
Abstract
Autosomal dominant optic atrophy (ADOA) is a neuro-ophthalmic condition characterized by bilateral degeneration of the optic nerves. Although heterozygous mutations in OPA1 represent the most common genetic cause of ADOA, a significant number of cases remain undiagnosed.
Here, we describe a family with a strong ADOA history with most family members spanning three generation having childhood onset of visual symptoms. The proband, in addition to optic atrophy, had neurological symptoms consistent with relapsing remitting multiple sclerosis. Clinical exome analysis detected a novel mutation in the AFG3L2 gene (NM_006796.2:c.1010G > A; p.G337E), which segregated with optic atrophy in family members. AFG3L2 is a metalloprotease of the AAA subfamily which exerts quality control in the inner mitochondrial membrane. Interestingly, the identified mutation localizes close to the AAA domain of AFG3L2, while those localized in the proteolytic domain cause dominant spinocerebellar ataxia type 28 (SCA28) or recessive spastic ataxia with epilepsy (SPAX5). Functional studies in patient fibroblasts demonstrate that the p.G337E AFG3L2 mutation strongly destabilizes the long isoforms of OPA1 via OMA hyper-activation and leads to mitochondrial fragmentation, thus explaining the family phenotype. This study widens the clinical spectrum of neurodegenerative diseases caused by AFG3L2 mutations, which shall be considered as genetic cause of ADOA.
Similar content being viewed by others

Introduction
ADOA is a genetic condition affecting the retinal ganglion cells (RGCs), whose axons form the optic nerve. It is a relatively common form of inherited optic neuropathy, with a prevalence of 3/100,000 in most populations worldwide. Patients are usually diagnosed during early childhood, because of bilateral visual loss related to optic disc pallor or atrophy, and typically in the context of a family history of ADOA.
Molecular diagnosis is provided by the identification of a mutation in the OPA1 gene (75% of ADOA patients) or in the OPA3 gene (1% of patients) [1]. However, many ADOA cases remain undiagnosed [2].
About 20% of patients with OPA1 mutations are known to develop additional co-morbidities of deafness, ophthalmoplegia, ataxia, myopathy and peripheral neuropathy [1].
OPA1 is a mitochondrial GTPase responsible for the fusion of the inner mitochondrial membrane (IMM), by which it regulates mitochondrial dynamics. OPA1 is transcribed in eight splicing isoforms which undergo constitutive processing in the IMM. At steady state, OMA1-operated cleavage at S1 site and/or YME1L1-operated cleavage at S2 site lead to the generation of long- (non-cleaved) and short- (cleaved) forms of OPA1 [3]. OPA1 long forms (L-OPA1) are the active mediators of mitochondrial fusion, and their processing to short, soluble forms (S-OPA1) limits fusion and can facilitate mitochondrial fragmentation [4, 5].
OPA1 processing is finely regulated by AFG3L2, a mitochondrial protein belonging to the AAA-protease subfamily (ATPases associated with various cellular activities) which exerts quality control in the IMM [6]. We previously demonstrated that loss of AFG3L2 induces hyper-activation of the stress-activated protease OMA1 and leads to excessive OPA1 processing, promoting mitochondrial fragmentation [7]. Mutations in the proteolytic domain of AFG3L2 cause dominant SCA28 and the rare recessive SPAX5, whose main clinical features are gait ataxia and lack of balance with cerebellar atrophy [8, 9].
In this report, we describe a family with ADOA. By clinical exome analysis we identified a novel mutation in AFG3L2 (p.G337E) segregating with optic atrophy within the family. In contrast to SCA28 causing-mutations, which mostly affect the proteolytic domain, this new mutation localizes close to the AAA domain of AFG3L2. Functional studies demonstrate that the p.G337E mutation abolishes AFG3L2 function and leads to a striking L-OPA1 destabilization, comparable to the one observed in Afg3l2 null cells. Our data disclose OMA1 hyper-activation, OPA1 enhanced processing and mitochondrial fragmentation as the pathogenic cascade of ADOA caused by AFG3L2 p.G337E mutation.
