
Abstract
Ependymoma is the third most common brain tumor in children, with well-described molecular characterization but poorly understood underlying germline risk factors. To investigate whether genetic predisposition to longer telomere length influences ependymoma risk, we utilized case–control data from three studies: a population-based pediatric and adolescent ependymoma case–control sample from California (153 cases, 696 controls), a hospital-based pediatric posterior fossa type A (EPN-PF-A) ependymoma case–control study from Toronto’s Hospital for Sick Children and the Children’s Hospital of Philadelphia (83 cases, 332 controls), and a multicenter adult-onset ependymoma case–control dataset nested within the Glioma International Case-Control Consortium (GICC) (103 cases, 3287 controls). In the California case–control sample, a polygenic score for longer telomere length was significantly associated with increased risk of ependymoma diagnosed at ages 12–19 (P = 4.0 × 10−3), but not with ependymoma in children under 12 years of age (P = 0.94). Mendelian randomization supported this observation, identifying a significant association between genetic predisposition to longer telomere length and increased risk of adolescent-onset ependymoma (ORPRS = 1.67; 95% CI 1.18–2.37; P = 3.97 × 10−3) and adult-onset ependymoma (PMR-Egger = 0.042), but not with risk of ependymoma diagnosed before age 12 (OR = 1.12; 95% CI 0.94–1.34; P = 0.21), nor with EPN-PF-A (PMR-Egger = 0.59). These findings complement emerging literature suggesting that augmented telomere maintenance is important in ependymoma pathogenesis and progression, and that longer telomere length is a risk factor for diverse nervous system malignancies.
Similar content being viewed by others

Introduction
Ependymoma is the third most common brain tumor in children, accounting for 5–10% of childhood brain tumors, with more than half of all cases occurring in children under five years old. Most pediatric ependymomas are intracranial in origin (90%), whereas a greater proportion of adult-onset ependymomas occur in the spinal cord (66%) [40]. The molecular characterization of ependymal tumors is well-described and may inform a new era of precision diagnostics and targeted therapies [25, 41]. Underlying germline risk factors that predispose individuals to develop ependymoma remain poorly understood, as the ability to perform genetic epidemiology studies of rare diseases is limited and traditional genome-wide association study (GWAS) approaches are underpowered. However, alternative analytic approaches that use polygenic scores or Mendelian randomization analyses to model genetic predisposition to “intermediate phenotypes” hold promise for advancing our understanding of genetic risk in rare diseases, including childhood cancers [6, 48]. Telomere length is perhaps the “intermediate phenotype” that has been best-characterized for its association with brain tumor risk, as genetic predisposition to longer telomeres increases risk of both adult glioma [52, 53] and meningioma [36]. Despite the known association between genetic predisposition to longer telomere length and certain adult-onset brain tumors, the association between ependymoma risk and telomere length has not been evaluated in either children or adults.
Telomeres are nucleoprotein structures that protect the ends of chromosomes during normal cellular DNA replication; however, telomeres shorten with each replicative cell division cycle until reaching a critically short length, at which point cellular senescence or apoptosis ensues [5, 16]. Telomere length is maintained during cellular replication by the enzyme telomerase, encoded by the TERT gene. Normally, telomerase is active in stem and progenitor cells, yet activity is repressed in normal somatic cells as an anti-proliferative mechanism [15]. An important hallmark of cancer is “enabling replicative immortality,” which is necessary for sustained malignant growth and which is often achieved by reactivating telomerase expression in immortalized cells [26]. Cancer cells are able to avoid senescence and apoptosis in part by maintaining telomere length indefinitely. This is typically achieved through telomerase reactivation or through a homologous recombination-associated process referred to as alternative lengthening of telomeres (ALT) [26, 37]. While dysregulated telomere biology has been implicated in ependymoma progression and prognosis [46, 50, 51], the TERT promoter mutations associated with telomerase reactivation are uncommon in both childhood and adult ependymomas, as are the ATRX mutations associated with ALT [9]. Individuals who are genetically predisposed to longer telomere length or more efficient telomere maintenance are at increased risk of adult glioma and childhood neuroblastoma [53, 55], perhaps due to an enhanced capacity for pre-malignant cells to divide and acquire additional oncogenic mutations before their telomere reserves are depleted [2, 56]. However, it is unknown whether genetic predisposition to telomere length contributes to ependymoma risk.
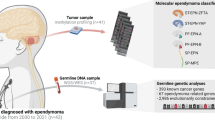
To investigate whether genetic predisposition to telomere length influences ependymoma risk, we examined the association between validated genetic instruments associated with longer leukocyte telomere length (LTL) and ependymoma risk in case–control analyses [14]. We utilized both polygenic scores modeling genetic predisposition to longer LTL and Mendelian randomization analyses to test for a causal association between longer LTL and ependymoma risk. Because a previously observed association between polygenic scores for telomere length and neuroblastoma risk implicated effect modification by age [55], we also performed age-stratified analyses in groupings defined a priori. The current study utilizes case–control data from three different collaborations, including: (1) a population-based pediatric and adolescent ependymoma case–control sample from California, (2) a hospital-based pediatric posterior fossa type A (PFA) ependymoma case–control study from Toronto’s Hospital for Sick Children and the Children’s Hospital of Philadelphia, and (3) a multicenter adult-onset ependymoma case–control dataset nested within the Glioma International Case-Control Consortium (GICC).
Methods
Ethics statement
The study was approved by Institutional Review Boards at The University of California, Berkeley, The University of California, San Francisco, the California Department of Public Health, The Children’s Hospital of Philadelphia, the University of Toronto Hospital for Sick Children, and Baylor College of Medicine.
California Cancer Record Linkage Project (CCRLP) case–control dataset
Blood samples from neonates born within the state of California are collected by the California Department of Public Health, Genetic Diseases Screening Branch for the purpose of disease screening, with remaining samples archived at − 20 °C since 1982 and made available for approved research. We linked statewide birth records from the California Department of Public Health for the years 1982–2009 to data from the California Cancer Registry (CCR) for diagnosis years 1988–2011. Cases were defined as patients diagnosed with ependymoma before age 20, per CCR record of 2014 ICD-O-3 codes 9391-9394. Controls were matched on race/ethnicity, sex, month and year of birth from the pool of children born in California during the same period and not reported to CCR as having any childhood cancer. Included in this analysis were 153 non-Hispanic white children with ependymoma and 696 controls, as previously described [62]. Subjects from other racial/ethnic backgrounds were not included due to the questionable performance of polygenic scores built with loci and effect estimates derived from European-ancestry populations and applied to Hispanic or African-American populations [24, 33], and further the lack of suitable GWAS of telomere length in these populations [10]. Details on the linkage and use of neonatal bloodspots for studying pediatric cancers have been reported previously [57].
