
Abstract
Avanafil (AV) is the phosphodiesterase (PDE) type 5 inhibitor drug used in erectile dysfunction, having pyrrolidine, pyrimidine, carboxamide, and chlorine as functional groups which can easily break by environmental changes and cause toxicity. Henceforth, in detail, HPLC stability study with the Quality-by-Design (QbD) approach is presented which leads to recommended storage conditions. The stability of AV was analyzed in hydrolysis, photolysis, and thermal and oxidative conditions. The application of the QbD approach during the stability method development comprises steps as screening and optimization. Quality target product profile (QTPP) was defined, and critical quality attributes (CQAs) were assigned to meet the QTPP requirements. Primary parameters obtained from the Ishikawa diagram were studied via Placket–Burman, and four critical factors were optimized through the central composite design (CCD). The finalized method includes mobile phase [10 mM ammonium acetate, pH 4.5 adjusted by acetic acid:ACN (60:40, v/v)] at 0.9-mL/min flow rate and 239-nm wavelength. A control strategy was set up to ensure that the method repeatedly meets the acceptance criteria. Overall, 16 degradation product peaks of AV in all conditions (solid and solution state) were identified with optimized method and evaluated by HPLC-PDA study. A comprehensive systemic optimization of AV stability study is stated for the first time, which reveals that AV is prone to degrade in sunlight, moisture, and temperature. Global regulators and manufacturers should take care of the packaging, handling, and labeling of AV. A fully validated LC–MS compatible stability method can be successfully applied to monitor AV stability from its formulation which can be wisely extrapolated to assess the AV from biological samples.

Graphical abstract
Similar content being viewed by others


Introduction
Stability testing is an important step in the drug approval process to assess a drug substance or drug product quality, which varies with time under the influence of environmental factors such as temperature, humidity, and light. An in-depth stability study for any new moiety is required for packaging, handling, retest period, and storage.
The common practice in the past was to develop a stability-indicating method using a trial and error approach called as one factor at a time (OFAT). The trials were taken to get resolved peaks until the best method was found. It was time-consuming and required a large number of experiments which often resulted in a non-robust performance (Maggio et al. 2013). Nowadays, in the application of experimental design, several experiments are carried out in which two or more variables are altered at the same time called the multiple factors at a time (MFAT) approach for method optimization (Patel and Kothari 2018). The stability-indicating method development using the Quality-by-Design (QbD) approach offers more accurate and robust results related to the OFAT approach, which comprises screening and optimization as steps (Schmidt and Molnár 2013; Tol et al. 2016, 2020; Karmarkar et al. 2011). Several reports focus on application of the QbD approach for analytical method development using design of experiment as one of the element (Hubert et al. 2014; Gavin and Olsen 2008). In addition, ICH has approved guideline Q14 with a detailed description of analytical method development using the QbD approach (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use n.d.-a). Hence, an attempt was made to develop a robust stability-indicating method with the QbD approach for Avanafil (AV).
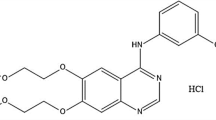
Chemically AV is [(s)-(4-[(3chloro-4-methoxybenzyl) amino]-2-[2-(hydroxymethyl)-1pyrrolidinyl]-N-(2 pyrimidinyl methyl)-5-pyrimidine carboxamide)] (Fig. 1). Pharmacologically, AV is a phosphodiesterase type 5 inhibitor developed by Mitsubishi Tanabe Pharma Corp. (Osaka, Japan) and used in the treatment of erectile dysfunction plus explored for the treatment in pulmonary hypertension. AV is soluble in acetonitrile (ACN), methanol, and dimethyl sulfoxide (DMSO), and is insoluble in water; it has a pKa value of 5.5.

Structure of Avanafil
A literature review of AV reveals that numerous reports were existing, like AV as a PDE 5 inhibitor (Ferguson and Carson 2013) and estimation by UV spectroscopy method (Savaliya et al. 2013). The simultaneous estimation of AV with dapoxetine was developed by a simple high-performance liquid chromatography (HPLC) method (Mangukiya et al. 2013) and UV-chemometrics, the HPLC–QbD approach (Patel and Kothari 2016). One report emphasized on AV biological estimation, tolerability, and pharmacokinetic study with liquid chromatography–mass spectrometry (LC–MS)/MS (Jung et al. 2010). The few reports focused on a stability method of AV. The report authored by Bhatt et al. (2015) developed a stability method which can identify merely the AV peak; hence, it does not fulfill the purpose of the stability-indicating analytical method (SIAM). The SIAM should detect all possible degradant and drug without interference. Another reported method authored by Kumar et al. (2017) discussed the stability study of AV for certain conditions using the gradient method. The method reveals the separation of drugs from the degradation product, but all degradation product peaks were not well separated. Recently published report focused on the identification of two related substances and one degradation product of AV by LC–TOF–MS (Can 2018). The reported method lacks in sensitivity and proper resolution as it identified only three degradation product peaks, and certain peaks were partially merged.
