
Abstract
Background
Edwardsiella tarda, the etiologic agent of edwardsiellosis, is a devastating fish pathogen prevailing in worldwide aquaculture industries and accounting for severe economic losses. There is a raising concern about E. tarda being a significant zoonotic pathogen, and it is urgent to develop a rapid detection of this pathogen. This is the first study to develop a test strip for rapid detection of E. tarda in turbot.
Results
Mouse monoclonal antibodies (MAbs) and rabbit polyclonal antibody (PAb) against E. tarda were generated from immunization of mice and rabbits with a virulent isolate of E. tarda EIB202. Two MAbs specific to isolates of E. tarda were obtained, and one of them (25C1) was selected to conjugate with colloidal gold as the detector antibody. Rabbit PAb was used as the capture antibody. It was found the strip had no cross-reactivity with non-E. tarda bacterial microbes and the limit of detection (LOD) was 1 × 105 colony-forming units (CFU)/ml. The detection could be visually observed by the naked eye within 5 min. This test strip was verified with a similar detection limit and much less analysis time compared with a dot blot immunoassay (1 × 105 CFU/ml for LOD and 120 min for reaction time). When the samples were mixed with turbot tissue homogenates, strong immunoreactivity was observed over 105 CFU/ml, which suggested that the turbot tissue homogenates did not affect the detection of the strip. Pre-enrichment with homogenized turbot tissue for 12 h could increase the detection limit of the E. tarda present in the sample up to 1 to 10 CFU/ml. In practice, in detecting 20 turbot ascite samples infected by E. tarda, the immunochromatographic test strip showed a high accuracy (100% positive).
Conclusions
The immunochromatographic test strip offers great promise for a rapid, simple, and economical method of E. tarda on-site detection, and with different antibodies, it might be used to detect other aquatic pathogens.
Similar content being viewed by others
Background
Edwardsiella tarda, the etiologic agent of edwardsiellosis, is a devastating fish pathogen prevailing in worldwide aquaculture industries and accounting for severe economic losses. E. tarda can cause systemic hemorrhagic septicemia and emphysematous putrefactive disease in more than 20 species of freshwater and marine fishes, including turbot, tilapia, eel, flounder, and catfish. There is a raising concern about E. tarda being a significant fish pathogen, and it is urgent to develop a rapid detection of this pathogen. The traditional diagnostic test available for identification of E. tarda in diseased fish is bacterial culture and identification using biochemical tests [1]. During the past decade, a number of immunoassay methods, including enzyme-linked immunosorbent assay (ELISA) and dot blot immunoassay, have been used to detect E. tarda in infected organs like the liver, spleen, and kidney of the diseased fish [2,3]. Polymerase chain reaction (PCR) and modified PCR methods such as loop-mediated isothermal amplification (LAMP) were also used in the diagnoses of E. tarda [1,4,5], and the LAMP method was proved to be successful for E. tarda detection with a limit of detection (LOD) of 10 colony-forming units (CFU) [6]. Although the above methods are effective for E. tarda diagnosis, they are time-consuming (2 h to 7 days) and depend on expensive instruments and highly skilled personnel. Colloidal gold-based immunochromatographic assay (ICA) is a user-friendly format with few simple procedures and has been widely applied as a diagnostic tool especially for on-site identification of pathogens. The aim of our research was to develop a portable one-step method for the rapid detection of E. tarda. With a strip, the infecting pathogen of diseased fish could be determined on-site in several minutes without any instrument.
Methods
Strains, media, and chemicals
The strains used in this study are listed in Table 1. E. tarda strains were grown in tryptic soy broth (TSB) (Difco, Detroit, MI, USA) or tryptic soy agar (TSA) (Difco, Detroit, MI, USA) at 28°C, while Escherichia coli strains were cultured in Luria-Bertani (LB) broth at 37°C. Edwardsiella ictaluri, Aeromonas hydrophila, and Chromobacterium violaceum were cultured at 30°C for 20 h in brain-heart infusion (BHI) broth, LB medium with 1% NaCl, and LB medium with NaCl 2.5%, respectively. Then, the bacteria were collected by centrifugation at 10,000× g for 5 min at 4°C, followed by wash for three times with pre-chilled phosphate-buffered saline (PBS; 10 mmol/l, pH 7.4). The bacterial suspension in PBS was enumerated with a UV-visible spectrophotometer and inactivated with 1% (v/v) formalin for 1 h at 30°C. The formalin-killed bacteria were washed three times with pre-chilled PBS, resuspended, and stored at −80°C for further use.