Case presentation
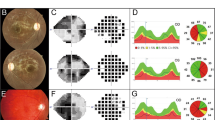
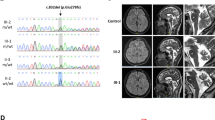
The proband was diagnosed with optic atrophy aged 4, when he was found to have reduced vision (right 3/60, left 2/60), poor color perception with Ishihara testing and mild optic atrophy. Electrophysiology investigation revealed poor amplitudes with visual evoked potentials and a normal electroretinogram. Brain Magnetic Resonance Imaging (MRI) at the age of 5 was normal. Optic atrophy slowly worsened with age, showing marked optic nerve pallor aged 20 (Fig. 1a). The proband also presented with an acute episode of cerebellar ataxia at the age of 18 and was diagnosed with relapsing remitting multiple sclerosis (MS). He fulfilled the McDonald criteria for diagnosis of MS and brain MRI demonstrated widespread demyelinating lesions in both cerebral, cerebellar hemispheres as well as the midbrain and cord (Fig. 1b). His cerebrospinal fluid (CSF) analysis showed oligoclonal bands. Anti-aquaporin 4 antibodies (Neuromyelitis optica-Immunoglobulin G - NMO IgG) testing was negative. His symptoms improved after plasma exchange and he is now stable on monthly Natalizumab infusions. There was a known history of mild ADOA in this family, with proband’s mother, maternal grandfather and multiple other maternal relatives being affected by optic atrophy but able to drive, with vision of at least 6/12. The proband’s younger brother was found to have a similar severe level of vision and optic atrophy aged 5 (Fig. 1c). None of the family members had symptoms of spinocerebellar ataxia.

Family clinical features and pedigree. a Fundus photos of proband age 20 showing bilateral optic nerve atrophy. b MRI Brain demonstrating numerous T2/FLAIR hyperintense lesions predominantly involving the periventricular white matter and the grey-white matter junction. c Pedigree demonstrating clear autosomal dominant inheritance of optic atrophy. The arrow indicates the proband. d AFG3L2 protein scheme with functional domains, reporting the mutation described here. e p.G337 AFG3L2 residue conservation among different AFG3L2 orthologues
Genetic testing
We identified a heterozygous missense mutation NM_006796.2(AFG3L2):c.1010G > A in exon 8 of the AFG3L2 gene in a family member with optic atrophy. This is a novel mutation, not reported in population databases such as gnomAD or in clinical cases, resulting in glycine to glutamic acid position 337 NP_006787.2(AFG3L2):p.G337E. This mutation segregates with optic atrophy in five family members in total and followed an autosomal dominant pattern of inheritance (Fig. 1c and d). p.G337E is very highly conserved and in silico softwares consistently predict it to be pathogenic (Fig. 1e).
Functional studies
To functionally assay the pathogenicity of the p.G337E mutation, we mutagenized an AFG3L2 WT-myc construct to obtain AFG3L2G337E-myc and overexpressed it in Afg3l2−/− MEFs. We analyzed by WB the levels and post translational processing of OPA1. In Afg3l2−/− MEFs OPA1 is processed at a higher rate compared to wt controls, leading to a striking reduction of L-OPA1, which inhibits fusion and triggers mitochondrial network fragmentation [7, 10]. We found that overexpression of AFG3L2G337E-myc does not restore, even partially, L-OPA1 in Afg3l2−/− MEFs. Conversely, L-OPA1 are recovered by AFG3L2 WT-myc overexpression, indicating that the p.G337E mutation completely abolishes AFG3L2 activity (Fig. 2a).

Overexpression of exogenous AFG3L2G337E-myc in a AFG3L2 null background does not rescue L-OPA1 and mitochondrial tubulation. a WB analysis and relative quantification of L-OPA1 after transient transfection of mt-YFP in combination with AFG3L2 WT-myc or AFG3L2G337E-myc in Afg3l2+/+ and Afg3l2−/− MEFs (ratio 1:3). c-MYC was used as transfection control. Bars represent means ± SEM of three independent experiments. Student’s t test: * p < 0.05; ** p < 0.01; *** p < 0.001. b Representative pictures of in live mitochondrial morphology after transient transfection of mt-YFP in combination with AFG3L2 WT-myc or AFG3L2G337E-myc in Afg3l2+/+ and Afg3l2−/− MEFs (ratio 1:3). The graph shows the morphometric analysis of mitochondrial morphology. At least 80 randomly selected cells were analyzed in each experiment. Chi-square analysis (two degrees of freedom): *** p < 0.001
We then analyzed mitochondrial morphology in Afg3l2−/− MEFs transfected with mt-YFP alone, or in combination with AFG3L2G337E-myc or AFG3L2 WT-myc. Strikingly, and in accordance with L-OPA1 enhanced processing, the mitochondrial network was comparable between Afg3l2−/− MEFs and Afg3l2−/− MEFs transfected with AFG3L2G337E-myc, with the highest percentage of cells showing fragmented mitochondria. Conversely, the overexpression of AFG3L2 WT-myc was able to restore mitochondrial tubulation in Afg3l2−/− MEFs (Fig. 2b).