Till date, published data for AV estimation seems that reported methods have several constraint for application in terms of identification of all possible degradation products, poor resolution, more run time, use of high-cost organic solvent, gradient method, and LC–MS incompatibility. Hence, the goal of the present study was to develop novel and fully validated stability-indicating analytical methods (SIAM) for AV (in API and pharmaceutical tablet formulation) using the QbD approach. Application of the QbD approach will fulfill stability method standards as appropriate resolution between peaks, robustness, selectivity, specificity, and precision, which is beneficial for manufacturers during the drug approval process and for labeling condition. To the best of our knowledge, this is the first detailed report of AV stability method development using the QbD approach in different stress conditions.
Materials and methods
Chemicals
Analytically pure (99.9 %) Avanafil (AV) was received as a gratis sample from Sunrise Remedies Pvt, Ltd. (Ahmedabad) and Om Laboratory (Ahmedabad) along with a certificate of analysis. Marketed tablet formulation (Avana) was procured as a gratis sample from Sunrise Remedies Pvt, Ltd. (Ahmedabad). Acetonitrile, ammonium acetate, and glacial acetic acid used were of HPLC grade procured from Merck, Mumbai, India. Hydrochloride acid, sodium hydroxide, and hydrogen peroxide (30% w/v) were used for solvent preparation and stress degradation studies were of analytical grade procured from CDH chemicals, Delhi, India.
Equipment
Fourier-transform infrared spectrometer (FTIR) (JASCO FT/IR-6100 series, Jasco, Japan) was used for identification. Agilent 1260 Infinity Quaternary HPLC equipped with a UV detector was used for the chromatographic separation. Agilent ChemStation software was used for data acquisition and analysis. Peak purity calculation was done by Jasco HPLC (Jasco analytical instruments, USA), which was equipped with a PDA (Photodiode array) detector. Labindia pH meter (Pico+) was used to measure the pH of the buffer. The membrane filters 0.22 μm (Axiva Sichem Biotech, Delhi) and syringe filters 0.45 μm were used for filtration. The Stat-Ease Design Expert Version 9.0.2® software aids in the method development through QbD by designing experiments (DoE), analysing and modelling the data, and establishing a design space.
Solution preparation
Preparation of standard solution
An accurately weighed 10 mg of AV was diluted with aliquot (1 mL) of ACN, sonicated for 5 min, and volume was made up to 10 mL with respective stressor (1000 μg/mL).
Preparation of working solution
From the standard solution of AV (10 mg in 10 mL) in respective stressor, 1 mL was collected at a specific time interval (as mentioned in Table 4) and made the volume up to 10 mL with ACN:water (50:50, v/v) to get 100 μg/mL concentration of the working solution.
Different stressors were used for different degradation conditions like in acidic hydrolysis hydrochloric acid (HCl), in an alkaline hydrolysis sodium hydroxide (NaOH), in neutral hydrolysis water, in oxidative degradation hydrogen peroxide (H2O2), in thermal and photolytic degradation ACN;water (50:50, v/v) was used as a stressor. Hydrolytic degradation (acid, alkaline, and neutral) was studied at different temperatures of 60, 70, and 80 °C. Oxidative degradation was studied by 1% H2O2, 6% H2O2, and 10% H2O2. For thermal (dry heat) degradation, samples in solid state were kept in the oven at 60 °C and in solution form (ACN:water, 50:50, v/v) were kept in the oven at 60 °C. For photolytic degradation, samples in solid-state and in solution form (ACN:water, 50:50, v/v) were kept in sunlight (as mentioned in Table 4).
Preparation of sample solution
Twenty AV tablets (Avana) were weighed and ground in a clean motor. The amount equivalent to 10 mg of AV was weighed and transferred into a 10-mL volumetric flask. To this powder, about 3 mL of ACN was added, and the flask was placed on a mechanical shaker for 10 min. The volume was made up to 10 mL with respective stressor to get 1000 μg/mL concentration. From that standard solution, 1 mL was collected at a specific time interval and made the volume up to 10 mL with ACN:water (50:50, v/v) to get 100 μg/mL concentration of the working solution.
