acids.
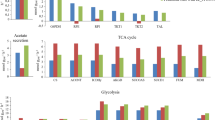
To simulate the cellular growth in carbon-limited medium, we set the cell growth to the maximal while constraining the glucose uptake rate based on the hypothesis that cells tend to maximal growth during exponential phase (Mishra et al. 2016). Moreover, the exchange fluxes of NH4+, phosphate, sulfite, H2O, and H+ were unconstrained to provide basic nutrients for cell growth. The non-growth-associated maintenance (NGAM) was set to 3 mmol/gDCW/h as observed for S. coelicolor (Borodina et al. 2005). As shown in Fig. 4, the prediction data from iZZ1342 matched reasonably with the data from the chemostat cultures. When the qs was changed from 0.5 to 1.5 mmol/gDCW/h, the prediction results of μ, qO2, and qCO2 between the in silico and in vivo were quite similar, indicating that the excellent performance of our new model across multiple environmental conditions to a large extent.

The result of the predicted and measured μ, qO2, and qCO2 for chemostat cultivation of S. erythraea NRRL23338. The NGAM used in simulation was 3 mmol ATP/gDCW h. Black represents simulated results of the GEMs iZZ1342, and red 13 represents the experimental data from our lab
Model validation by in vivo 13C fluxes
Cellular metabolic flux is a significant and direct indicator of the physiological state (Nielsen 2003). Nowadays, GEM models could be used to predict the cellular reaction fluxes due to the fact that GEM model contains the total reactions that can be carried out in the strain. Furthermore, it can avoid any biases caused by lumping reactions or omitting pathways that cannot be prejudged (Saratram and Maranas 2015). However, there is still a possibility to exit obvious discrepancies between the in vivo calculated flux and the simulated flux in silico (Damiani et al. 2015). Therefore, we compared the in vivo calculated flux acquired from the 13C Metabolic Flux Analysis (13C MFA) technology and the simulated flux acquired from FBA to further evaluate the prediction accuracy of our model iZZ1342.
In order to evaluate the prediction accuracy of the model, first we used the 13C-labeled technique to get the flux distribution of analyzed specific metabolic pathways (Hong et al. 2016), and then compared with the flux distribution simulated from our model in silico (Additional file 7). The main aim of this work is to predict how well fluxes gained from analysis of our constraint-based GEM model reflect the real flux distribution.
When analyzing the cellular 13C metabolic flux, we combined the corrected mass isotopomer distributions, the extracellular fluxes as well as the metabolic network. At the same time, a software which is used to calculate the central carbon metabolic flux called INCA was used to iteratively calculate the absolute flux solution that described the data exactly (Young 2014). The central carbon metabolic fluxes identified by INCA and simulation are shown in Fig. 5. As shown in Fig. 5a, the metabolic profiles in the FBA simulations agreed well with the observed experimentally. In Fig. 5b, the correlation coefficient between the simulated fluxes and the calculated 13C fluxes is shown to be 0.97, indicating the good performance of iZZ1342.

The distribution of the central metabolism flux in the cellular. a Metabolic flux profiles of the central metabolism of S. erythraea. The upper number represents the flux acquired from the 13C MFA and the lower number represents the flux simulated from our model iZZ1342. b Consistent changes in fluxes can be found both in the calculated 13C fluxes as well as FBA calculation using iZZ1342
Essential genes target prediction in silico for strain design
The molecular mechanisms in traditional mutation and screening approach for improving the production of erythromycin are still poorly understood. However, this information is quite significant for designing the rational strategies for high-yield strain (Peano et al. 2012). In this study, we used iZZ1342 to find the essential gene targets and give reliable information for making strain design.