The goat anti-mouse IgG was purchased from Tiangen Biotech (Beijing, China). Freund’s complete adjuvant and Freund’s incomplete adjuvant, HAuCl4·3H2O, sodium citrate, and bovine serum albumin (BSA) were from Sigma (St. Louis, MO, USA). Nitrocellulose membranes were purchased from Millipore (Billerica, MA, USA). PVC sheets, absorbent paper, and glass paper were purchased from Shanghai Goldbio Tech (Shanghai, China). Other reagents were of analytical purity, and Milli-Q (Millipore purification system)-purified water was used for all experiments.
Preparation of outer membrane proteins
The outer membrane proteins (OMPs) of E. tarda EIB202 were extracted as described by Darwish et al. [17] with minor modifications. Briefly, E. tarda EIB202, isolated from diseased turbot, was cultured in LB medium containing 1% glucose at 30°C for 20 h. The bacteria were collected by centrifugation at 10,000× g for 5 min at 4°C and then washed three times with pre-chilled PBS. The pelleted bacteria were suspended in 10 ml of HEPES buffer (10 mmol/l, pH 7.4) with 2 mmol/l phenylmethanesulfonyl fluoride and sonicated at 350 W for 120 times (4 s once with 6-s interval). Intact cells and large debris were removed by centrifugation at 10,000× g for 15 min at 4°C. Supernatants were pelleted and centrifuged at 100,000× g for 1 h at 4°C. The precipitate was resuspended with 5 ml HEPES buffer, and the same volume of HEPES buffer containing 2% sodium lauroyl sarcosine was added to solubilize inner membrane proteins. Thirty minutes later, the pellets were ultra-centrifuged at 100,000× g for 1 h at 4°C. The purified OMPs were suspended in 1 ml PBS and aliquoted in small portions and stored at −80°C.
Production of antibodies against E. tarda
Monoclonal antibody (MAb) specific to E. tarda was produced according to previously described methods [18]. E. tarda was cultured in TSB at 28°C for 16 h, and the bacteria were then collected and resuspended in sterile water for deoxycholate hydrogen sulfide lactose (DHL) agar plate counting. It was inactivated at 37°C for 48 h in 1% formaldehyde and then used as the immunogen after washing three times with PBS to remove the formaldehyde. Five BALB/c mice were immunized by intraperitoneal injection of the 200 μl suspension containing 108 inactivated bacteria per ml or OMPs mixed with an equal volume of Freund’s adjuvant (complete Freund’s adjuvant for the first immunization and incomplete for the second and third immunization) for three times at 2-week intervals. One week after the third immunization, serum antibody titers of immunized mice were determined by indirect ELISA on inactivated E. tarda or OMP-coated plates. Mice producing the highest ELISA titer were given an intravenous boost (OMP, 50 μg) without adjuvant. One week after the intravenous boost, two BALB/c mice were sacrificed for the fusion with SP2/0-Ag14 myeloma cells. The dispersed spleen lymphocytes were prepared and fused with myeloma cells (SP2/0-Ag-14) to produce hybridomas. For the screen of MAbs against the somatic antigen of E. tarda, indirect ELISA was processed using supernatants of hybridomas with formalin-killed whole cells of E. tarda coating on plates. Two of the strongly reacting MAbs (designated as 25C1 and 15H10) were used for further characterizations and applications in immunoassays. Positive hybridoma cells selected by indirect ELISA and cloned by limiting dilution were cultured and injected intraperitoneally into the BALB/c mice with 5 × 106 cells each mouse for ascite production. After 7 to 14 days, ascite was collected. MAb in ascite fluid was precipitated with 50% ammonium sulfate, centrifuged, and dialyzed against PBS before being purified using a protein G agarose column.
New Zealand white rabbits were subcutaneously and intramuscularly injected with a total of 20 ml suspension containing 108 inactivated bacteria per ml mixed with an equal volume of Freund’s adjuvant (complete Freund’s adjuvant for the first immunization and incomplete for the second and third immunization) for four times at 3-week intervals, and then the rabbit antisera were collected by ear bleeding. The sensitivity and specificity of the antisera were determined by dot blotting and Western blotting. High-titer and highly specific antisera were pooled and purified using a protein A agarose column.
