
Abstract
Last decade witnessed an enormous progress in generating authentic infectious prions or PrPSc in vitro using recombinant prion protein (rPrP). Previous work established that rPrP that lacks posttranslational modification is able to support replication of highly infectious PrPSc with assistance of cofactors of polyanionic nature and/or lipids. Unexpectedly, previous studies also revealed that seeding of rPrP by brain-derived PrPSc gave rise to new prion strains with new disease phenotypes documenting loss of a strain identity upon replication in rPrP substrate. Up to now, it remains unclear whether prion strain identity can be preserved upon replication in rPrP. The current study reports that faithful replication of hamster strain SSLOW could be achieved in vitro using rPrP as a substrate. We found that a mixture of phosphatidylethanolamine (PE) and synthetic nucleic acid polyA was sufficient for stable replication of hamster brain-derived SSLOW PrPSc in serial Protein Misfolding Cyclic Amplification (sPMCA) that uses hamster rPrP as a substrate. The disease phenotype generated in hamsters upon transmission of recombinant PrPSc produced in vitro was strikingly similar to the original SSLOW diseases phenotype with respect to the incubation time to disease, as well as clinical, neuropathological and biochemical features. Infrared microspectroscopy (IR-MSP) indicated that PrPSc produced in animals upon transmission of recombinant PrPSc is structurally similar if not identical to the original SSLOW PrPSc. The current study is the first to demonstrate that rPrP can support replication of brain-derived PrPSc while preserving its strain identity. In addition, the current work is the first to document that successful propagation of a hamster strain could be achieved in vitro using hamster rPrP.
Similar content being viewed by others

Introduction
Prion diseases or transmissible spongiform encephalopathies represent a class of lethal, transmissible neurodegenerative disorders of humans and animals [53]. The key event underlying prion diseases involves the conformational change of the α-helical, native, cellular form of the prion protein (PrPC) expressed by a host on a cell surface into a self-replicating, β-sheet rich, transmissible form (PrPSc) [52]. PrPC is posttranslationally modified with a glycophosphatidylinositol (GPI) anchor and up to two N-linked glycans; these modifications are carried over upon conversion of PrPC into PrPSc [58, 59, 62]. Prions spread between organisms or from cell to cell by recruiting host-encoded PrPC and replicating their disease-specific misfolded structures via a template-assisted mechanism [14]. According to this mechanism, PrPSc template recruits PrPC expressed by a host and converts them into a new PrPSc with the folding pattern faithfully repeating that of the PrPSc template [14]. While prions can propagate indefinitely via serial passaging in wild type hosts or cultured cells, generating infectious prions in vitro de novo from recombinant PrP (rPrP) has been a challenge [5].
In the absence of cellular cofactors, rPrP readily adopts self-propagating β-sheet rich states including amyloid fibrils [7, 8]. While rPrP amyloid fibrils propagate well in vitro [9], they display miniscule specific infectivity in animals, as they do not recruit PrPC effectively [15, 29, 39, 43]. When inoculated into wild type hosts, rPrP amyloid fibrils initiate a process of synthetic strain evolution that eventually lead to emergence of authentic PrPSc and clinical prion disease upon serial passaging [43,44,45,46]. Evolution of synthetic strains in vivo involves a transformation from the PrP folding patterns specific to rPrP amyloid fibrils, which do not accommodate well PrP with posttranslational modifications, to the folding pattern specific to PrPSc that can effectively recruit PrPC with GPI anchor and N-linked glycans [42].
In the past decade, several studies demonstrated that authentic PrPSc infectious to wild type host could be generated using rPrP in vitro with assistance of cofactors of polyanionic nature and/or lipids using serial protein misfolding cyclic amplification (sPMCA) [21, 22, 63, 67]. Ma and colleagues showed that mouse recombinant PrPSc, infectious to wild type mice, could be produced in sPMCA de novo using rPrP in the presence of the anionic phospholipid 1-palmitoyl-2-oleolyl-sn-glycero-3-phospho(1′-rac-glycerol) and total liver RNA [63, 67]. Supattapone and colleagues showed that phosphatidylethanolamine (PE) can be used as a sole cofactor for generating recombinant mouse PrPSc with high infectivity titers in sPMCA reactions seeded with mouse prion strains [21, 22]. Castilla and colleagues reported generation of several new strains in bank voles upon transmission of sPMCA-derived materials produced with assistance of several cofactors of polyanionic nature and bank vole rPrP as a substrate [23]. These studies established that highly infectious recombinant PrPSc could be generated from rPrP mixed with non-PrP components. In addition, these studies provided illustration that presence of different cofactors gave rise to different synthetic prion strains. However, it still remains unclear whether strain-specific features could be imposed on rPrP by brain-derived PrPSc seeds and, if so, can be faithfully replicated by rPrP, i.e. whether prion strain identity can be preserved by rPrP [60].