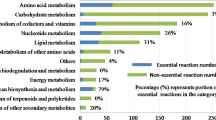
During the reconstruction of our GEM model, we established the relationship of genes, proteins, and reactions (GPRs), so that we can use the GPRs to predict the genotype efficiently. In this process, we used the Single-Gene Deletion function of the Cobra Toolbox v2.0 to predict the essential genes, the partially essential genes and the non-essential genes (Additional file 8). When the minimal chemically defined medium was used to cultivate strains, the result of the simulation shows that 318 genes are essential genes (Fig. 6). These genes are mainly distributed in the TCA cycle, amino acids biosynthesis and metabolism, energy metabolism, and so on. However, when the optimized chemically defined medium was adopted, the number of essential genes has declined markedly, from 318 to 186 (Fig. 6). That is because abundant nitrogen sources were added into the new optimized medium and that resulted in replacing the synthesis pathways of some amino acids. Furthermore, 89 genes were identified as partially essential genes (Additional file 9), and the important characteristics of these genes is that knockout of these genes has a subtle impact on cell growth. However, these genes may play a crucial role in the synthesis of products. They are important targets for subsequent strain design because the yield of product synthase may increase with a slowdown of cell growth (Pan and Qiang 2012). In order to verify the effect of the target gene, some targets which are included in the 89 partially essential genes have been validated by knockout experiments in our lab and on the other published papers, including SACE_5639 (Chen et al. 2016; Weber et al. 2012), SACE_0728 (Mironov et al. 2004), SACE_0731 (Minas et al. 1998), and SACE_6669 (Hong et al. 2017). The knockout results can be found in Table 4. To evaluate the other gene targets, further knockout experiments are needed to validate the prediction results of iZZ1342.

Results of single-gene deletion research with iZZ1342. a The gene expression data and the categories of the expressed gene in KEGG with iZZ1342 (red is the necessary gene, blue is the semi-essential gene, and yellow is the non-essential gene). b The relative growth rate changes of S. erythraea between minimal chemically defined medium and optimized chemically defined medium
Conclusion
We have currently reconstructed and evaluated the genome-scale metabolic model (GEMs) of S. erythraea, called iZZ1342, which contains the latest gene annotation information, physiological parameters, and detailed GPRs relationships. Furthermore, we have also checked the mass and charge balance of all the reactions and metabolites. For those metabolic pathways that lack certain key reactions, we integrated the pathways by filling the gaps. The new model iZZ1342 contains 1614 metabolites and 1684 reactions, in which 1441 reactions are annotated with genes. Comparing with the previous model, the new model has a lot of improvements, mainly including the following aspects: firstly, the balance of mass and electrical charge have been checked; secondly, the ineffective reactions have been deleted and the lacking metabolic pathways have been filled; thirdly, the gene-protein-reactions (GPRs) relationship has been constructed so that we can use our model to design strain by gene target prediction. Finally, we have validated the accuracy and predictive power of the model by comparing the biomass growth parameters, metabolic fluxes, and other physiological parameters with the simulation results.
We validated the new model in several aspects. Firstly, we tested the sensitivity, also called robustness of the model. The result shows good consistency with the simulated result that used E. coli GSMM for simulation when changing the content of every component in our model. Secondly, we tested the model by measuring availability of different carbon and nitrogen sources. The result shows an excellent predictive power of our model: the accuracy of prediction when using different carbon sources is 77.8 and 87.9% when using different nitrogen sources. Thirdly, we tested the model using physiological growth parameters. When we use the glucose as the only carbon sources, the simulation results show a positive correlation with the experimental data. Finally, we validated the model by in vivo 13C fluxes. The result of comparison shows that in the main metabolic pathway, they have a quite good identity and the R2 of fluxes between MFA and GEM model is 0.9638. However, in the other few pathways, the results highlight that further attention should be paid to promote our model.
We employed our model to find all the partially essential genes, and these genes are important targets for subsequent strain designing. According to the published studies, four genes are in the range of successful knockout. However, the other gene targets emphasize that further knockout experiments are needed to validate the prediction results of iZZ1342.