CCRLP DNA extraction and genotyping
Details on the use of neonatal bloodspots for DNA extraction and genotyping have been reported previously [61]. In brief, DNA was extracted from a one-third portion of a 12-mm dried blood spot using the QIAamp DNA Investigator Kit (Qiagen), followed by addition of 280 μL of Buffer ATL and 20 μL of Proteinase K to each sample. Samples were vortexed and then incubated in a dry-bath shaker at 900 rpm and 56 °C for one hour. Samples were then briefly centrifuged, after which the lysate solution was transferred to a new 2 mL microcentrifuge tube, and the solid remnants discarded. 1 μL of 1 ng/μL carrier RNA was added to the lysate, briefly vortexed, and placed in the Qiagen Qiacube automated work station for DNA isolation, yielding a purified DNA sample in ATE buffer. DNA was genotyped on the Affymetrix Axiom World Array (LAT), with average genotype concordance > 99% between duplicate samples on the same plates. Quality-control procedures included call-rate filtering for single nucleotide polymorphisms (SNPs) and samples, performed iteratively by removing SNPs with call rates < 92%, then samples with call rates < 95%, then SNPs with call rates < 97%, then samples with call rates < 96%. SNPs with significant departure from Hardy–Weinberg equilibrium (P < 1.0 × 10−5) among controls were excluded. Samples with mismatched reported versus genotyped sex were also excluded. Identity-by-descent (IBD) analyses were performed in PLINK on cases and controls, with exclusion of one member of any sample pair that had an identity-by-descent proportion > 0.18 [44]. Using SNP array data from the HapMap phase III European reference panel samples, we calculated ancestry-informative principal components (PCs) and removed any sample showing evidence of non-European ancestry (> 3 SDs from mean CEPH values on PCs 1–3). We performed haplotype phasing and imputation with SHAPEIT v2.79029 and Minimac3 software using phased genotype data from the 2016 release of the Haplotype Reference Consortium [17, 34]. SNPs with imputation quality (INFO) scores < 0.60 or posterior probabilities < 0.90 were excluded.
Toronto case-control dataset
A total of 83 pediatric ependymoma patients (median age 3 years) and 332 control children of non-Hispanic white ethnicity were genotyped on the Illumina OmniExpress array at the Center for Applied Genomics (CAG) at the Children’s Hospital of Philadelphia (CHOP). All ependymoma patients were recruited onto study at The Hospital for Sick Children at The University of Toronto and had posterior fossa type A (PF-EPN-A) tumors, as determined by integrated analysis of DNA methylation, copy-number, gene expression, and clinical parameters, as previously described [41]. DNA was extracted from blood where available, but a subset of patient DNA specimens were extracted from tumor specimen to increase sample size. Because PF-EPN-A are genetically bland and rarely harbor either point mutations or copy-number alterations, including in the regions of chromosomes 2, 3, 4, 5, 10, 17, 19, and 20 where SNPs used as genetic instruments for telomere length are located, deviation from constitutive genotypes appeared minimal [41]. GWAS data underwent quality-control procedures as previously described [19, 32]. Genotypes were phased using SHAPEIT2 [18] and whole-genome imputation was performed using IMPUTE2 [29] with 1000 Genomes Phase 3 release as the imputation reference panel [22]. Case–control comparisons were performed using the frequentist test with an additive model and score method to deal with uncertainty as implemented in SNPTEST v2.4.1 [32], with adjustment for the ten five ancestry-informative PCs.
Glioma International Case-Control dataset (GICC)
GICC is the largest glioma study to-date including biospecimens and blood samples, conducted by the Genetic Epidemiology of Glioma International Case-Control Consortium [1]. Individual-level genotype and phenotype data are available for download from the Database of Genotypes and Phenotypes (dbGaP, Study Accession phs001319.v1.p1) after review and approval by the NCI Data Access Committee. From the GICC data, a subset of 103 adult ependymoma cases age 18–72 (with three cases < age 20) and 3287 controls were selected. Subject recruitment and control selection has previously been described in detail [1].
Single SNP and polygenic score analyses
We investigated the individual effect of eight telomere-length associated SNPs on ependymoma risk in the CCRLP, Toronto, and GICC datasets. We also assessed the combined effect of these SNPs in CCRLP cases and controls, where individual-level genotype data were available (for Toronto and GICC subjects, only SNP-level summary statistics were available). SNPs were chosen based on strong prior evidence of association with LTL in previous GWAS publications demonstrating genome-wide significant associations (P < 5 × 10−8) and excluding linked SNPs (R2 < 0.05) in order to avoid “double-counting” risk loci [14]. Although the proportion of variation explained by the eight SNPs is individually small (~ 2%) [14], the genotypically-estimated relative LTL across individuals ranged from 140 to 943 base pairs. This 803 base-pair range corresponds to > 25 years of age-related telomere attrition (based on an average LTL attrition rate of 20–40 base pairs/year) [21]. We first assessed single LTL SNP associations with ependymoma risk using logistic regression, assuming an allelic additive model for 0, 1 or 2 copies of the allele for longer LTL. For CCRLP subjects, we also constructed polygenic scores for longer LTL by calculating the weighted sum of the number of alleles corresponding to longer LTL (up to 16 alleles from 8 unlinked SNPs) for each individual, in which the weight was taken as the effect estimate from the LTL GWAS from the ENGAGE Consortium Telomere Group [14]. We performed a logistic regression analysis of the standardized polygenic scores for longer LTL and ependymoma risk, adjusting for sex and the top 10 principal components. Resulting beta estimates are interpreted as the difference in ependymoma risk per one standard deviation increase in the LTL score. We also assessed the differences in LTL and ependymoma association stratified by age (< 12 years old vs. ≥ 12 years old) and tumor location (spinal vs. intracranial) in CCRLP data. We used PLINK to complete both the single SNP and polygenic score association analyses.
Mendelian randomization analyses
Mendelian randomization (MR) is a causal inference method in which genetic variants are used as instrumental variables, i.e. proxies for a risk factor of interest, to evaluate the causal relationship between the risk factor and an outcome of interest. In the two-sample summary data MR approach, summary statistics for SNP-exposure associations are obtained from a different set of samples from those for the SNP-outcome association, assuming both samples are drawn from the same underlying population [11]. The polygenic score association analysis can be considered a form of MR analysis in which the score is considered an instrumental variable [12], assuming that all variants contributing to the score are valid instruments that do not violate any of the three MR assumptions. However, a formal application of the MR method, along with various sensitivity analyses, are necessary as prior studies show that risk score association analyses can suffer from higher false positive rates due to horizontal pleiotropy, a violation of the MR assumptions [28, 45]. Furthermore, in the absence of individual-level data SNP data, the two-sample summary data MR method can be useful for approximating the association between a genetic score and an outcome of interest using summary statistics. Our MR analyses used summary statistics for the association with ependymoma risk of the same 8 SNPs used in our polygenic score model, adjusted for sex and 10 principal components. We used the inverse-variance weighted (IVW) method, along with the (1) MR-Egger method, which provides consistent estimates in the presence of horizontal pleiotropy given that pleiotropic effects are independent of instrument strength across all variants; [7], (2) the weighted median method [8], which provides consistent estimates even when up to 50% of the information comes from invalid instruments; and (3) the mode-based method [27], which provides consistent estimates even when a majority of instruments are invalid, to assess the causal association between LTL and ependymoma risk in CCRLP data, Toronto data, and GICC data. We used the MendelianRandomization R package for these analyses [59].
Results
From the CCRLP dataset, a total of 153 non-Hispanic white pediatric ependymoma patients and 696 controls were available for analyses after linkage, newborn bloodspot DNA extraction, genotyping, QC procedures, and SNP imputation. Demographics and clinical features of the CCRLP ependymoma cases are shown in Additional file 1: Table S1. Demographic details on the Toronto (n = 83 cases, 332 controls) and the GICC (n = 103 cases, 3287 controls) case–control datasets have been previously described [1, 62].