Reactivity and characterization of MAbs
Indirect ELISA tests were performed to assess the reactivity of MAbs to the formalin whole cells of E. tarda isolates. ELISA plates were coated with 50 μl per well of formalin-killed whole bacterial cells (108 CFU/ml) in carbonate-bicarbonate buffer (pH 9.6) and incubated overnight at 4°C. All tests were performed in duplicate. The indirect ELISA tests were performed as Kumar et al. described [18]. The isotyping of MAbs was carried out with a mouse monoclonal antibody isotyping kit (Sigma, St. Louis, MO, USA). Indirect ELISA was also used to test the sensitivity of the MAbs to detect formalin-killed whole cells of E. tarda. Briefly, ELISA plates were coated and dried overnight with 50 μl of tenfold diluted solutions of whole bacterial cells (108 to 10 CFU/ml). The subsequent steps were similar as mentioned above.
Preparation of test strip
The MAb specific to E. tarda was conjugated to colloidal gold particles (diameter = 40 nm) as the method described by Frens [19] and dispersed onto a glass fiber membrane as the conjugate pad. The rabbit polyclonal antibody (2 mg/ml) and goat anti-mouse IgG (1 mg/ml) were micro-sprayed with BioDot XYZ3050 (BioDot, Irvine, CA, USA) onto a nitrocellulose membrane separately at the position that would become the test line (T) at the upper position and control line (C) around the bottom of the membrane as the analytical membrane. A glass fiber membrane and absorbent paper were used as the sample pad and absorbent paper, respectively. The sample pad, conjugate pad, analytical membrane, and absorbent pad were assembled with 2 mm overlapping between each component on a plastic adhesive backing. The assembled master card was then cut into strips of 5 mm wide with a numerical controlled slitter (Shanghai Goldbio Tech, Shanghai, China) and stored in a desiccated plastic bag.
Specificity and sensitivity of test strip
In order to determine the cross-reactivity of the test strip, five strains of E. tarda, two strains of E. ictaluri, and ten other bacterial strains (Table 1) were diluted in the same volume of test to a concentration of 106 CFU/ml with PBS and applied to the test strips.
The sensitivity of the strip was determined qualitatively. The serially diluted E. tarda of several concentrations (1 × 106, 5 × 105, 1 × 105, and 5 × 104 CFU/ml) in sample buffer was used in sensitivity detection. The strip sensitivity results were compared with the results from dot blotting. All of the detections were performed in triplicate.
Dot blotting
The bacterial suspension used to determine the specificity of the test strips and E. tarda EIB202 of serial dilutions were spotted onto a nitrocellulose membrane (2 μl/spot), dried at 37°C, and quenched in 5% Blotto (5% nonfat dry milk, 0.05% Tween-20 in PBS) for 2 h. After being incubated with MAb at 37°C for 1 h, the membrane was extensively washed three times with PBST (0.05% Tween-20 in PBS) and incubated in goat anti-mouse IgG antibody horseradish peroxidase conjugate (GAM-HRP) at 37°C for 1 h. Then, the membrane was washed three times and incubated in TMB membrane substrate for 5 min. After revealing dark gray spots, the reaction was terminated by incubation of the membrane in water.
Detection of E. tarda in artificially contaminated fish tissue
Spleen tissues from fresh turbot were dissected and homogenized in TSB supplemented with 2% NaCl at a ratio of 1:9 (w/v). The turbot homogenate (10 ml) was spiked with E. tarda EIB202 to various original concentrations from 1 to 107 CFU/ml by tenfold serial dilutions. One milliliter from each tube was collected at 0, 6, 12, and 24 h and immediately heat killed at 60°C for 30 min and stored at −20°C before testing with the test strip. For the strip test, 400 μl from each sample was mixed with PBS at a ratio of 1:1, and 100 μl of the mixture was applied to the test strip.
On-site detection with test strip
Twenty ascite samples of diseased turbot from an aquaculture farm (Shandong Province, China) was prepared and applied directly to the test strip. Simultaneously, 10 μl ascite sample was spread on a DHL plate. The colonies with a black center (indicating H2S production) were counted and identified to be E. tarda with the commercial API 20E kit (bioMérieux, Marcy-l’Étoile, France) and 16S rRNA sequencing.