Abbreviations
- S. erythraea :
-
Saccharopolyspora erythraea
- GEM:
-
Genome-Scale Metabolic Model
- GPRs:
-
gene-protein-reaction associations
- NGAM:
-
non-growth-associated maintenance
- GAM:
-
growth-associated ATP maintenance
- D:
-
dilution ratio
- DO:
-
dissolved oxygen
- DCW:
-
dry cell weight
- FBA:
-
flux-balanced analysis
- μ :
-
specific growth rate
- ORF:
-
open reading frame
References
Alam MT, Merlo ME, Hodgson DA, Wellington EM, Takano E, Breitling R (2010) Metabolic modeling and analysis of the metabolic switch in Streptomyces coelicolor. BMC Genomics 11(1):1–9
Alam MT, Medema MH, Takano E, Breitling R (2011) Comparative genome-scale metabolic modeling of actinomycetes: the topology of essential core metabolism. FEBS Lett 585(14):2389–2394
Albert DB, Martens CS (1997) Determination of low-molecular-weight organic acid concentrations in seawater and pore-water samples via HPLC. Mar Chem 56(1–2):27–37
Bibb MJ (2005) Regulation of secondary metabolism in streptomycetes. Curr Opin Microbiol 8(2):208–215
Bordbar A, Monk JM, King ZA, Palsson BO (2014) Constraint-based models predict metabolic and associated cellular functions. Nat Rev Genet 15(2):107–120
Borodina I, Krabben P, Nielsen J (2005) Genome-scale analysis of Streptomyces coelicolor A3(2) metabolism. Genome Res 15(6):820–829
Bushell ME, Smitht J, Lynch HC (1997) A physiological control model for erythromycin production in batch and cyclic fed batch culture. Microbiology 143(2):475–480
Carata E, Peano C, Tredici SM, Ferrari F, Talà A, Corti G, Alifano P (2009) Phenotypes and gene expression profiles of Saccharopolyspora erythraea rifampicin-resistant (rif) mutants affected in erythromycin production. Microb Cell Fact 8(1):18
Carreras C, Frykman S, Ou S, Cadapan L, Zavala S, Woo E, Patel S (2002) Saccharopolyspora erythraea-catalyzed bioconversion of 6-deoxyerythronolide B analogs for production of novel erythromycins. J Biotechnol 92(3):217–228
Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Mueller LA (2012) The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res 36:623–631
Chen C, Hong M, Chu J, Huang M, Ouyang L, Tian X, Zhuang Y (2016) Blocking the flow of propionate into TCA cycle through a mutB knockout leads to a significant increase of erythromycin production by an industrial strain of Saccharopolyspora erythraea. Bioprocess Biosyst Eng 40:1–9
Damiani AL, He QP, Jeffries TW, Wang J (2015) Comprehensive evaluation of two genome-scale metabolic network models for Scheffersomyces stipitis. Biotechnol Bioeng 112(6):1250–1262
Donachie WD, Begg KJ (1970) Growth of the bacterial cell. Nature 227(5264):1220–1224
El-Enshasy HA, Mohamed NA, Farid MA, El-Diwany AI (2008) Improvement of erythromycin production by Saccharopolyspora erythraea in molasses based medium through cultivation medium optimization. Bioresour Technol 99(10):4263–4268
Ellis DI, Goodacre R (2012) Metabolomics-assisted synthetic biology. Curr Opin Biotechnol 23(1):22–28
Feist AM, Al E (2009) Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol 7(2):129–143
Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Palsson BØ (2007) A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol 3(1):1–18
Gehlenborg N, O’Donoghue SI, Baliga NS, Goesmann A, Hibbs MA, Kitano H, Tenenbaum D (2010) Visualization of omics data for system biology. Nat Methods 7(3):56–68
Ghojavand Bonakdarpour H, Heydarian B, Hamedi SM (2011) The inter-relationship between inoculum concentration, morphology, rheology and erythromycin productivity in submerged cultivation of Saccharopolyspora erythraea. Braz J Chem Eng 28(4):565–574
Hollywood K, Brison DR, Goodacre R (2006) Metabolomics: current technologies and future trends. Proteomics 6(17):4716–4723
Hong M, Huang M, Chu J, Zhuang Y, Zhang S (2016) Impacts of proline on the central metabolism of an industrial erythromycin-producing strain Saccharopolyspora erythraea via 13C labeling experiments. J Biotechnol 231:1–8
Kanehisa M, Goto S, Hattori M, Aokikinoshita KF, Itoh M, Kawashima S, Hirakawa M (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34:354–357