A total of eight SNPs previously associated with LTL at genome-wide significant levels and independently replicated by Codd et al. [14] were successfully genotyped in all three ependymoma datasets: rs11125529 (ACYP2), rs10936599 (TERC), rs7675998 (NAF1), rs2736100 (TERT), rs9420907 (OBFC1), rs3027234 (CTC1), rs8105767 (ZNF208), and rs755017 (RTEL1). Two nominally significant associations (Punadjusted < 0.05) were observed in the CCRLP age-stratified analysis for patients ≥ 12 years old at diagnosis at rs10936599 in TERC (OR = 1.98; 95% CI = 1.06, 4.06; P = 0.043) and at rs7675998 in NAF1 (OR = 2.15; 95% CI = 1.09, 4.74; P = 0.039), but no associations were observed for CCRLP patients < 12 (Additional file 1: Table S2). One nominally significant association (P < 0.05) was observed in the Toronto PF-A case–control sample at rs9420907 in OBFC1 (OR = 1.71; 95% CI = 1.07, 2.73; P = 0.026), but no single-SNP associations were observed in the GICC adult ependymoma data (Additional file 1: Table S3).
The association between a polygenic score for longer LTL and pediatric ependymoma risk was assessed using logistic regression, adjusting for sex and 10 PCs. Among all California cases and controls age ≤ 19, a non-significant association was observed (OR = 1.12; 95% CI 0.94–1.34; P = 0.207, Table 1). When cases were stratified by age at diagnosis, using cutoffs defined a priori based on previous observations in neuroblastoma [55], a significant association was observed between longer LTL score and increased risk of ependymoma in CCRLP patients diagnosed at ≥ 12 years of age (OR = 1.67; 95% CI 1.18–2.37; P = 3.97 × 10−3). However, the LTL score was not associated with ependymoma risk in patients < 12 years old (OR = 1.12; 95% CI 0.94–1.34; P = 0.21). In a case–case comparison to test for etiologic heterogeneity by age at diagnosis, the LTL score was significantly higher in adolescent-onset ependymoma patients (12–19 years of age) compared with childhood-onset ependymoma patients (0–12 years of age) (P = 0.021). When further stratified by tumor location, longer LTL was associated with increased risk of both intracranial and spinal ependymoma diagnosed at ≥ 12 years of age (P = 0.048 and 0.024, respectively). However, longer LTL was not associated with increased risk of intracranial ependymoma in children diagnosed before age 12 (P = 0.53), and longer LTL was inversely associated with risk of spinal ependymoma in children under 12 (P = 8.9 × 10−3) (Additional file 1: Table S4). When intracranial ependymoma were further restricted to supratentorial tumors, longer LTL was again significantly associated with increased risk in those diagnosed at ≥ 12 years of age (P = 0.024), but no among those diagnosed before 12 years of age (P = 0.86) (Additional file 1: Table S4).
Formal Mendelian randomization analyses were conducted to make causal inferences about the association between telomere length and ependymoma risk in all three datasets. The MR results complemented the polygenic score analyses in CCRLP data, with IVW MR estimates suggesting a significant causal association between longer LTL and increased ependymoma risk in patients diagnosed at ≥ 12 years of age (PIVW = 6.3 × 10−3). Sensitivity analyses supported this link, finding no evidence of directional pleiotropy with MR Egger tests (PMR-intercept = 0.67). A similar positive association was observed in GICC adult ependymoma data using the MR Egger estimate (PMR-Egger = 0.042), where a non-zero intercept term suggested the presence of directional pleiotropy (PMR-intercept = 0.059). Weighted-median and mode-based MR analyses also show positive associations in CCRLP adolescent (age ≥ 12) and in GICC adult ependymoma, although effect sizes were smaller and several confidence intervals included the null (Table 2).
In contrast to the CCRLP adolescent and the GICC adult ependymoma patients, no associations between LTL and ependymoma risk were observed for the CCRLP patients < 12 years of age or among the Toronto pediatric PF-A patients (median age, 3 years) in any of the MR estimates. We visualized the per-allele association of ependymoma risk (y-axis) plotted against the per-allele association with LTL (x-axis), with the slopes of the fitted lines equal to the IVW and MR Egger estimates (Fig. 1). The intercept is fixed at the origin for the IVW method and unconstrained for MR Egger. The slope is relatively flat for Toronto PF-A patients (Fig. 1a) and the CCRLP patients < 12 (Fig. 1b) using both IVW and MR Egger estimates, but is clearly positive in CCRLP patients age ≥ 12 for both IVW and MR Egger (Fig. 1c). The slope for the adult ependymoma analysis is relatively flat for the IVW estimate, but a significant positive slope was observed for the MR-Egger estimate (Fig. 1d).

Per-allele association of ependymoma risk (y-axis) and leukocyte telomere length (x-axis) at eight SNPs known to influence telomere length, with the slopes of fitted lines equal to the inverse-variance weighted (IVW) Mendelian randomization estimate (solid line) and the MR-Egger estimate (dashed line) in a Toronto EPN-PF-A case–control data; b CCRLP childhood-onset (< 12 years) ependymoma case–control data; c CCRLP adolescent-onset (12–19 years) ependymoma case–control data; d Glioma International Case-Control Consortium (GICC) adult-onset ependymoma case–control data
Discussion
Our results suggest that a genetic predisposition to longer telomere length increases the risk of adolescent and adult-onset ependymoma, but not risk of ependymoma diagnosed in children younger than 12 or in very young children with the EPN-PF-A molecular subtype. In addition to polygenic score analyses, the MR results in particular suggest that longer telomere length is a causal risk factor underlying adolescent ependymoma development. Although intracranial tumors are more common in children and spinal ependymomas are more common in adults [30], we observed that the association between genetic predisposition to longer LTL and adolescent-onset ependymoma risk was consistent in both the intracranial and spinal subgroups, as well as among the supratentorial tumors. Our findings contribute to the growing literature implicating longer telomere length as a risk factor for nervous system tumors in both children and adults [53, 55], and in a number of other non-nervous system cancers including lung, melanoma, chronic lymphocytic leukemia, and osteosarcoma [38, 52, 60].
Although the histopathologic distribution of primary CNS tumors differs in children and adults, childhood and adult-onset brain tumors likely share some common etiologic factors. Although adult-onset cancers are often linked to lifestyle and environmental risk factors while heritable and in utero exposures predominate in pediatric cancers, very few non-genetic risk factors have been identified for CNS malignancies [39]. While pediatric ependymomas have distinct genomic profiles from their adult counterparts, most notably in the epigenetically-driven EPN-PF-A subgroup that was not associated with telomere length in our analyses, telomerase reactivation has been observed to varying degrees across ependymoma subgroups and ages of onset [23, 30, 35].
We previously proposed that longer telomere length increases cancer risk by augmenting capacity for sustained cellular replication, allowing pre-malignant cells to accumulate the mutations necessary to resist apoptosis and enable replicative immortality [54], mutations such as hypermethylation of the TERT promoter [13, 23]. Our observation that genetic predisposition to longer LTL is associated with adolescent-onset and adult-onset ependymoma, but not childhood-onset ependymoma, supports this multi-step model of tumorigenesis in patients ages 12 and up. Longer telomere length may be an important mediator of ependymoma risk in adolescents and adults, where the tumor is more dependent upon acquiring somatic driver mutations and does not arise from an epigenetically dysregulated developmental cell lineage [26, 35].