We then evaluated the effect of the p.G337E mutation in a more physiopathologic context, by analyzing primary fibroblasts from the proband (III-2) and his affected mother (II-2). We firstly assayed by WB the effect of the p.G337E mutation on the stability of AFG3L2 monomer and we found that the whole amount of AFG3L2 is comparable between patient and control fibroblasts (Fig. 3a). On the contrary, we observed that L-OPA1 are strongly destabilized by an enhanced processing in patient fibroblasts compared to controls, leading to a striking reduction of L-OPA1 and to an accumulation of S-OPA1 (Fig. 3b). We also proved that this is due to the hyper-activation of OMA1, as demonstrated by a significant reduction in its amount when compared to controls (Fig. 3b). Indeed, we previously showed that OMA1 is hyper-activated in the absence of AFG3L2 and undergoes autocatalytic degradation [7, 11]. We also evaluated possible compensatory effects of YME1L1, but the levels of this protease resulted comparable between patients and controls (Fig. 3b).

Patient fibroblasts show enhanced L-OPA1 processing via OMA1 hyper-activation. WB analysis and relative quantification of (a) AFG3L2 and (b) OPA1 total amount, YME1L1, L-OPA1, S-OPA1, OMA1 in human primary fibroblasts. G337E_1 is the proband and G337E_2 is patient II-2 of the pedigree. Bars represent means ± SEM of three independent experiments. Student’s t test: * p < 0.05; ** p < 0.01; *** p < 0.001. (c) Representative pictures of mitochondrial morphology in human primary fibroblasts infected with mtDsRed2 and visualized by live imaging microscopy. The graph shows the morphometric analysis of mitochondrial morphology. At least 100 randomly selected cells were analyzed in each experiment. Chi-square analysis (two degrees of freedom): *** p < 0.001
Live imaging evaluation of mitochondrial network showed a highly fused and interconnected mitochondria in control cells, while patient fibroblasts carrying the p.G337E mutation presented shorter and more isolated organelles, consistent with OPA1 abnormal processing (Fig. 3c).
Discussion and conclusions
Here we describe a family in which a novel heterozygous mutation in AFG3L2 co-segregates with ADOA. Interestingly, the identified p.G337E mutation localizes close to the AAA domain of AFG3L2, in contrast with those causing SCA28 and SPAX5, which clusterize in the proteolytic domain of the protein. Functional studies of the p.G337E mutation demonstrate its pathogenicity, as it strongly affects the processing of L-OPA1 leading to aberrant mitochondrial fragmentation. The defective mitochondrial dynamics is in line with most optic neuropathies exhibiting mitochondrial dysfunction in RGCs as underlying mechanism [12].
Other heterozygous/compound heterozygous mutations in AFG3L2 have been described in isolated cases with nonsyndromic optic atrophy (although not supported by functional studies) [13, 14], and in familial syndromic and non-syndromic optic atrophy [15, 16]. The present study further demonstrates that heterozygous mutations in AFG3L2 shall be considered as a genetic cause for ADOA in OPA1 and OPA3- negative cases.
The family described here has features of ADOA with no clinical evidence of spinocerebellar ataxia in any of the affected members but the proband, who experienced an acute episode of cerebellar ataxia. As proband’s clinical features, MRI and CSF investigations were consistent with relapsing remitting MS and the demyelinating lesions (including the cerebellar lesions) settled on treatment with Natalizumab, his cerebellar symptoms were most likely due to MS. However, it remains to be determined if these demyelinating neurological symptoms could be directly related to the AFG3L2 mutation. Although mouse studies have shown that deletion of AFG3L2 (either constitutive or in mature mouse oligodendrocytes) can cause myelin abnormalities [17, 18], no such phenotype has been described in humans before. Comorbidity cannot therefore be excluded in the proband.