Kim TY, Sohn SB, Kim YB, Kim WJ, Lee SY (2012) Recent advances in reconstruction and applications of genome-scale metabolic models. Curr Opin Biotechnol 23(4):617–623
Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Shoemaker BA (2016) PubChem substance and compound databases. Nucleic Acids Res 44:1202–1213
Kjeldsen KR, Nielsen J (2009) In silico genome-scale reconstruction and validation of the Corynebacterium glutamicum metabolic network. Biotechnol Bioeng 102(2):583
Kumar VS, Dasika MS, Maranas CD (2007) Optimization based automated curation of metabolic reconstructions. BMC Bioinform 8(1):1–16
Li YY, Xiao C, Yu WB, Hao L, Ye ZQ, Hui Y, Ye BC (2013) Systems perspectives on erythromycin biosynthesis by comparative genomic and transcriptomic analyses of S. erythraea E3 and NRRL23338 strains. BMC Genomics 14(1):523
Licona-Cassani C, Marcellin E, Quek LE, Jacob S, Nielsen LK (2012) Reconstruction of the Saccharopolyspora erythraea genome-scale model and its use for enhancing erythromycin production. Antonie Van Leeuwenhoek 102(3):493–502
Markowitz VM, Chen IM, Palaniappan K (2012) IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res 40:115–122
Martin SM, Bushell ME (1996) Effect of hyphal morphology on bioreactor performance of antibiotic-producing Saccharopolyspora erythraea cultures. Microbiology 142(7):1783–1788
Mcdermott JF, Lethbridge G, Bushell ME (1993) Estimation of the kinetic constants and elucidation of trends in growth and erythromycin production in batch and continuous cultures of Saccharopolyspora erythraea using curve-fitting techniques. Enzyme Microb Technol 15(8):657
Medema MH, Trefzer A, Kovalchuk A, van den Berg M, Müller U, Heijne W, Nierman WC (2010) The sequence of a 1.8-mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol Evol 2(1):212–224
Minas W, Brünker P, Kallio PT, Bailey JE (1998) Improved erythromycin production in a genetically engineered industrial strain of Saccharopolyspora erythraea. Biotechnol Prog 14(4):561–566
Hong M, Han M, Liu X, Huang M, Ju C (2017) 13C-assisted metabolomics analysis reveals the positive correlation between specific erythromycin production rate and intracellular propionyl-CoA pool size in Saccharopolyspora erythraea. Bioprocess Biosyst Eng 40(4):1–12
Mironov VA, Sergienko OV, Nastasiak IN, Danilenko VN (2004) Biogenesis and regulation of biosynthesis of erythromycins in Saccharopolyspora erythraea: a review. Prikl Biokhim Mikrobiol 40(6):613
Mishra P, Park GY, Lakshmanan M, Lee HS, Lee H, Chang MW, Lee DY (2016) Genome-scale metabolic modeling and in silico analysis of lipid accumulating yeast Candida tropicalis for dicarboxylic acid production. Biotechnol Bioeng 113(9):1993
Nielsen J (2003) It is all about metabolic fluxes. J Bacteriol 185(24):7031–7035
O’Brien EJ, Lerman JA, Chang RL, Hyduke DR, Palsson BØ (2013) Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol Syst Biol 9:1–13
Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Leadlay PF (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25(4):447–453
Orth JD, Thiele I, Palsson BO (2010) What is flux balance analysis? Nat Biotechnol 28(3):245–248
Pan P, Qiang H (2012) Reconstruction and in silico analysis of metabolic network for an oleaginous yeast, Yarrowia lipolytica. PloS ONE 7(12):1–11
Peano C, Talà A, Corti G, Pasanisi D, Durante M, Mita G, Alifano P (2012) Comparative genomics and transcriptional profiles of Saccharopolyspora erythraea NRRL 2338 and a classically improved erythromycin over-producing strain. Microb Cell Fact 11(1):32
Rostamza M, Noohi A, Hamedi J (2008) Enhancement in production of erythromycin by Saccharopolyspora erythraea by the use of suitable industrial seeding media. Daru 16(1):13–17
Saratram G, Maranas CD (2015) 13C metabolic flux analysis at a genome-scale. Metab Eng 32:12–22
Schellenberger J, Que R, Fleming RMT, Thiele I, Orth JD, Feist AM, Rahmanian S (2011) Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc 6(9):1290
Stefanovic E, Fitzgerald G, Mcauliffe O (2017) Advances in the genomics and metabolomics of dairy lactobacilli: a review. Food Microbiol 61:33–49