In addition to polygenic score tests, we performed formal MR analyses to assess potential causal associations. These approaches are particularly useful for investigating the genetic epidemiology of pediatric malignancies given sample size limitations that hinder traditional GWAS approaches. Polygenic score analyses test genetic propensity for a phenotype, rather than a single variant, and have improved power over both single-SNP analyses and MR approaches [42, 43, 45]. Importantly, formal MR analyses can confirm the relationship between polygenic scores (longer LTL) and the outcome of interest (ependymoma) and provides a test of causal inference under valid assumptions that can be evaluated with various sensitivity analyses. MR analyses implied a causal relationship between longer telomere length and adolescent-onset ependymoma and there was no evidence of directional pleiotropy based on the null MR Egger intercept test. Our MR results lend support to a causal relationship between longer LTL and adolescent-onset ependymoma, complementing the polygenic score analyses.
In the analysis of GICC adult-onset ependymomas, longer LTL was positively associated with ependymoma risk in all MR models, although the IVW, weighted-median, and mode-based MR associations were not statistically significant. However, the MR Egger intercept test suggested that directional pleiotropy may be present. This directional pleiotropy, wherein genetic variants have pleiotropic effects that—on average—differ from zero, can result in biased IVW, weighted median, and mode-based estimates [7, 27]. Importantly, the MR Egger estimate, which is unbiased in the case of directional pleiotropy, suggested a positive causal association with LTL, consistent with the polygenic score and MR associations in adolescent-onset patients. Thus, the MR Egger estimate results suggest that genetic predisposition to longer LTL may be a risk factor for adult-onset ependymoma, although causal inference is complicated by the apparent presence of directional pleiotropy. Based on Fig. 1d, the primary source of this directional pleiotropy appears to be rs3027234 in CTC1. CTC1 is one of three members of the CST complex that binds to single-stranded DNA and is required to protect telomeres from DNA degradation. In addition to a role in telomere protection, the CST complex has a more general role in DNA metabolism at non-telomeric sites and was shown to protect DNA double-strand breaks from end resection, leading to repair by non-homologous end joining rather than homologous recombination [3].
Of note are the extreme magnitudes of the MR effect estimates in our analyses, which resulted in odds ratios (ORs) ranging from 91 to 368 per standard deviation of longer LTL score, with correspondingly large 95% confidence intervals. MR estimates are useful for testing causal relationships, but have limited utility in determining the exact size of a causal effect [47]. Our MR effect estimates may be inflated due to various reasons, such as age-specific variation in SNP-exposure or SNP-outcome associations, or under-estimation of genetic associations with the exposure compared to the outcome. Importantly, the large ORs observed in our analysis could also be a result of unstable estimates due to small sample sizes. However, a previous comprehensive MR study across multiple cancer types suggests that larger MR associations tend to be seen for tissue sites with lower rates of stem cell division, such as the brain, with the largest such estimate observed for glioma (OR, 5.27). Thus, we do not rule out the possibility that longer LTL may have a large magnitude of effect on ependymoma risk in adolescents and adults.
The role of telomere maintenance has been extensively investigated in ependymoma, although our study appears to be the first to investigate germline modifiers of telomere length in ependymoma etiology. The telomerase enzyme is normally expressed in stem and progenitor cells to maintain telomere length, but is suppressed in somatic tissues. Telomerase activity is reactivated in many cancer subtypes, including ependymomas [4, 9, 13, 23, 31]. Telomerase activity has been linked to ependymoma progression, recurrence, and survival, and has been implicated as an important prognostic marker and therapeutic target [23, 50, 51], where telomerase inhibition has demonstrated anti-tumorigenic effects in in vitro and xenograft models of pediatric ependymoma [4, 58]. Telomere dysfunction has also been linked to chromothrypsis, a form of genomic instability characterized by tens to hundreds of clustered DNA rearrangements, which was previously associated with greater telomere length in medulloblastoma and ependymoma [20]. TERT promotor mutations that reactivate telomerase in glioblastoma have occasionally been identified in adult ependymoma, but not in children [4, 9, 31]. In pediatric ependymoma, hypermethylation of the TERT promotor has consistently been associated with telomerase reactivation [13, 23], indicating that epigenetic mechanisms of telomere maintenance may also enable replicative immortality in ependymoma cells. Germline variants, including methylation quantitative trait loci (meQTLs), may accelerate or even enable such epigenetic reactivation. Our results build upon this body of literature by demonstrating that constitutively longer telomere length/inherently better telomere maintenance is associated with ependymoma predisposition in adolescents and adults.
A strength of this study is that our age-stratified findings in the CCRLP dataset were supported by independent datasets of molecularly-subgrouped pediatric cases (Toronto) and adult-onset cases (GICC). However, our study has limitations. Ependymoma is a rare malignancy, so our study is limited by its relatively modest sample size. Despite being better powered to observe a significant association in the childhood-onset ependymoma cases, we still detected a significant association in the smaller subset of adolescent-onset ependymoma cases. There are also limitations to our MR analyses, including the issue of horizontal pleiotropy, discussed earlier, and potential violations of MR assumptions. The assumptions include: (1) a consistent log-linear association between telomere length and cancer risk; (2) that LTL-associated variants have similar associations in ependymal cells; and (3) that the LTL variants derived from an adult population have similar associations in children and adolescents. Violations of any of these assumptions would likely result in a bias toward the null, so the significant associations observed in our data are more likely to be attenuated than to be inflated. However, we cannot rule out the possibility that LTL variants used in this study are less associated in a pediatric population [49], resulting in a null association among the Toronto EPN-PF-A and CCRLP < 12 subsets.
In summary, we leverage a polygenic score and MR framework to examine whether longer telomere length may be a risk factor for ependymoma across age strata. Our findings indicate that genetic predisposition to longer LTL is associated with increased risk of adolescent- and adult-onset ependymoma, but not with childhood ependymoma, including EPN-PF-A. These findings complement emerging literature suggesting that dysregulated telomere maintenance is important for ependymoma pathogenesis and that longer telomere length is a risk factor for several different nervous system malignancies. Future studies should work to incorporate germline data into genomic and epigenomic profiling of ependymoma tumors to explore the relationship between heritable variation and telomerase activity in somatic cells.
Availability of data and materials
This study used biospecimens from the California Biobank Program. Any uploading of genomic data and/or sharing of these biospecimens or individual data derived from these biospecimens has been determined to violate the statutory scheme of the California Health and Safety Code Sections 124980(j), 124991(b), (g), (h), and 103850 (a) and (d), which protect the confidential nature of biospecimens and individual data derived from biospecimens. Certain aggregate results may be available from the authors by request.