Results and discussion
Preparation and characteristics of antibodies against E. tarda
In order to obtain MAb, BALB/c mice were immunized with the formalin-killed bacteria and purified OMPs of E. tarda, respectively. Compared with the antibody titer of anti-serum against OMP, the antibody titer of anti-serum against the formalin-killed bacteria was lower even after the fifth immunization and not suitable for hybridization. It might be attributed to the characteristics of E. tarda as an intracellular pathogen. MAbs against the OMP of E. tarda have been reported in detection of E. tarda from different geographic locations [18]. Therefore, OMP was used as the immunogen in this study. Out of the 1,536 wells of culture supernatants from two fusions, two hybridomas generated a strongly positive signal in indirect ELISA. Stable hybridoma cell lines 25C1 and 15H10 were obtained following three rounds of limiting dilution and the cloning procedure.
The isotyping results classified the two anti-E. tarda MAbs as subclass IgG2α. The reactivity of MAbs with different strains of E. tarda and with other bacteria was investigated using indirect ELISA. The two MAbs, 25C1 and 15H10, reacted strongly with formalin-killed whole bacterial cells from all isolates of E. tarda but not with the whole bacterial cells of E. coli, E. ictaluri, Vibrio anguillarum, Vibrio parahaemolyticus, Vibrio alginolyticus, Vibrio harveyi, Chromobacterium violaceum, and Aeromonas hydrophila (Figure 1A). The results showed that MAb 15H10 reacted with E. ictaluri more than MAb 25C1 (Figure 1A). The sensitivity of MAb 25C1 to detect whole cells of E. tarda was up to a level of 1 × 104 CFU/ml (Figure 1B). According to the results above, the cell line 25C1 produced an IgG2α subtype of MAb with high specificity to E. tarda which was selected to prepare MAb used in subsequent experiments. Rabbit anti-E. tarda EIB202 anti-sera were purified through a protein A column which demonstrated specificity to all of the tested E. tarda isolates and could be used as the capture antibody in test strip production.

Reactivity and sensitivity of MAbs to E. tarda and test bacterial strains in indirect ELISA. (A) A fixed quantity of formalin-killed whole cells of E. tarda (108 CFU/ml) were coated to the plate, and 1:200 dilution of 25C1 and 15H10 MAbs was used as the primary antibody. (B) The ELISA plate was coated with tenfold diluted solutions (108 to 10 CFU/ml) of formalin-killed whole cells of E. tarda EIB202, and 1:200 dilution of 25C1 MAb was used as the primary antibody.
Preparation of test strip
The test strip was assembled in a sandwich format. MAb 25C1 was used for conjugation with colloidal gold and served as the detection antibody. The rabbit anti-E. tarda polyclonal antibody (PAb) was used as the capture antibody. Transmission electron microscope (TEM) images revealed that the optimal diameter of colloidal gold particles ranged from 36 to 42 nm, with a mean diameter of 40 nm, to produce a distinguishable red color. The optimal concentrations of antibodies used to form the positive line and test line were determined by signal visualization. A clear bright-red display emerged with 6.0 μg/ml of gold-labeled MAb 25C1 and 1.0 μg/ml of rabbit anti-E. tarda PAb (data not shown).
Specificity and sensitivity of test strip
According to the recently described method, the detections of all the E. tarda strains used in this study produced positive results when 100 μl bacterial suspension at a concentration of 1 × 106 CFU/ml was added on the strip. There were not any cross-reactions of the strip with E. ictaluri, V. anguillarum, E. coli, C. violaceum, A. hydrophila, V. parahaemolyticus, V. alginolyticus, and V. harveyi (Figure 2). The test strip showed a high specificity to E. tarda and supposed to detect different isolates of E. tarda.

Sensitivity evaluation of the test strip prepared with monoclonal antibody 25C1 against E. tarda The standard bacterial suspension of E. tarda EIB202 was enumerated and serially diluted to 1 × 109, 1 × 108, 1 × 107, 1 × 106, 5 × 106, 5 × 105, 1 × 105, and 5 × 104 CFU/ml and directly applied to the test strips. The detection limit of the test strip was determined by the diminishing of red color at the test line.
To determine the sensitivity of the ICA test strip, different concentrations of bacterial suspension of E. tarda EIB202 were applied to the ICA strip and tested in triplicate. The sensitivity of the ICA test strip showed a positive correlation with bacterial concentration when the bacterial concentration was between 5 × 104 and 1×109 CFU/ml. The color intensity of the test line increased with E. tarda concentration. When the suspension with a concentration of 1 × 105 CFU/ml was added on the strip, the intensity of the test line was significantly weaker (within 5 min) than that of the control line (Figure 3), and the test line at 5 × 104 CFU/ml bacteria was visible just at about 60 min. Therefore, 1 × 105 CFU/ml of E. tarda could be considered as the low limit of the ICA test strip for visual detection (within 5 min).