Thiele I, Palsson BØ (2010) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc 5(1):93–121
Tomàs-Gamisans M, Ferrer P, Albiol J (2016) Integration and validation of the genome-scale metabolic models of Pichia pastoris: a comprehensive update of protein glycosylation pathways, lipid and energy metabolism. PloS ONE 11(1):e0148031
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Weber JM, Wierman CK, Hutchinson CR (1985) Genetic analysis of erythromycin production in Streptomyces erythreus. J Bacteriol 164(1):425–433
Weber JM, Cernota WH, Gonzalez MC, Leach BI, Reeves AR, Wesley RK (2012) An erythromycin process improvement using the diethyl methylmalonate responsive (Dmr) phenotype of the Saccharopolyspora erythraea mutB strain. Appl Microbiol Biotechnol 93(4):1575–1583
Wentzel A, Sletta H, Consortium S, Ellingsen TE, Bruheim P (2012) Intracellular metabolite pool changes in response to nutrient depletion induced metabolic switching in Streptomyces coelicolor. Metabolites 2(1):178–194
Young JD (2014) INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30(9):1333–1335
Zou X, Hang HF, Chu J, Zhuang YP, Zhang SL (2009) Enhancement of erythromycin A production with feeding available nitrogen sources in erythromycin biosynthesis phase. Bioresour Technol 100(13):3358–3365
Authors’ contributions
ZZ carried out the model reconstruction and validation. JC and MH participated in the design and coordination of the study. AM helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was financially supported by a grant from the Major State Basic Research, National Natural Science Foundation of China (No. 201276081), and partially supported by NWOMoST Joint Program (2013DFG32630).
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the results of this work are included within the article and its additional files.
Consent for publication
All authors have read and approved the manuscript before submitting it to bioresources and bioprocessing.
Ethics approval and consent to participate
This article did not contain any studies with human participants or animals performed by any of the authors.
Funding
The Major State Basic Research Development Program of China (973 Program, No. 2012CB721006), National Natural Science Foundation of China (No. 21276081), the National Scientific and Technological Major Special Project (Significant Creation of New drugs, No. 2011ZX09203-001-03), and NWOMoST Joint Program (2013DFG32630) provided funding support.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1.
Information of databases used in this study.
Additional file 2.
Biomass composition of S. erythraea NRRL23338.
Additional file 3.
iZZ1342 reactions associated with gene annotations. Excel file with the list of S. erythraea iZZ1342 model reactions and other information about the reactions.
Additional file 4.
iZZ1342 in SBML format.
Additional file 5.
Metabolites in iZZ1342. Excel file with the total list of metabolites in iZZ1342 and other detailed information about the metabolites.
Additional file 6.
The expressed genes from transcriptomics analysis result of S. erythraea NRRL23338.
Additional file 7.
The central carbon metabolic fluxes simulated with iZZ1342 and from 13C labeled experiments. Flux values (in mmol/gDCW/h) for reactions that produce in S. erythraea NRRL23338 metabolism which are simulated by GEM iZZ1342 and from experiments. Reactions names can be traced in reaction list in Additional file 3.
Additional file 8.
Essential genes and partially essential genes predicted by iZZ1342. Excel file with the list of Essential genes and partially essential genes predicted by iZZ1342.
Additional file 9.
The detailed information of the 89 partially essential genes (except for the four targets which are list in the text).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhuang, Z., Huang, M. & Chu, J. In silico reconstruction and experimental validation of Saccharopolyspora erythraea genome-scale metabolic model iZZ1342 that accounts for 1685 ORFs. Bioresour. Bioprocess. 5, 26 (2018). https://doi.org/10.1186/s40643-018-0212-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-018-0212-x