References
Amirian ES, Armstrong GN, Zhou R, Lau CC, Claus EB, Barnholtz-Sloan JS, Il’yasova D, Schildkraut J, Ali-Osman F, Sadetzki S, Johansen C, Houlston RS, Jenkins RB, Lachance D, Olson SH, Bernstein JL, Merrell RT, Wrensch MR, Davis FG, Lai R, Shete S, Amos CI, Scheurer ME, Aldape K, Alafuzoff I, Brannstrom T, Broholm H, Collins P, Giannini C, Rosenblum M, Tihan T, Melin BS, Bondy ML (2016) The Glioma International Case-Control Study: a report from the Genetic Epidemiology of Glioma International Consortium. Am J Epidemiol 183:85–91. https://doi.org/10.1093/aje/kwv235
Aviv A, Anderson JJ, Shay JW (2017) Mutations, cancer and the telomere length paradox. Trends Cancer 3:253–258. https://doi.org/10.1016/j.trecan.2017.02.005
Barazas M, Annunziato S, Pettitt SJ, de Krijger I, Ghezraoui H, Roobol SJ, Lutz C, Frankum J, Song FF, Brough R, Evers B, Gogola E, Bhin J, van de Ven M, van Gent DC, Jacobs JJL, Chapman R, Lord CJ, Jonkers J, Rottenberg S (2018) The CST complex mediates end protection at double-strand breaks and promotes PARP inhibitor sensitivity in BRCA1-deficient cells. Cell Rep 23:2107–2118. https://doi.org/10.1016/j.celrep.2018.04.046
Barszczyk M, Buczkowicz P, Castelo-Branco P, Mack SC, Ramaswamy V, Mangerel J, Agnihotri S, Remke M, Golbourn B, Pajovic S, Elizabeth C, Yu M, Luu B, Morrison A, Adamski J, Nethery-Brokx K, Li XN, Van Meter T, Dirks PB, Rutka JT, Taylor MD, Tabori U, Hawkins C (2014) Telomerase inhibition abolishes the tumorigenicity of pediatric ependymoma tumor-initiating cells. Acta Neuropathol 128:863–877. https://doi.org/10.1007/s00401-014-1327-6
Blackburn EH, Epel ES, Lin J (2015) Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science 350:1193–1198. https://doi.org/10.1126/science.aab3389
Blanco-Gomez A, Castillo-Lluva S, Del Mar Saez-Freire M, Hontecillas-Prieto L, Mao JH, Castellanos-Martin A, Perez-Losada J (2016) Missing heritability of complex diseases: enlightenment by genetic variants from intermediate phenotypes. BioEssays 38:664–673. https://doi.org/10.1002/bies.201600084
Bowden J, Davey Smith G, Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44:512–525. https://doi.org/10.1093/ije/dyv080
Bowden J, Davey Smith G, Haycock PC, Burgess S (2016) Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 40:304–314. https://doi.org/10.1002/gepi.21965
Brugger F, Dettmer MS, Neuenschwander M, Perren A, Marinoni I, Hewer E (2017) TERT promoter mutations but not the alternative lengthening of telomeres phenotype are present in a subset of ependymomas and are associated with adult onset and progression to ependymosarcoma. J Neuropathol Exp Neurol 76:61–66. https://doi.org/10.1093/jnen/nlw106
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, Suveges D, Vrousgou O, Whetzel PL, Amode R, Guillen JA, Riat HS, Trevanion SJ, Hall P, Junkins H, Flicek P, Burdett T, Hindorff LA, Cunningham F, Parkinson H (2019) The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47:D1005–D1012. https://doi.org/10.1093/nar/gky1120
Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG, Consortium E-I (2015) Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol 30:543–552. https://doi.org/10.1007/s10654-015-0011-z
Burgess S, Thompson SG (2013) Use of allele scores as instrumental variables for Mendelian randomization. Int J Epidemiol 42:1134–1144. https://doi.org/10.1093/ije/dyt093
Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, Zhukova N, Walker EJ, Martin D, Merino D, Wasserman JD, Elizabeth C, Alon N, Zhang L, Hovestadt V, Kool M, Jones DT, Zadeh G, Croul S, Hawkins C, Hitzler J, Wang JC, Baruchel S, Dirks PB, Malkin D, Pfister S, Taylor MD, Weksberg R, Tabori U (2013) Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol 14:534–542. https://doi.org/10.1016/S1470-2045(13)70110-4
Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Hottenga JJ, Fischer K, Esko T, Surakka I, Broer L, Nyholt DR, Mateo Leach I, Salo P, Hagg S, Matthews MK, Palmen J, Norata GD, O’Reilly PF, Saleheen D, Amin N, Balmforth AJ, Beekman M, de Boer RA, Bohringer S, Braund PS, Burton PR, de Craen AJ, Denniff M, Dong Y, Douroudis K, Dubinina E, Eriksson JG, Garlaschelli K, Guo D, Hartikainen AL, Henders AK, Houwing-Duistermaat JJ, Kananen L, Karssen LC, Kettunen J, Klopp N, Lagou V, van Leeuwen EM, Madden PA, Magi R, Magnusson PK, Mannisto S, McCarthy MI, Medland SE, Mihailov E, Montgomery GW, Oostra BA, Palotie A, Peters A, Pollard H, Pouta A, Prokopenko I, Ripatti S, Salomaa V, Suchiman HE, Valdes AM, Verweij N, Vinuela A, Wang X, Wichmann HE, Widen E, Willemsen G, Wright MJ, Xia K, Xiao X, van Veldhuisen DJ, Catapano AL, Tobin MD, Hall AS, Blakemore AI, van Gilst WH, Zhu H, Yuvi CA, Erdmann J, Reilly MP, Kathiresan S, Schunkert H, Talmud PJ, Pedersen NL, Perola M, Ouwehand W, Kaprio J, Martin NG, van Duijn CM, Hovatta I, Gieger C, Metspalu A, Boomsma DI, Jarvelin MR, Slagboom PE, Thompson JR, Spector TD, van der Harst P, Samani NJ (2013) Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet 45:422–427, 427e421–422. https://doi.org/10.1038/ng.2528
Cong YS, Wright WE, Shay JW (2002) Human telomerase and its regulation. Microbiol Mol Biol Rev 66:407–425. https://doi.org/10.1128/mmbr.66.3.407-425.2002
d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426:194–198. https://doi.org/10.1038/nature02118
Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, Schlessinger D, Stambolian D, Loh PR, Iacono WG, Swaroop A, Scott LJ, Cucca F, Kronenberg F, Boehnke M, Abecasis GR, Fuchsberger C (2016) Next-generation genotype imputation service and methods. Nat Genet 48:1284–1287. https://doi.org/10.1038/ng.3656
Delaneau O, Marchini J, Zagury JF (2011) A linear complexity phasing method for thousands of genomes. Nat Methods 9:179–181. https://doi.org/10.1038/nmeth.1785
Diskin SJ, Capasso M, Schnepp RW, Cole KA, Attiyeh EF, Hou C, Diamond M, Carpenter EL, Winter C, Lee H, Jagannathan J, Latorre V, Iolascon A, Hakonarson H, Devoto M, Maris JM (2012) Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat Genet 44:1126–1130. https://doi.org/10.1038/ng.2387
Ernst A, Jones DT, Maass KK, Rode A, Deeg KI, Jebaraj BM, Korshunov A, Hovestadt V, Tainsky MA, Pajtler KW, Bender S, Brabetz S, Grobner S, Kool M, Devens F, Edelmann J, Zhang C, Castelo-Branco P, Tabori U, Malkin D, Rippe K, Stilgenbauer S, Pfister SM, Zapatka M, Lichter P (2016) Telomere dysfunction and chromothripsis. Int J Cancer 138:2905–2914. https://doi.org/10.1002/ijc.30033
Fitzpatrick AL, Kronmal RA, Kimura M, Gardner JP, Psaty BM, Jenny NS, Tracy RP, Hardikar S, Aviv A (2011) Leukocyte telomere length and mortality in the Cardiovascular Health Study. J Gerontol A Biol Sci Med Sci 66:421–429. https://doi.org/10.1093/gerona/glq224