Specificity evaluation of the test strip prepared with monoclonal antibody 25C1 against E. tarda. Bacteria were diluted to 1 × 106 CFU/ml with PBS and applied to the sample pad of the test strip.
Comparison to dot blot hybridization
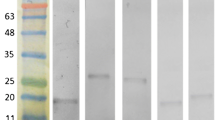
As an easy technique, dot blot hybridization is currently used for pathogen detection. However, the dot blotting procedure is laboratory-dependent and time-consuming which needs long result turn-around time (approximately 6 h) [20,21]. In this study, the dot blotting using MAb 25C1 described earlier as a binding antibody was performed to determine the specificity and sensitivity of this method. The results showed that Mab 25C1 could recognize all isolates of E. tarda used in the study and had no cross-reaction with all the bacterial strains except for E. tarda, which displayed the perfect specificity of MAb 25C1. The LOD of dot blotting was 1 × 105 CFU/ml, which is similar to that of the ICA method (Figure 4). Compared with that of the dot blotting, the detection process of the ICA method was simple and time-saving (5 min vs 6 h). Although the dot blotting produced a similar LOD to that of the ICA strip, it was reported that the limited capacity of the nitrocellulose membrane might reduce the detection sensitivity of this method because the high protein in the sample could compete with the bacterial antigen [22]. The test strip offered similar detection sensitivity to that of the dot blotting and would be a convenient tool for direct identification of fish pathogen E. tarda.

Dot blotting detection with monoclonal antibody 25C1 against E. tarda. Bacterial strains used for the cross-reactivity test at a concentration of 1 × 106 CFU/ml and E. tarda EIB202 of serial dilutions were spotted onto the nitrocellulose membranes (2 μl/spot) and processed dot blotting using MAb 25C1. Row 1: E. tarda EIB202 (a) 1 × 106 CFU/ml, (b) 5 × 105 CFU/ml, (c) 1 × 105 CFU/ml, (d) 5 × 104 CFU/ml, (e) 1 × 104 CFU/ml, (f) 5 × 103 CFU/ml, (g) 1 × 103 CFU/ml. Row 2: (a) E. tarda EIB202, (b) E. tarda WY37, (c) E. tarda ATCC15947, (d) E. tarda ATCC23692, (e) E. tarda DT, (f) E. ictaluri LH-51, (g) E. ictaluri 33202. Row 3: (a) V. anguillarum MVM425, (b) V. anguillarum VIB72, (c) V. anguillarum AV-1, (d) E. coli CC118, (e) E. coli DH5α, (f) C. violaceum CV026, (g) A. hydrophila. Row 4: (a) V. parahaemolyticus, (b) V. alginolyticus EPGS, (c) V. harveyi VIB647, (d) blank.
Detection of E. tarda in artificially contaminated fish tissue
The application of test strips to detect the targeted microorganisms in the fish tissue samples was evaluated. When mixed with homogenized turbot tissue, a strong immunoreactivity was observed in the samples inoculated with E. tarda EIB202 over 105 CFU/ml, which is similar to that of the sample in TSB. The results suggested that the existence of turbot tissue homogenates did not affect the detection of the strip. After pre-enrichment with the tissue of turbot for 6 h, a moderate immunoreactivity was observed in the turbot homogenate samples inoculated with E. tarda over 103 CFU/ml (Figure 5A). After pre-enrichment for 12 and 24 h, a moderate positive observation could be detected at the inoculate of 10′ CFU/ml in TSB. Therefore, pre-enrichment of the sample for 12 h could increase the detection limit of the E. tarda present in the sample up to 1 to 10 CFU/ml.

Strip test of E. tarda EIB202 with turbot tissue homogenates and on-site detection of turbot ascite. (A) E. tarda EIB202 at various concentrations (1 to 107 CFU/ml) were pre-enriched with turbot tissue homogenates for 0 to 24 h before being applied to the strip. PBS was used as the blank sample. (B) Ascites of diseased turbot collected from an aquaculture farm (Shandong Province, China) numbered 1 to 20 were prepared and applied directly to the test strip. The concentrations of E. tarda in ascites ranged from 5.1 × 105 to 7.8×107 CFU/ml by DHL agar counting. PBS was used as the blank sample.