Implementation of systematic quality by design approach (Garg et al. 2015; Michael et al. 2015; Ren et al. 2016; Kurmi et al. 2014; Dharani et al. 2018; Prajapati and Agrawal 2015; Mohamed et al. 2016; Orlandini et al. 2016; Bhandi et al. 2016; Kalariya et al. 2015a; International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use n.d.-b, c)
The QbD is a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control. Systematic optimization of stability-indicating method includes three stages, viz., primary parameter selection, secondary parameter screening, and method optimization with consideration of degradant in forced degraded samples (Patel and Kothari 2018; Schmidt and Molnár 2013; Tol et al. 2016, 2020). AV (100 μg/mL) solution was exposed to different degradation conditions. During the initial trial, considerable degradation (< 20% degradation of AV to avoid secondary degradation) was found in acidic condition with respect to other degradation conditions. Hence, stability method was optimized using acid degraded solution (1N HCl at 60 °C for 9 h; 100 μg/mL). Resolution between drug peak and all degradation peaks were key criteria for developing stability method; hence, it was chosen as the response variable.
Screening for stability method
All quality target product profiles (QTPPs) and critical quality attributes (CQAs) for stability method were listed out. The selection of parameters was done using a quality risk management tool as per ICH Q9. From the Ishikawa diagram and risk assessment tool total, eleven primary parameters were selected and subjected to secondary parameter screening by Plackett–Burman design. Resolution between all peaks was considered as a response variable since it is a very significant criterion for selectivity and specificity of SIAM. Analytical runs were performed as per Plackett–Burman design. Further half-normal plots and Pareto charts were derived to determine significant factors.
Optimization for stability method (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use n.d.-b; Kalariya et al. 2015b, c)
The factor having a significant effect on the resolution of peaks was further identified as a critical method variable during secondary parameter selection. Central composite design (CCD) was applied for selected significant factors. A total of 30 experimental runs were performed and analyzed against the response variable (resolution between peaks). By the use of Design expert 9.0.2®, statistical calculations, models, polynomial equations, p value, F value, contour plot, 3D plot, and desirability plot were derived. At last, control strategy was decided, which shows a planned set of controls, derived from current product and process understanding that assure the process performance. An optimized chromatographic condition was used to check the degradation of the drug in all degradation conditions.
Application of the method to marketed formulation
The forced degradation condition and developed stability-indicating method was applied to the marketed tablet formulation. The assay was performed to check the selectivity of the developed method for the estimation of AV in tablet formulation (AVANA).
Method validation (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use n.d.-c)
The developed stability-indicating method was validated as per ICH guidelines Q2 (R1) (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use n.d.-c). The system suitability of the developed method was checked by evaluating percentage of relative standard deviation (% RSD) values of system suitability parameters like area, asymmetry, theoretical plate, and retention time of five standard replicates. Linearity was estimated by preparing serial dilutions in a concentration range between 10 and 70 μg/mL from the stock solution. The analysis was done, six times (n = 6) for each concentration. The precision (Interday and intraday) were assessed at three different concentrations; 10 μg/mL, 30 μg/mL, and 70 μg/mL in triplicates on different three times/days. Accuracy was performed at three different concentrations (80%, 100%, and 120% of the intended content of active ingredient) at three-time intervals, followed by the addition of a known amount of drug to the sample and % recovery was calculated for each concentration. Accuracy concentration levels were 54 μg/mL, 60 μg/mL, and 66 μg/mL for AV. Each concentration level solution was prepared and injected in triplicate. The percentage of recovery was calculated. LOD (limit of detection) and LOQ (limit of quantification) values were calculated with the standard deviation method. Serial dilutions of standard solutions were prepared and injected in descending order. Specificity study was used to determine the ability of the method to determine analyte in the presence of different other components (degradant, excipients, impurities, etc.).
Results and discussion
Quality by Design instability method
Identification of QTPP and CQAs
It is considered as the first step in QbD-based stability method development. Various QTPPs, CQAs, and process parameters for stability-indicating HPLC method were identified and listed as in Supplemental Table 1. The systematic QbD approach includes primary parameter screening, secondary screening, and optimization. Primary parameter screening was done by the Ishikawa diagram (Fig. 2) and risk assessment (Fig. 3). A systematic risk assessment is performed to identify critical method parameters and critical process parameters whose variability may affect potential CQAs. The relative risk due to each parameter was ranked as high, medium, or low. High risk means those quality attributes that could have a high impact on the stability method. Low risk means those quality attributes that had a low impact on the stability method. The high-risk factors were necessary to study for the development of the robust method.