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR (2015) A global reference for human genetic variation. Nature 526:68–74. https://doi.org/10.1038/nature15393
Gojo J, Lotsch D, Spiegl-Kreinecker S, Pajtler KW, Neumayer K, Korbel P, Araki A, Brandstetter A, Mohr T, Hovestadt V, Chavez L, Kirchhofer D, Ricken G, Stefanits H, Korshunov A, Pfister SM, Dieckmann K, Azizi AA, Czech T, Filipits M, Kool M, Peyrl A, Slavc I, Berger W, Haberler C (2017) Telomerase activation in posterior fossa group A ependymomas is associated with dismal prognosis and chromosome 1q gain. Neuro Oncol 19:1183–1194. https://doi.org/10.1093/neuonc/nox027
Grinde KE, Qi Q, Thornton TA, Liu S, Shadyab AH, Chan KHK, Reiner AP, Sofer T (2019) Generalizing polygenic risk scores from Europeans to Hispanics/Latinos. Genet Epidemiol 43:50–62. https://doi.org/10.1002/gepi.22166
Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, Johann PD, Balasubramanian GP, Segura-Wang M, Brabetz S, Bender S, Hutter B, Sturm D, Pfaff E, Hubschmann D, Zipprich G, Heinold M, Eils J, Lawerenz C, Erkek S, Lambo S, Waszak S, Blattmann C, Borkhardt A, Kuhlen M, Eggert A, Fulda S, Gessler M, Wegert J, Kappler R, Baumhoer D, Burdach S, Kirschner-Schwabe R, Kontny U, Kulozik AE, Lohmann D, Hettmer S, Eckert C, Bielack S, Nathrath M, Niemeyer C, Richter GH, Schulte J, Siebert R, Westermann F, Molenaar JJ, Vassal G, Witt H, Project IP-S, Project IM-S, Burkhardt B, Kratz CP, Witt O, van Tilburg CM, Kramm CM, Fleischhack G, Dirksen U, Rutkowski S, Fruhwald M, von Hoff K, Wolf S, Klingebiel T, Koscielniak E, Landgraf P, Koster J, Resnick AC, Zhang J, Liu Y, Zhou X, Waanders AJ, Zwijnenburg DA, Raman P, Brors B, Weber UD, Northcott PA, Pajtler KW, Kool M, Piro RM, Korbel JO, Schlesner M, Eils R, Jones DTW, Lichter P, Chavez L, Zapatka M, Pfister SM (2018) The landscape of genomic alterations across childhood cancers. Nature 555:321–327. https://doi.org/10.1038/nature25480
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hartwig FP, Davey Smith G, Bowden J (2017) Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 46:1985–1998. https://doi.org/10.1093/ije/dyx102
Hemani G, Bowden J, Davey Smith G (2018) Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet 27:R195–R208. https://doi.org/10.1093/hmg/ddy163
Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR (2012) Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44:955–959. https://doi.org/10.1038/ng.2354
Kilday JP, Rahman R, Dyer S, Ridley L, Lowe J, Coyle B, Grundy R (2009) Pediatric ependymoma: biological perspectives. Mol Cancer Res 7:765–786. https://doi.org/10.1158/1541-7786.MCR-08-0584
Koelsche C, Sahm F, Capper D, Reuss D, Sturm D, Jones DT, Kool M, Northcott PA, Wiestler B, Bohmer K, Meyer J, Mawrin C, Hartmann C, Mittelbronn M, Platten M, Brokinkel B, Seiz M, Herold-Mende C, Unterberg A, Schittenhelm J, Weller M, Pfister S, Wick W, Korshunov A, von Deimling A (2013) Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol 126:907–915. https://doi.org/10.1007/s00401-013-1195-5
Marchini J, Howie B, Myers S, McVean G, Donnelly P (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39:906–913. https://doi.org/10.1038/ng2088
Martin AR, Gignoux CR, Walters RK, Wojcik GL, Neale BM, Gravel S, Daly MJ, Bustamante CD, Kenny EE (2017) Human demographic history impacts genetic risk prediction across diverse populations. Am J Hum Genet 100:635–649. https://doi.org/10.1016/j.ajhg.2017.03.004
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, Luo Y, Sidore C, Kwong A, Timpson N, Koskinen S, Vrieze S, Scott LJ, Zhang H, Mahajan A, Veldink J, Peters U, Pato C, van Duijn CM, Gillies CE, Gandin I, Mezzavilla M, Gilly A, Cocca M, Traglia M, Angius A, Barrett JC, Boomsma D, Branham K, Breen G, Brummett CM, Busonero F, Campbell H, Chan A, Chen S, Chew E, Collins FS, Corbin LJ, Smith GD, Dedoussis G, Dorr M, Farmaki AE, Ferrucci L, Forer L, Fraser RM, Gabriel S, Levy S, Groop L, Harrison T, Hattersley A, Holmen OL, Hveem K, Kretzler M, Lee JC, McGue M, Meitinger T, Melzer D, Min JL, Mohlke KL, Vincent JB, Nauck M, Nickerson D, Palotie A, Pato M, Pirastu N, McInnis M, Richards JB, Sala C, Salomaa V, Schlessinger D, Schoenherr S, Slagboom PE, Small K, Spector T, Stambolian D, Tuke M, Tuomilehto J, Van den Berg LH, Van Rheenen W, Volker U, Wijmenga C, Toniolo D, Zeggini E, Gasparini P, Sampson MG, Wilson JF, Frayling T, de Bakker PI, Swertz MA, McCarroll S, Kooperberg C, Dekker A, Altshuler D, Willer C, Iacono W, Ripatti S, Soranzo N, Walter K, Swaroop A, Cucca F, Anderson CA, Myers RM, Boehnke M, McCarthy MI, Durbin R, Haplotype Reference C (2016) A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48:1279–1283. https://doi.org/10.1038/ng.3643
Michealraj KA, Kumar SA, Kim LJY, Cavalli FMG, Przelicki D, Wojcik JB, Delaidelli A, Bajic A, Saulnier O, MacLeod G, Vellanki RN, Vladoiu MC, Guilhamon P, Ong W, Lee JJY, Jiang Y, Holgado BL, Rasnitsyn A, Malik AA, Tsai R, Richman CM, Juraschka K, Haapasalo J, Wang EY, De Antonellis P, Suzuki H, Farooq H, Balin P, Kharas K, Van Ommeren R, Sirbu O, Rastan A, Krumholtz SL, Ly M, Ahmadi M, Deblois G, Srikanthan D, Luu B, Loukides J, Wu X, Garzia L, Ramaswamy V, Kanshin E, Sanchez-Osuna M, El-Hamamy I, Coutinho FJ, Prinos P, Singh S, Donovan LK, Daniels C, Schramek D, Tyers M, Weiss S, Stein LD, Lupien M, Wouters BG, Garcia BA, Arrowsmith CH, Sorensen PH, Angers S, Jabado N, Dirks PB, Mack SC, Agnihotri S, Rich JN, Taylor MD (2020) Metabolic regulation of the epigenome drives lethal infantile ependymoma. Cell. https://doi.org/10.1016/j.cell.2020.04.047
Muskens IS, Hansen HM, Smirnov IV, Molinaro AM, Bondy ML, Schildkraut JM, Wrensch M, Wiemels JL, Claus EB (2019) Longer genotypically-estimated leukocyte telomere length is associated with increased meningioma risk. J Neurooncol 142:479–487. https://doi.org/10.1007/s11060-019-03119-w
O’Sullivan RJ, Karlseder J (2010) Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol 11:171–181. https://doi.org/10.1038/nrm2848
Ojha J, Codd V, Nelson CP, Samani NJ, Smirnov IV, Madsen NR, Hansen HM, de Smith AJ, Bracci PM, Wiencke JK, Wrensch MR, Wiemels JL, Walsh KM, Group ECT (2016) Genetic variation associated with longer telomere length increases risk of chronic lymphocytic leukemia. Cancer Epidemiol Biomark Prev 25:1043–1049. https://doi.org/10.1158/1055-9965.EPI-15-1329
Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA, Turner MC, Walsh KM, Wrensch MR, Barnholtz-Sloan JS (2014) The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol 16:896–913. https://doi.org/10.1093/neuonc/nou087
Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C, Barnholtz-Sloan JS (2019) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol 21:v1–v100. https://doi.org/10.1093/neuonc/noz150
Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, Wani K, Tatevossian R, Punchihewa C, Johann P, Reimand J, Warnatz HJ, Ryzhova M, Mack S, Ramaswamy V, Capper D, Schweizer L, Sieber L, Wittmann A, Huang Z, van Sluis P, Volckmann R, Koster J, Versteeg R, Fults D, Toledano H, Avigad S, Hoffman LM, Donson AM, Foreman N, Hewer E, Zitterbart K, Gilbert M, Armstrong TS, Gupta N, Allen JC, Karajannis MA, Zagzag D, Hasselblatt M, Kulozik AE, Witt O, Collins VP, von Hoff K, Rutkowski S, Pietsch T, Bader G, Yaspo ML, von Deimling A, Lichter P, Taylor MD, Gilbertson R, Ellison DW, Aldape K, Korshunov A, Kool M, Pfister SM (2015) Molecular classification of ependymal tumors across All CNS Compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. https://doi.org/10.1016/j.ccell.2015.04.002
Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, Davey Smith G, Sterne JA (2012) Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res 21:223–242. https://doi.org/10.1177/0962280210394459
Pierce BL, Ahsan H, Vanderweele TJ (2011) Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol 40:740–752. https://doi.org/10.1093/ije/dyq151
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575. https://doi.org/10.1086/519795
Richardson TG, Harrison S, Hemani G, Davey Smith G (2019) An atlas of polygenic risk score associations to highlight putative causal relationships across the human phenome. Elife 8:e43657. https://doi.org/10.7554/eLife.43657
Ridley L, Rahman R, Brundler MA, Ellison D, Lowe J, Robson K, Prebble E, Luckett I, Gilbertson RJ, Parkes S, Rand V, Coyle B, Grundy RG, Children’s C, Leukaemia Group Biological Studies C (2008) Multifactorial analysis of predictors of outcome in pediatric intracranial ependymoma. Neuro Oncol 10:675–689. https://doi.org/10.1215/15228517-2008-036
Schooling CM, Au Yeung SL, Freeman G (2013) Mendelian randomization estimates may be inflated. J Am Coll Cardiol 61:1931. https://doi.org/10.1016/j.jacc.2012.12.049
Semmes EC, Zhang C, Walsh KM (2018) Intermediate phenotypes underlying osteosarcoma risk. Oncotarget 9:37345–37346. https://doi.org/10.18632/oncotarget.26476
Stathopoulou MG, Petrelis AM, Buxton JL, Froguel P, Blakemore AI, Visvikis-Siest S (2015) Genetic determinants of leucocyte telomere length in children: a neglected and challenging field. Paediatr Perinat Epidemiol 29:146–150. https://doi.org/10.1111/ppe.12173
Tabori U, Ma J, Carter M, Zielenska M, Rutka J, Bouffet E, Bartels U, Malkin D, Hawkins C (2006) Human telomere reverse transcriptase expression predicts progression and survival in pediatric intracranial ependymoma. J Clin Oncol 24:1522–1528. https://doi.org/10.1200/JCO.2005.04.2127
Tabori U, Wong V, Ma J, Shago M, Alon N, Rutka J, Bouffet E, Bartels U, Malkin D, Hawkins C (2008) Telomere maintenance and dysfunction predict recurrence in paediatric ependymoma. Br J Cancer 99:1129–1135. https://doi.org/10.1038/sj.bjc.6604652
Telomeres Mendelian Randomization C, Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, Bowden J, Wade KH, Timpson NJ, Evans DM, Willeit P, Aviv A, Gaunt TR, Hemani G, Mangino M, Ellis HP, Kurian KM, Pooley KA, Eeles RA, Lee JE, Fang S, Chen WV, Law MH, Bowdler LM, Iles MM, Yang Q, Worrall BB, Markus HS, Hung RJ, Amos CI, Spurdle AB, Thompson DJ, O’Mara TA, Wolpin B, Amundadottir L, Stolzenberg-Solomon R, Trichopoulou A, Onland-Moret NC, Lund E, Duell EJ, Canzian F, Severi G, Overvad K, Gunter MJ, Tumino R, Svenson U, van Rij A, Baas AF, Bown MJ, Samani NJ, van t’Hof FNG, Tromp G, Jones GT, Kuivaniemi H, Elmore JR, Johansson M, McKay J, Scelo G, Carreras-Torres R, Gaborieau V, Brennan P, Bracci PM, Neale RE, Olson SH, Gallinger S, Li D, Petersen GM, Risch HA, Klein AP, Han J, Abnet CC, Freedman ND, Taylor PR, Maris JM, Aben KK, Kiemeney LA, Vermeulen SH, Wiencke JK, Walsh KM, Wrensch M, Rice T, Turnbull C, Litchfield K, Paternoster L, Standl M, Abecasis GR, SanGiovanni JP, Li Y, Mijatovic V, Sapkota Y, Low SK, Zondervan KT, Montgomery GW, Nyholt DR, van Heel DA, Hunt K, Arking DE, Ashar FN, Sotoodehnia N, Woo D, Rosand J, Comeau ME, Brown WM, Silverman EK, Hokanson JE, Cho MH, Hui J, Ferreira MA, Thompson PJ, Morrison AC, Felix JF, Smith NL, Christiano AM, Petukhova L, Betz RC, Fan X, Zhang X, Zhu C, Langefeld CD, Thompson SD, Wang F, Lin X, Schwartz DA, Fingerlin T, Rotter JI, Cotch MF, Jensen RA, Munz M, Dommisch H, Schaefer AS, Han F, Ollila HM, Hillary RP, Albagha O, Ralston SH, Zeng C, Zheng W, Shu XO, Reis A, Uebe S, Huffmeier U, Kawamura Y, Otowa T, Sasaki T, Hibberd ML, Davila S, Xie G, Siminovitch K, Bei JX, Zeng YX, Forsti A, Chen B, Landi S, Franke A, Fischer A, Ellinghaus D, Flores C, Noth I, Ma SF, Foo JN, Liu J, Kim JW, Cox DG, Delattre O, Mirabeau O, Skibola CF, Tang CS, Garcia-Barcelo M, Chang KP, Su WH, Chang YS, Martin NG, Gordon S, Wade TD, Lee C, Kubo M, Cha PC, Nakamura Y, Levy D, Kimura M, Hwang SJ, Hunt S, Spector T, Soranzo N, Manichaikul AW, Barr RG, Kahali B, Speliotes E, Yerges-Armstrong LM, Cheng CY, Jonas JB, Wong TY, Fogh I, Lin K, Powell JF, Rice K, Relton CL, Martin RM, Davey Smith G (2017) Association between telomere length and risk of cancer and non-neoplastic diseases: a Mendelian randomization study. JAMA Oncol 3:636–651. https://doi.org/10.1001/jamaoncol.2016.5945
Walsh KM, Codd V, Rice T, Nelson CP, Smirnov IV, McCoy LS, Hansen HM, Elhauge E, Ojha J, Francis SS, Madsen NR, Bracci PM, Pico AR, Molinaro AM, Tihan T, Berger MS, Chang SM, Prados MD, Jenkins RB, Wiemels JL, Group ECT, Samani NJ, Wiencke JK, Wrensch MR (2015) Longer genotypically-estimated leukocyte telomere length is associated with increased adult glioma risk. Oncotarget 6:42468–42477. https://doi.org/10.18632/oncotarget.6468
Walsh KM, Rice T, Decker PA, Kosel ML, Kollmeyer T, Hansen HM, Zheng S, McCoy LS, Bracci PM, Anderson E, Hsuang G, Wiemels JL, Pico AR, Smirnov I, Molinaro AM, Tihan T, Berger MS, Chang SM, Prados MD, Lachance DH, Sicotte H, Eckel-Passow JE, Wiencke JK, Jenkins RB, Wrensch MR (2013) Genetic variants in telomerase-related genes are associated with an older age at diagnosis in glioma patients: evidence for distinct pathways of gliomagenesis. Neuro Oncol 15:1041–1047. https://doi.org/10.1093/neuonc/not051