On-site detection of E. tarda infection with test strip
E. tarda is an important fish pathogen and has caused huge losses to the aquaculture industry throughout the world. Outbreaks of edwardsiellosis appear first in the form of infections affecting only a few fish, and these diseased fish are able to survive only for a few days. When early detection and proper measurement are not undertaken and the diseased fish not treated as soon as possible, the infection spreads to the surrounding areas to create an edwardsiellosis epidemic [6]. Since early detection is critical for controlling this disease, rapid and accurate methods for edwardsiellosis diagnosis would be very valuable. To evaluate the method developed in this work, the immunochromatographic test strip was practically utilized to detect E. tarda in an industrial turbot farm (Yantai, Shandong Province, China). Diseased turbot with typical symptoms of edwardsiellosis such as eye tumefaction, inflammation, hemorrhages, and ascites were sampled. Each of 20 ascite samples of diseased turbot was collected and applied directly to the test strip for E. tarda on-site detection, and PBS was used as blank control due to the absence of ascite in healthy turbot. Among the 20 ascite samples, 20 samples (100%) were positively detected, and the test lines were clearly indicated (Figure 5B). The concentrations of E. tarda in ascites ranged from 5.1 × 105 to 7.8 × 107 CFU/ml by DHL agar counting. The major colonies in the 20 ascite samples were further measured using API 20E and 16S rDNA sequencing to confirm that the diseased turbot were infected with E. tarda rather than other possible pathogens. Fortunately, the complete genome of E. tarda is available [23]. The results of 16S rRNA sequencing displayed that all the colonies isolated from the ascite samples belong to the same E. tarda strain with 99% identity to E. tarda FL6-60 [GenBank:CP002154.1] and EIB202 [GenBank:CP001135.1]. It was suggested that the diseased turbot from the aquaculture farm might be infected by the same E. tarda strain. In the conventional bacterial culture test, the procedure for isolation and identification of E. tarda from diseased fish samples usually requires 2 to 3 days, including bacterial enrichment, plating onto DHL agars, sequential subculture, and biochemical characteristic test, which is time-consuming and tedious. Therefore, the test strip is effective for the on-site detection of E. tarda in ascite samples.
Conclusions
The results in this work indicated that the detection limit, simplicity, and specificity of the test strips are sufficient for on-site use. In conclusion, the one-step immunodiagnostic test presented here could prove to be a useful tool for the inexpensive, rapid, and convenient on-site detection of E. tarda in clinical samples.
References
Baird KD, Chikarmane HM, Smolowitz R, Uhlinger KR (2003) Detection of Edwardsiella infections in Opsanus tau by polymerase chain reaction. Biol Bull 205:235–236
Swain P, Mukherjee SC, Sahoo PK, Das BK, Pattnaik P, Murjani G, Nayak SK (1999) Dot enzyme linked immunosorbent assay (Dot-ELISA) for diagnosis of Edwardsiella tarda infection in fish. Asian Fish Science 14:89–93
Bai FF, Lan JX, Wang Y, Han Y, Zhang XH (2009) Indirect enzyme-linked immunosorbent assay (ELISA) for rapid detection of Edwardsiella tarda. J Fishery Sci of China 16:619–625
Chen JD, Lai SY (1998) PCR for direct detection of Edwardsiella tarda from infected fish and environmental water by application of the hemolysin gene. Zool Stud 37:169–176
Lan J, Zhang XH, Wang Y, Chen J, Han Y (2008) Isolation of an unusual strain of Edwardsiella tarda from turbot and establish a PCR detection technique with the gyrB gene. J Appl Microbiol 105:644–651
Savan R, Igarashi A, Matsuoka S (2004) Sensitive and rapid detection of edwardsiellosis in fish by a loop-mediated isothermal amplification method. Appl Environ Microbiol 70:621–624
Xiao J, Wang Q, Liu Q, Wang X, Liu H, Zhang Y (2009) Isolation and identification of fish pathogen Edwardsiella tarda from mariculture in China. Aquac Res 40:13–17
Wu H, Ma Y, Zhang Y, Zhang H (2004) Complete sequence of virulence plasmid pEIB1 from the marine fish pathogen Vibrio anguillarum strain MVM425 and location of its replication region. J Appl Microbiol 97:1021–1028