The functional studies we conducted on the p.G337E mutation clearly prove its pathogenicity. Exogenous expression of p.G337E AFG3L2 in an Afg3l2 null background indicates that the mutant protein has no residual function. Indeed, the complete loss of L-OPA1 and mitochondrial fragmentation in this condition were comparable to those previously observed in Afg3l2 null MEFs [10]. In agreement, we found swollen mitochondria with altered cristae in the optic nerve of Afg3l2 null mice (VB and FM unpublished observation). Investigations in patient fibroblasts also revealed faster abnormal processing of OPA1 despite the heterozygous state of the mutation, with strong reduction of L-OPA1, accumulation of S-OPA1 and altered mitochondrial fusion. Interestingly, the decrease in L-OPA1 is significantly more pronounced in the proband compared to his affected mother, in line with the more severe phenotype, indicating that OPA1 processing might be considered as an outcome of disease severity in this form of ADOA. We also demonstrated that OPA1 processing is caused by strong OMA1 hyper-activation, which is reduced in amount in patients versus controls because of its faster autocatalysis. On the contrary, YME1L1 levels were not altered in patients, thus excluding a compensatory upregulation of YME1L1 on the final outcome on mitochondrial dynamics.
OPA1 processing is more severely compromised in this family compared to what we previously described in SCA28 and SPAX5-patient fibroblasts, where the levels of L-OPA1 were moderately reduced compared to controls, the accumulation of S-OPA1 was not appreciated, and the mitochondrial network presented shorter tubules, but not evident fragmentation [7]. Interestingly, the mutation we identified localizes close to the AAA domain, while most of those associated with SCA28 or SPAX5 affect the proteolytic domain, suggesting that mutations in different domains of this protein could differently affect its molecular function. Mutations localizing in the AAA domain of AFG3L2 can abolish ATP binding/hydrolysis and impact more severely on proteolytic activity, in agreement with a recent work in which ATPase and proteolytic activity of AFG3L2 carrying different mutations were assessed in vitro [19].
SCA28 and SPAX5 predominantly affect the cerebellum, while this novel AFG3L2 mutation predominantly affects the optic nerve. The aberrant OPA1 processing and severe mitochondrial fragmentation we observed, together with the fact that most ADOA patients carry OPA1 mutations, indicates that a fine control of mitochondrial dynamics is crucial for RGC survival. We may speculate that AFG3L2 mutations that predominantly and severely impact on OPA1 processing affect specifically RGCs, while those mostly impinging on other AFG3L2-related functions (oxidative phosphorylation and mitochondrial calcium homeostasis) affect Purkinje neurons in the cerebellum [10, 20, 21]. Purkinje neurons are selectively vulnerable to these defects, since they are characterized by a high oxidative metabolism and experience elevated calcium fluxes due to massive glutamatergic stimulations [22, 23].
In conclusion, our study broadens the spectrum of neurodegenerative diseases associated with AFG3L2 mutations and expands the genetic causes leading to ADOA, enforcing aberrant OPA1 processing as common mechanism for this disease.
Availability of data and materials
All data generated or analyzed during this study are included in this article.
Abbreviations
- AAA:
-
ATPase associated with a variety of cellular activities
- ADOA:
-
Autosomal dominant optic atrophy
- CSF:
-
Cerebrospinal fluid
- IMM:
-
Inner mitochondrial membrane
- L-OPA:
-
Long OPA1 forms
- MEF:
-
Murine embryonic fibroblast
- MRI:
-
Magnetic resonance imaging
- MS:
-
Multiple sclerosis
- NMO-IgG:
-
Neuromyelitis optica-Immunoglobulin G
- RGC:
-
Retinal ganglion cell
- SCA28:
-
Spinocerebellar ataxia type 28
- SPAX5:
-
Spastic ataxia 5
- S-OPA1:
-
Short OPA1
- WB:
-
Western blot
- WT:
-
Wild type
- YFP:
-
Yellow fluorescent protein
References
Lenaers G et al (2012) Dominant optic atrophy. Orphanet J Rare Dis 7:46
Cohn AC et al (2007) Autosomal dominant optic atrophy: penetrance and expressivity in patients with OPA1 mutations. Am J Ophthalmol 143(4):656–662
MacVicar T, Langer T (2016) OPA1 processing in cell death and disease - the long and short of it. J Cell Sci 129(12):2297–2306
Anand R et al (2014) The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204(6):919–929
Ban T et al (2017) Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat Cell Biol 19(7):856–863
Quiros PM, Langer T, Lopez-Otin C (2015) New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol 16(6):345–359
Tulli S et al (2019) Pathogenic variants in the AFG3L2 proteolytic domain cause SCA28 through haploinsufficiency and proteostatic stress-driven OMA1 activation. J Med Genet 56(8):499–511
Di Bella D et al (2010) Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet 42(4):313–321
Pierson TM et al (2011) Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet 7(10):e1002325
Maltecca F et al (2012) Respiratory dysfunction by AFG3L2 deficiency causes decreased mitochondrial calcium uptake via organellar network fragmentation. Hum Mol Genet 21(17):3858–3870
Consolato F et al (2018) m-AAA and i-AAA complexes coordinate to regulate OMA1, the stress-activated supervisor of mitochondrial dynamics. J Cell Sci 131(7):jcs213546. https://doi.org/10.1242/jcs.213546. PMID: 29545505.