Ishikawa diagram for a cause–effect relationship

Risk assessment for stability method of AV
From the Ishikawa diagram and risk assessment, a total of eleven primary parameters were selected which are called primary critical parameters for SIAM. Selected primary parameters were subjected to secondary screening with the application of the Plackett–Burman design. The selected response variable for the stability method is resolution between peaks. The Plackett–Burman design was applied for 11 selected factors at two levels (low, high) as shown in Table 1. The levels of factor (parameter) were decided based on preliminary trials and from the literature review. The mobile phase ratio was studied at two levels (50:50, v/v and 70:30, v/v). The organic phase was studied for two widely used organics ACN and MeOH for method development. The temperature of the column oven can affect the viscosity of the mobile phase; hence, it was studied at 10 °C and 30 °C. The degassing of the mobile phase during HPLC method development is required to reduce the interference of air bubbles. During initial trials, slight baseline drift was found due to air bubble entrapment; hence, degassing time was studied for 10 min and 25 min. Further, the effect of mobile phase pH on the method was studied at 3 and 5. Flow rate was studied at 0.5 mL/min and 1.2 mL/min, and injection sample volume was studied for 5 μL and 20 μL. The selection of column (C18 and C8) was based on column chemistry and availability. The different buffer ammonium acetate (AA) and ammonium format (AF) were selected for study with strength at 5-mM and 10-mM levels. The detection wavelength selection was based on wavelength maxima of AV and studied for 230 nm and 250 nm (Table 1).
The statistical calculation for Plackett–Burman design was done by a Pareto chart and half normal plot; from these, significant secondary parameters were decided as shown in Fig. 4. Factors which cross t limit were considered as significant variable for all responses [Resolution (1–2) (I), (2–3) (II), (3–4) (III), (4–5) (IV), (5–6) (V), and (6–7) (VI)]. The Pareto charts were derived, and it was observed that values for Organic phase type (B), Degase time (D), Sample volume (G), Column type (H), Buffer (I), Buffer strength (J), and Detection wavelength (K) were taken as constant for further analysis, and mobile phase ratio (A), the temperature of column oven (C), pH of buffer (E), and flow rate (F) were significant factors so they need to optimize. The selected four parameters (factors) were further used for the optimization of the method by design expert 9.0.2®.

Pareto and half-normal plots for stability method of AV
Method optimization by CCD
Response surface methodology can be used for optimization of four critical factors at more than three levels to reduce the number of experimental runs without loss of orthogonal nature of the model; hence, central composite design (CCD) was designed by Design expert 9.0.2® and studied for n = 30 experiments as shown in Table 2. The objective of designing these experiments was to achieve optimal separation between the critical pairs in the shorter possible run time. Levels of values for design and responses were found as below. The level of four independent factors was decided as discussed below. Initially, ACN with water in different ratios was tried, but the better separation was achieved by the addition of buffer. In chromatography, the buffer pH should be ± 2 of pKa as between that range 100% drug is in ionized or unionized form which gives better separation. Here, AV has pKa value 5.54, and for better separation, studied buffer pH range was between ± 2 of pKa. Subsequently, change in organic phase ratio affects retention time of degradation peaks. And the temperature affects elution time due to change in viscosity of mobile phase. All the listed parameters had individual and interaction effects during SIAM development. The SIAM focused on selectivity and specificity of analyte without interference, which can estimate by resolution; hence, resolutions between degradation peaks were considered as response variables (Table 2).
The four critical factors (mobile phase ratio, pH of the buffer, flow rate, and temperature of column) were studied at two levels (Low, High) using CCD. The decided levels for factors were mobile phase ratio, ACN:buffer [40:60, (v/v); 80:20, (v/v)], pH of buffer (2.5, 6.5), flow rate (0.25 mL/min, 1.25 mL/min), and temperature (0 °C, 40 °C). The levels for mobile phase ratio, flow rate, and column temperature were chosen from the preliminary trials. The level for buffer pH was studied according to the AV pKa value. The preliminary study emphasized on the selection of acidic degradation condition due to the required % degradation of drug and presence of more merged degradation peaks with respect to other degradation conditions. Hence, the scrutinized dependent variables were the resolution between the merged peaks, where Rs (1–2) [resolution between DP I and DP II], Rs (2–3), Rs (3–4), Rs (4–5), Rs (5–6), and Rs (6–7) were chosen.
The mathematical model was generated for each response, and statistical calculations were done for all factors from ANOVA results (Supplemental Table 2). The statistical values as p, R2, and adjusted R2 were studied, and the polynomial equation was derived for each factor. Calculated R2 for all CQAs were ranged between 0.752 and 0.999, indicating that a high proposition of variation in the responses could be attributed to the models. The mathematical model (quartic and quadratic) summary data indicated a reasonably good agreement between the adjusted R2 and R2 values for all response variables. The magnitude of the coefficients in the polynomial equation and the p value indicate that the studied four factors have a significant effect. The polynomial equation expresses the response variables in terms of the addition and subtraction of factors.