Wang Q, Yang M, Xiao J, Wu H, Wang X, Lv Y, Xu L, Zheng H, Wang S, Zhao G, Liu Q, Zhang Y (2009) Genome sequence of the versatile fish pathogen Edwardsiella tarda provides insights into its adaptation to broad host ranges and intracellular niches. PLoS One 4, e7646
Ye J, Ma Y, Liu Q, Zhao D, Wang Q, Zhang Y (2008) Regulation of Vibrio alginolyticus virulence by the LuxS quorum-sensing system. Journal of Fish Diseases 31:161–169
Rui H, Liu Q, Wang Q, Ma Y, Liu H, Shi C, Zhang Y (2009) Role of alkaline serine protease, asp, in Vibrio alginolyticus virulence and regulation of its expression by luxO-luxR regulatory system. J Microbiol Biotechnol 19:431–438
Pang L, Zhang X, Zhong Y, Chen J, Li Y, Austin B (2006) Identification of Vibrio harveyi using PCR amplification of the toxR gene. Lett Appl Microbiol 43:249–255
Ravn L, Christensen A, Molin S, Givskov M, Gram L (2001) Methods for detecting acylated homoserine lactones produced by gram-negative bacteria and their application in studies of AHL-production kinetics. J Microbiol Methods 44:239–251
Wang Y, Xu Z, Jia A, Chen J, Mo Z, Zhang X (2009) Genetic diversity between two Vibrio anguillarum strains exhibiting different virulence by suppression subtractive hybridization. Acta Microbiologica Sinica 49:363–371
Dennis J, Zylstra G (1998) Plasposons: modular self-cloning minitransposon derivatives for rapid genetic analysis of gram-negative bacterial genomes. Appl Environ Microbiol 64:2710–2715
Winzer K, Sun Y, Green A, Delory M, Blackley D, Hardie K, Baldwin T, Tang C (2002) Role of Neisseria meningitidis luxS in cell-to-cell signaling and bacteremic infection. Infection and Immunology 70:2245–2248
Darwish AM, Newton JC, Plumb JA (2001) Effect of incubation temperature and salinity on expression of the outer membrane protein profile of Edwardsiella tarda. J of Aquaculture Animal Health 13:269–75
Kumar G, Rathore G, Sengupta U, Kapoor D, Lakra WS (2010) Production of monoclonal antibodies specific to major outer membrane protein of Edwardsiella tarda. Comp Immunol Microbiol Infect Dis 33:133–144
Frens G (1973) Controlled nucleation for the regulation of the particle size in monodisperse gold suspension. Nature, Physics Science 241:20–21
Chaivisuthangkura P, Senapin S, Wangman P, Longyant S, Sithigorngul P (2013) Simple and rapid detection of infectious myonecrosis virus using an immunochromatographic strip test. Arch Virol 158:1925–1930
Giagkas DC, Choli-Papadopoulou T, Pantazaki AA (2013) Development of an antibody for detection of rhamnolipids characterized as a major bacterial virulence factor. Antibodies 2:501–516
Parin C, Chalinan P, Siwaporn L, Paisarn S (2013) Evaluation of monoclonal antibody based immunochromatographic strip test for direct detection of Vibrio cholerae O1 contamination in seafood samples. J Microbiol Methods 2:304–311
Wang X, Wang Q, Xiao J, Liu Q, Wu H, Xu L, Zhang Y (2009) Edwardsiella tarda T6SS component evpP is regulated by esrB and iron, and plays essential roles in the invasion of fish. Fish and Shellfish Immunology 27:469–477
Acknowledgements
This work was supported by grants from the Ministry of Agriculture of China (Nos. CARS-50 and nyhyzx-201303047), Shanghai Rising-Star Program (No. 13QA1401000), and the Project of Marine Public Welfare Profession (No. 201405003-2).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HL carried out the preparation of the monoclonal antibody targeting E. tarda and development of the test strip. YW carried out the on-site detection of the test strip and helped in the manuscript preparation. JX, QW, QL, and YZ have made substantive intellectual contributions to this study and substantial contributions to the design of the study. All authors read and approved the final manuscript and helped design the whole study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, H., Wang, Y., Xiao, J. et al. An immunochromatographic test strip for rapid detection of fish pathogen Edwardsiella tarda . Bioresour. Bioprocess. 2, 20 (2015). https://doi.org/10.1186/s40643-015-0047-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-015-0047-7