Walsh KM, Whitehead TP, de Smith AJ, Smirnov IV, Park M, Endicott AA, Francis SS, Codd V, Group ECT, Samani NJ, Metayer C, Wiemels JL (2016) Common genetic variants associated with telomere length confer risk for neuroblastoma and other childhood cancers. Carcinogenesis 37:576–582. https://doi.org/10.1093/carcin/bgw037
Walsh KM, Wiencke JK, Lachance DH, Wiemels JL, Molinaro AM, Eckel-Passow JE, Jenkins RB, Wrensch MR (2015) Telomere maintenance and the etiology of adult glioma. Neuro Oncol 17:1445–1452. https://doi.org/10.1093/neuonc/nov082
Wiemels JL, Walsh KM, de Smith AJ, Metayer C, Gonseth S, Hansen HM, Francis SS, Ojha J, Smirnov I, Barcellos L, Xiao X, Morimoto L, McKean-Cowdin R, Wang R, Yu H, Hoh J, DeWan AT, Ma X (2018) GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat Commun 9:286. https://doi.org/10.1038/s41467-017-02596-9
Wong VC, Morrison A, Tabori U, Hawkins CE (2010) Telomerase inhibition as a novel therapy for pediatric ependymoma. Brain Pathol 20:780–786. https://doi.org/10.1111/j.1750-3639.2010.00372.x
Yavorska OO, Burgess S (2017) MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol 46:1734–1739. https://doi.org/10.1093/ije/dyx034
Zhang C, Hansen HM, Semmes EC, Gonzalez-Maya J, Morimoto L, Wei Q, Eward WC, DeWitt SB, Hurst JH, Metayer C, de Smith AJ, Wiemels JL, Walsh KM (2020) Common genetic variation and risk of osteosarcoma in a multi-ethnic pediatric and adolescent population. Bone 130:115070. https://doi.org/10.1016/j.bone.2019.115070
Zhang C, Morimoto LM, de Smith AJ, Hansen HM, Gonzalez-Maya J, Endicott AA, Smirnov IV, Metayer C, Wei Q, Eward WC, Wiemels JL, Walsh KM (2018) Genetic determinants of childhood and adult height associated with osteosarcoma risk. Cancer 124:3742–3752. https://doi.org/10.1002/cncr.31645
Zhang C, Ostrom QT, Hansen HM, Gonzalez-Maya J, Hu D, Ziv E, Morimoto L, de Smith AJ, Muskens IS, Kline CN, Vaksman Z, Hakonarson H, Diskin SJ, Kruchko C, Barnholtz-Sloan JS, Ramaswamy V, Ali-Osman F, Bondy ML, Taylor MD, Metayer C, Wiemels JL, Walsh KM (2020) European genetic ancestry associated with risk of childhood ependymoma. Neuro Oncol. https://doi.org/10.1093/neuonc/noaa130
Acknowledgements
The biospecimens and/or data used in this study were obtained from the California Biobank Program, (SIS request numbers #597 and #311)” Section 6555(b), 17 CCR. The California Department of Public Health is not responsible for the results or conclusions drawn by the authors of this publication. This study used birth-year, newborn sex, and parental race/ethnicity data obtained from the State of California Center for Health Statistics and Informatics. The California Department of Public Health is not responsible for the analyses, interpretations, or conclusions drawn by the authors regarding the birth-year, sex, or parental race/ethnicity data used in this publication. The collection of cancer incidence data used in this study was supported by the California Department of Public Health pursuant to California Health and Safety Code Section 103885; CDC’s National Program of Cancer Registries, under cooperative agreement 5NU58DP006344; the National Cancer Institute’s Surveillance, Epidemiology and End Results Program under contract HHSN261201800032I awarded to the University of California, San Francisco, contract HHSN261201800015I awarded to the University of Southern California, and contract HHSN261201800009I awarded to the Public Health Institute, Cancer Registry of Greater California. The ideas and opinions expressed herein are those of the author(s) and do not necessarily reflect the opinions of the State of California, Department of Public Health, the NCI, and the CDC or their Contractors and Subcontractors.
Funding
This work was supported by the Pediatric Brain Tumor Foundation (KMW), The Sontag Foundation (KMW), The National Institutes of Health T32 CA151022-06 (CZ), R01 CA194189 (KMW, JLW), P50 CA190991 (KMW), R01 CA155461 (JLW), ‘A’ Awards from The Alex’s Lemonade Stand Foundation (AJdS, KMW), and the Cancer Prevention and Research Institute of Texas RP160097T (QTO). The GICC was supported by grants from the National Institutes of Health (R01CA139020, R01CA52689, P50097257, and P30CA125123). Additional support was provided by the McNair Medical Institute and the Population Sciences Biorepository at Baylor College of Medicine.
Author information
Authors and Affiliations
Consortia
Contributions
Conception or design of the work (CZ, MDT, VR, KMW), acquisition, analysis, or interpretation of data (CZ, QTO, ECS, HMH, LM, AJdS, MP, ZV, HH, SJD, CM, GICC, MDT, JLW, MLB, KMW), drafted the work or substantively revised it (CZ, ECS, AJdS, CM, JLW, KMW). All authors have approved the submitted version and have agreed both to be personally accountable for the author’s own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics statement
The study was approved by Institutional Review Boards at The University of California, Berkeley, The University of California, San Francisco, the California Department of Public Health, The Children’s Hospital of Philadelphia, the University of Toronto Hospital for Sick Children, and Baylor College of Medicine.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests or conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The Glioma International Case-Control Study (GICC): Complete GICC membership listed in Supplementary Note.
Supplementary information
Additional file 1:
Supplementary note: The Glioma International Case Control Study membership. Supplementary Table 1. Demographic and clinical data for pediatric ependymoma cases (ages 0-19) from the California Cancer Record Linkage Project (CCRLP). Supplementary Table 2. Single SNP associations between LTL-associated variants and ependymoma risk in the CCRLP case-control dataset, stratified by age at diagnosis. Supplementary Table 3. Single SNP associations between LTL-associated variants and ependymoma risk in the Toronto and GICC case-control datasets. Supplementary Table 4. Association between polygenic score for longer leukocyte telomere length and risk of ependymoma in the California Cancer Records Linkage Project (CCRLP)a, stratified by tumor site (spinal vs. intracranial) and age (<12, ≥12). Associations are adjusted for sex and first 10 principal components
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, C., Ostrom, Q.T., Semmes, E.C. et al. Genetic predisposition to longer telomere length and risk of childhood, adolescent and adult-onset ependymoma. acta neuropathol commun 8, 173 (2020). https://doi.org/10.1186/s40478-020-01038-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-020-01038-w