The response surface plots (contour plot and 3D plots) were generated for resolution between each peak to study individual effect and interaction effects (Supplemental Figure 1). As a brief of response surface plots, there are increases in the pH of mobile phase (A: 3.5 to 5.5) and mobile phase composition (B: 50:50 to 70:30, v/v) and resolution increases of up to optimum level (A: 4.5, B: 60:40) than resolution decreases. To achieve the desirability (di), the response criteria were established as “maximum” for the resolutions. The Derringer’s desirability was calculated from contour and 3D plots as shown in Supplemental Figure 1. Design expert software suggests three predictions (global solutions) from the data of desirability and surface graphs. The high desirability value indicates maximum value of response. All three software-suggested predictions were experimentally checked. The optimum conditions were derived as mobile phase composition, buffer:ACN (60:40, v/v), pH 4.5, the flow rate 0.90 mL/min, and column oven temperature 20 °C, and desirability 0.956. The same desirability results were achieved by contour and 3D plots (Supplemental Figure 2). The overlay of all experimental runs gives the working design space as shown in Fig. 5. To locate the optimum analytical conditions in the design space, numerical optimization was carried out by applying constraints on the variables. This region considered as the most robust and operable region to work to develop the SIAM for AV. From all statistical calculations and experimental work, a risk assessment table was updated. Updated risk assessment shows the application of a systemic approach, factors with high severity converted into less severity (Supplemental Figure 3). The software-suggested method and predictions for resolution were checked experimentally; the found resolution was greater than 2 for all studied response variables [Rs (1–2), Rs (2–3), Rs (3–4), Rs (4–5), Rs (5–6), and Rs (6–7)].

Overlay plot for stability method of AV
Control strategy
The control strategy was decided as a final step in QbD, as shown in Table 3. It summarizes the proven acceptable ranges for each analysis by stability and critical process parameters for AV.
The optimized condition was derived as nucleosil, C18 column (250 × 4.6 mm, 5 μm) and mobile phase composed of 10 mM ammonium acetate buffer:ACN (60:40, v/v), pH 4.5, a flow rate of 0.90 mL/min, and column oven temperature of 20 °C. The optimized chromatographic condition with quality by approach was used to check the degradation of the drug in all degradation conditions. Overlay chromatograms of AV in acid hydrolysis, alkaline hydrolysis, peroxide degradation, photolytic degradation, and thermal degradation are shown in Fig. 6. In different degradation conditions, various DPs were found. Nomenclature of formed degradation products in different condition were given based on their Rt (retention time). The optimized condition resolves the AV from sixteen degradation products in different degradation conditions with required system suitability parameters as resolution (> 2), theoretical plates (> 2000), and tailing factor (≤ 1.5), which is crucial for the chemical stability method.

Overlay chromatogram of AV (100 μg/mL) in (a) acid hydrolysis, alkaline hydrolysis and peroxide and (b) photolytic and thermal degradation in the optimized chromatographic condition
The optimized method was applied for all degradation conditions and found DPs were in the acid condition DPs I, III, IV, VIII, XI, XIII, and XIV; in alkaline condition DPs I, III, IV, VIII, XI, XIII, XIV, and XV; in neutral degradation of the drug formation of DPs I, III, IV, VIII, XIII, and XIV; in oxidative degradation of the drug formation of DPs VI, XII, XIII, and XIV; in photolytic degradation DPs II, V, VII, X, XIII, XIV, and XVI; and in thermal degradation DPs II, V, IX, XIII, and XIV. Found common DPs for acid and alkaline hydrolysis were I, III, IV, VIII, XI, XIII, and XIV as shown in Fig. 6.
Stress degradation behavior of AV
The stress degradation studies for all conditions were performed to demonstrate that the developed method is specific, selective, robust, and precise. The % AV degradation obtained in different conditions listed in Table 4. From the result of stress degradation of AV, it was found that AV is sensitive towards thermal, photolytic, oxidative, and hydrolytic conditions. It was confirmed from the additional peaks achieved in the respective chromatography and also by the reduction in the area of AV in each condition when compared with their area at zero time. The mass balance was calculated from the responses obtained in AV and all the degradation products obtained after stress studies. Mass balance was found greater than 90 for alkaline, photolytic, and thermal degradation. For acid and oxidative degradation, mass balance was 89 and 82, respectively, due to loss of certain mass during degradation. AV % degradation in different degradation condition is as shown in Table 4.
Based on the degradation data, the order of degradation behavior observed was
-
Thermal > Photolytic > Oxidative > Hydrolytic (acid, alkaline, and neutral)
From the result of the degradation behavior of AV, recommended labeling condition was “Store in a cool and dry place. Protect from light and moisture”.
Marketed formulation degradation
The marketed tablet formulation of AV was subjected to acid, alkaline, photolytic, thermal, and oxidative degradation. The degraded drug product samples were studied with the optimized method. The marketed formulation degradation results reveal that drug product degradation pattern was found alike as in API (Fig. 7), which was confirmed by the RRt parameter and peak purity data.