Pilz YL, Bass SJ, Sherman J (2017) A review of mitochondrial optic neuropathies: from inherited to acquired forms. J Optom 10(4):205–214
Charif M et al (2015) A novel mutation of AFG3L2 might cause dominant optic atrophy in patients with mild intellectual disability. Front Genet 6:311
Colavito D et al (2017) Non-syndromic isolated dominant optic atrophy caused by the p.R468C mutation in the AFG3 like matrix AAA peptidase subunit 2 gene. Biomed Rep 7(5):451–454
Magri S et al (2018) Concurrent AFG3L2 and SPG7 mutations associated with syndromic parkinsonism and optic atrophy with aberrant OPA1 processing and mitochondrial network fragmentation. Hum Mutat 39(12):2060–2071
Caporali L et al (2020) ATPase domain AFG3L2 mutations alter OPA1 processing and cause optic neuropathy. Ann Neurol https://doi.org/10.1002/ana.25723.
Maltecca F et al (2008) The mitochondrial protease AFG3L2 is essential for axonal development. J Neurosci 28(11):2827–2836
Wang S et al (2016) The mitochondrial m-AAA protease prevents demyelination and hair greying. PLoS Genet 12(12):e1006463
Puchades C et al (2019) Unique structural features of the mitochondrial AAA+ protease AFG3L2 reveal the molecular basis for activity in health and disease. Mol Cell 75(5):1073–1085 e6
Maltecca F et al (2015) Purkinje neuron Ca2+ influx reduction rescues ataxia in SCA28 model. J Clin Invest 125(1):263–274
Konig T et al (2016) The m-AAA protease associated with Neurodegeneration limits MCU activity in mitochondria. Mol Cell 64(1):148–162
Hartmann J, Konnerth A (2005) Determinants of postsynaptic Ca2+ signaling in Purkinje neurons. Cell Calcium 37(5):459–466
Patron M, Sprenger HG, Langer T (2018) M-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res 28(3):296–306
Acknowledgements
We thank the family for collaborating with this study. We thank the Medical photography Unit of the Royal Victorian Eye and Ear Hospital, Melbourne. We are also grateful to Martina Brughera for technical assistance.
Funding
This project was supported by Italian Ministery of Health # RF-2016-02361610 and National Ataxia Foundation (FM). The Centre for Eye Research Australia receives Operational Infrastructure Support from the Victorian Government.
Author information
Authors and Affiliations
Contributions
FM and AH conceived the study, interpreted the results and wrote the manuscript. JS assisted with writing the manuscript and was involved in clinical care of the family. MF and BAT performed exome sequencing. VB performed the functional experiments, analyzed data and helped writing the manuscript. JBR and LSK are family ophthalmologist and orthoptist, respectively. All authors critically discussed the data.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Subjects’ consent for the study was obtained according to the Declaration of Helsinki and was approved by the ethical committee of The Royal Melbourne Hospital.
Consent for publication
The patients signed a consent for publication approved by the ethical committee of The Royal Melbourne Hospital.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Baderna, V., Schultz, J., Kearns, L.S. et al. A novel AFG3L2 mutation close to AAA domain leads to aberrant OMA1 and OPA1 processing in a family with optic atrophy. acta neuropathol commun 8, 93 (2020). https://doi.org/10.1186/s40478-020-00975-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-020-00975-w