Overlay chromatogram of AV and its marketed formulation in different stress conditions
Validation
The developed method was validated as per ICH guidelines Q2 (R1) and results listed in Table 5. The system suitability of the developed method was checked (10 μg/mL) by evaluating peak area, asymmetry, and theoretical plate and fulfills acceptance criteria. The correlation coefficient was found to be 0.997 for AV in the range of 10–70 μg/mL which confirms the linearity of the method. All the degradation peaks in chromatograms showed resolution (> 2) indicating peaks were well separated having individual peak purity from 0.990–0.999. The found LOD and LOQ values were 0.360 μg/mL and 1.150 μg/mL, respectively. The interday and intraday precision was evaluated at three different concentrations, i.e., 10, 30, and 50 μg/mL (each in triplicate) on three consecutive days and the same day, respectively. The % RSD values for interday and intraday were found to be 1.680–1.800 and 0.326–0.697, respectively. Accuracy of the AV was performed by using a standard sample at three different levels 80%, 100%, and 120% (54, 60, 66 μg/mL), respectively. The % recovery ± RSD was found to be 99.280 ± 0.239 to 101.420 ± 0.092 and the % assay ± RSD was found to be (99.430–102.040) ± 0.997. Obtained working space after optimization was considered as a robust region. In that region, parameters changed deliberately, showing assay value deviating not more than 2%.
Avana tablets were applied to different degradation conditions with an optimized method, showing a similar separation pattern to that of API, which confirms method selectivity. Hence, developed methods can be further used for estimation of AV in tablet formulation without the interference of excipients and degradation product peaks.
Conclusion
Nowadays, the term quality is highly focused by regulatory authorities; hence, continuous monitoring of each parameter during every step becomes more crucial for pharmaceutical companies. The product quality can affect due to drug degradation with respect to exposure of different environmental conditions during handling, manufacturing, and storing. Hence, the report discussed a novel stability-indicating method development by QbD in terms of drug stability study and separation of all possible degradation peaks from the drug. A systematic QbD approach was applied to derive full-proof chemical stability of AV using screening and optimization as steps. The primary parameter screening was done by the Ishikawa diagram and risk assessment. Selected eleven primary parameters were studied via the application of Plackett–Burman design, and from that, four were listed out as critical. The critical factors (mobile phase composition, pH, column oven temperature, and flow rate) were optimized by central composite design (CCD) to achieve better resolution between merged degradation peaks. The statistical calculations, surface plots, desirability values and overlay plot were derived from design expert software. The optimized method was practically checked, and the control strategies were decided. The final method reveals a robust method, better resolution between peaks, less time for method development, and minimum solvent usage. The degradation peaks of AV in different stress conditions were evaluated by the HPLC-PDA study. It showed separation of total of 16 degradation products in different degradation conditions. The chemical stability study of AV reveals that AV is prone to degrade in thermal, photolytic (sunlight), and hydrolytic (acid, alkaline, and neutral) conditions. A detailed stability-indicating method with the QbD approach for AV is reported for the first time, which concludes that the AV and its tablet formulation require protection from sunlight, moisture, and temperature during manufacturing, packaging, and handling. The required storage conditions for AV are “Store in a cool and dry place” and “Protect from light and moisture”. The presented study benefited for the drug quality management during synthesis, shelf life, and expiry date determination. The fully validated method can be successfully applied to monitor the stability of AV and its tablet formulation during pharmaceutical process. As the developed method is LC–MS compatible, it can be wisely extended for AV estimation from biological samples.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AV:
-
Avanafil
- CQAs:
-
Critical quality attributes
- DP:
-
Degradation product
- DMSO:
-
Dimethyl sulfoxide
- ED:
-
Erectile dysfunction
- FTIR:
-
Fourier-transform infrared spectrometer
- HPLC:
-
High performance liquid chromatography
- LC-MS:
-
Liquid chromatography mass spectroscopy
- MFAT:
-
Multiple factors at a time
- OFAT:
-
One factor at a time
- PDE:
-
Phosphodiesterase
- QTPP:
-
Quality target product profile
- RSD:
-
Relative standard deviation
- SIAM:
-
Stability indicating analytical method
References
Bhandi MM, Borkar RM, Shankar G, Raut S, Nagesh N, Srinivas R. Identification and characterization of stressed degradation products of rabeprazole using LC-ESI/MS/MS and 1H-NMR experiments: in vitro toxicity evaluation of major degradation products. RSC Adv. 2016;6:10719–35. https://doi.org/10.1039/C5RA27965H.
Bhatt B, Raval K, Buddhadev S. Stability indicating analytical method development and validation for the estimation of Avanafil in pharmaceutical dosage form. Inter J of Pharma Quality Assu. 2015;3.6:181–94 Access from: www.ijpda.com.
Can NO. Development of validated and stability-indicating LC-DAD and LC-MS/MS methods for determination of Avanafil in pharmaceutical preparations and identification of novel degradation product by LCMS-IT-TOF. Molecule. 2018;23:1–14.
Dharani S, Sogra F, Ali B, Afrooz H, Bhattacharya R, Khan M, Rahman Z. Quality and in-use stability comparison of brand and generics of extended-release phenytoin sodium capsules. J Pharm Sci. 2018;108:1808–17. https://doi.org/10.1016/j.xphs.2018.12.022.
Ferguson JE, Carson CC. Phosphodiesterase type 5 inhibitors as a treatment for erectile dysfunction: current information and new horizons. Arab J Urol. 2013;11:222–9. https://doi.org/10.1016/j.aju.2013.07.009.
Garg LK, Sait SS, Krishnamurthy T, Kumar CH. Quality by Design (QbD): a practical experimental design approach by blocking and varying certain factors of a stability-indicating HPLC method for simultaneous determination of omeprazole and ketoprofen. J Liq Chromatogr Relat Technol. 2015;38:677–86. https://doi.org/10.1080/10826076.2014.951766.
Gavin PF, Olsen BA. A quality by design approach to impurity method development for atomoxetine hydrochloride (LY139603). J Pharm Biomed Anal. 2008;1100.46:431–41. https://doi.org/10.1016/j.jpba.2007.10.037.
Hubert C, Lebrun P, Houari S, Ziemons E, Rozet E, Hubert P. Improvement of a stability-indicating method by Quality-by-Design versus Quality-by-Testing: a case of a learning process. J Pharm Biomed Anal. 2014;88:401–9. https://doi.org/10.1016/j.jpba.2013.09.026.
ICH Expert working group, ICH guideline Q8(R2) Pharmaceutical Development. International Conference on Harmonisation. https://www.ich.org/products/guidelines/quality/article/quality-guidelines.html. Accessed 21 Dec 2018.
ICH Expert working group, ICH guideline Q2(R1) Validation of analytical methods definitions and terminology. International conference on Harmonisation. https://www.ich.org/products/guidelines/quality/article/quality-guidelines.html. Accessed 21 Dec 2018.
ICH Expert working group, ICH guideline Q14 Analytical procedure development and revision of Q2 (R1) analytical validation. International conference on Harmonisation. https://www.ich.org/products/guidelines/quality/article/quality-guidelines.html. Accessed 21 Dec 2018.
Jung J, Choi S, Cho SH, Ghim JL, Hwang A, Kim U, Kim BS, Koguchi A, Miyoshi S, Okabe H, Bae KS, Lim HS. Tolerability and pharmacokinetics of avanafil, a phosphodiesterase type 5 inhibitor: a single- and multiple-dose, double-blind, randomized, placebo-controlled, dose-escalation study in healthy Korean male volunteers. Clin Ther. 2010;32:1178–87. https://doi.org/10.1016/j.clinthera.2010.06.011.
Kalariya PD, Patel PN, Sharma M, Garg P, Srinivas R, Talluri MV. Characterization of stress degradation products of blonanserin by UPLC-QTOF-tandem mass spectrometry. RSC Adv. 2015c;5:69273–88. https://doi.org/10.1039/c5ra10641a.
Kalariya PD, Patel PN, Srinivas R, Talluri MV. Quality by design-based development of a selective stability-indicating UPLC method of dolutegravir and characterization of its degradation products by UPLC-QTOF-MS/MS. New J Chem. 2015b;39:6303–14. https://doi.org/10.1039/C5NJ00698H.
Kalariya PD, Sharma M, Garg P, Thota JR, Ragampeta S, Talluri MV. Characterization of stress degradation products of mirabegron using UPLC-QTOF-MS/MS and in silico toxicity predictions of its degradation products. RSC Adv. 2015a;5:31024–38. https://doi.org/10.1039/C5RA01711D.
Karmarkar S, Garber R, Genchanok Y, George S, Yang X, Hammond R. Quality by design (QbD) based development of a stability-indicating HPLC method for drug and impurities. J Chromatogr Sci. 2011;49:439–46.
Kumar N, Sangeetha D, Kalyanraman L, Sainath K. Stability indicating HPLC method for simultaneous determination of degradation products and process-related impurities of Avanafil in Avanafil tablets. Acta Chromatogram. 2017. p. 1–6.
Kurmi M, Kumar S, Singh B, Singh S. Implementation of design of experiments for optimization of forced degradation conditions and development of a stability-indicating method for furosemide. J Pharm Biomed Anal. 2014;96:135–43. https://doi.org/10.1016/j.jpba.2014.03.035.
Maggio RM, Vignaduzzo SE, Kaufman TS. Practical and regulatory considerations for stability-indicating methods for the assay of bulk drugs and drug formulations. Trends Anal Chem. 2013;49:57–70. https://doi.org/10.1016/j.trac.2013.05.008.
Mangukiya M, Rathod B, Dhaduk D, Maniya J. Development and validation of a reversed-phase HPLC method for simultaneous determination of Avanafil and Dapoxetine hydrochloride in combined pharmaceutical formulation. Inven Rapid Pharm Anal Qual Assur. 2013.
Michael AM, Rezk MR, Lotfy HM, Shehata MA. Comparative study of multivariate and univariate determination of zolmitriptan in the presence of its degradation products. Anal Chem Lett. 2015;5:73–84. https://doi.org/10.1080/22297928.2015.1039942.
Mohamed AM, Abdel Wadood HM, Mousa HS. Dual design spaces for micro-extraction together with the core-shell chromatographic determination of dorzolamide and timolol in rabbit plasma: an example of quality by design method development. New J Chem. 2016;40:8424–37. https://doi.org/10.1039/C5NJ03719K.
Orlandini S, Pasquini B, Caprini C, Del Bubba M, Squarcialupi L, Colotta V, Furlanetto S. A comprehensive strategy in the development of a cyclodextrin-modified microemulsion electrokinetic chromatographic method for the assay of diclofenac and its impurities: mixture process variable experiments and quality by design. J Chromatogr A. 2016;1466:189–98. https://doi.org/10.1016/j.chroma.2016.09.013.
Patel MN, Kothari CS. Multivariate approaches for simultaneous determination of Avanafil and Dapoxetine by UV chemometrics and HPLC-QbD in binary mixtures and pharmaceutical product. J AOAC Int. 2016;99:649–63.
Patel MN, Kothari CS. Review on implementation of multivariate approach for forced degradation study and impurity profiling with regulatory considerations. Chromatographia. 2018;81(1):105–25.
Prajapati P, Agrawal YK. Development of a green method for separation and identification of the degradation impurity of isoniazid by SFC-MS/MS. Anal Methods. 2015;7:7776–83. https://doi.org/10.1039/C5AY01852H.
Ren Z, Zhang X, Wang H, Jin X. Using an innovative quality-by-design approach for the development of a stability-indicating UPLC/Q-TOF-ESI-MS/MS method for stressed degradation products of imatinib mesylate. RSC Adv. 2016;6:13050–62. https://doi.org/10.1039/C5RA26456A.
Savaliya SD, Chaudhary AB, Rathod BG, Dobariya TD, Mohan S. Development and validation of UV spectrophotometric method for estimation of Avanafil in tablet dosage form. Invent Ra Pharm Anal Qual Assur. 2013.
Schmidt AH, Molnár I. Using an innovative quality-by-design approach for the development of a stability-indicating UHPLC method for ebastine in the API and pharmaceutical formulations. J Pharm Biomed Anal. 2013;78:65–74. https://doi.org/10.1016/j.jpba.2013.01.032.
Tol T, Kadam N, Raotole N, Desai A, Samanta G. A simultaneous determination of related substances by high performance liquid chromatography in a drug product using quality by design approach. J Chromatogr A. 2016;1432:26–38.
Tol T, Tawde H, Gorad S, Jagdale A, Kulkarni A, Kasbale A. Optimization of a liquid chromatography method for the analysis of related substances in daclatasvir tablets using design of experiments integrated with the steepest ascent method and Monte Carlo simulation. J Pharm Biomed Anal. 2020;178:112943. https://doi.org/10.1016/j.jpba.2019.112943.
Acknowledgements
The authors would like to thank the Institute of Pharmacy, Nirma University, India for providing necessary facilities and senior research fellowship (SRF) for this work. Presented work is a part of the Doctor of Philosophy (Ph.D.) research work of Mital Patel, submitted to Nirma University, Ahmedabad, India.
Funding
Institute of Pharmacy, Nirma University, India provided necessary facilities, junior research fellowship (JRF), and senior research fellowship (SRF) (NU/IP/Ph.D./FT/Stipend/14-15/003) for this work.
Author information
Authors and Affiliations
Contributions
MP performed, analyzed and interpreted the analytical data, while CK guided and supervised the project. Both MP and CK authors contributed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
All the authors report no conflict of interest. The authors alone are responsible for the content and writing of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplement Table 1.
QTPP, CQAs and process parameters for SIAM method by HPLC. Supplement Table 2. Response variable ANOVA results. Supplement Figure 1. Contour and 3 D plots for CCD optimization. Supplement Figure 2. Desirability contour plot and 3D Plot for AV stability method. Supplemental Figure 3. Updated risk assessment for AV stability method.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patel, M., Kothari, C. Comprehensive stability-indicating method development of Avanafil Phosphodiesterase type 5 inhibitor using advanced Quality-by-Design approach. J Anal Sci Technol 11, 29 (2020). https://doi.org/10.1186/s40543-020-00228-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40543-020-00228-4