It has been well established that molecular features responsible for prion strain identity are well preserved when crude brain homogenates containing PrPC is used for replicating prion strains in sPMCA [12, 28]. However, this is not the case, when rPrP is used for in vitro conversion. Studies using quaking-induced conversion assay revealed lack of infectivity and strain identity in rPrP conversion products generated by seeding of rPrP with a diverse range of prion strains originating from a number of species [65]. Surprisingly, supplementing PE as a sole cofactor to mouse rPrP conversion assays restored high titer of prion infectivity, yet did not preserve strain identity [22]. In fact, seeding of rPrP and PE mixtures with three mouse strains gave rise to recombinant PrPSc that produced a new strain in wild type mice with the same diseases phenotype regardless of the original strain used for the seeding [22]. Moreover, similar results were obtained using hamster PrPC purified from hamster brains as a substrate and synthetic polyA as a sole cofactor [18]. Regardless of whether sPMCA reactions mixtures consisting of purified hamster PrPC and polyA were seeded with hamster strains Sc237 or 139H or conducted as non-seeded reactions, the newly produced PrPSc gave rise to the same disease phenotype in hamsters [18].
What are the minimal molecular requirements for a faithful replication of a prion strain in vitro? Can faithful replication of a prion strain be achieved using rPrP that lacks posttranslational modifications? What is the minimal set of cofactors sufficient for a faithful replication of a prion strain in vitro? Do prions from different species rely on different sets of cofactors? The current study reports that faithful replication of hamster strain SSLOW could be achieved in vitro using rPrP as a substrate. We found that a mixture of PE and polyA was sufficient for stable replication of hamster brain-derived SSLOW PrPSc in sPMCA that use hamster rPrP as a substrate. The disease phenotype generated in hamsters upon transmission of recombinant PrPSc produced in vitro was strikingly similar to the original SSLOW diseases phenotype with respect to the incubation time to disease, clinical, neuropathological, biochemical and structural features of PrPSc, as indicated by infrared microspectroscopy. The current study is the first to demonstrate that rPrP can support replication of brain-derived PrPSc while preserving its strain identity.
Materials and methods
Brain material
Hyper and Drowsy scrapie brain materials were kindly provided by Richard Bessen (Colorado State University, Fort Collins, CO); 263K was kindly provided by Robert Rohwer (Veterans Affair Maryland Health Care System, Baltimore, MD); one 263K scrapie hamster brain used for preparation and FT-IR analysis of highly purified PrPSc was taken from the prion tissue archive at the Robert Koch-Institute; SSLOW scrapie brain homogenate was prepared using animals from the 4th passage of SSLOW [45]; atypical PrPres was generated from brain material in vitro as described [49]. Ten percent (wt/vol) brain homogenates (10% BH) were prepared in PBS, pH 7.4, using glass/Teflon homogenizers attached to a cordless 12 V compact drill (Ryobi) as previously described [45]. To seed PMCA, 10% BH was diluted in PMCA conversion buffer [44] and briefly sonicated immediately before use.
Expression and purification of rPrP
Syrian hamster full-length rPrP encompassing residues 23–231 was expressed and purified according to a previously described procedure [9] with minor modifications [43]. Immediately before use, lyophilized rPrP was dissolved in 10 mM Na acetate, pH 5.0, filtered through 0.45 μm syringe filter and the rPrP concentration was measured. For the formation of rPrPresPolyA, lyophilized rPrP was dissolved in 5 mM MES, pH 6.0.
Cofactors
L-α-phosphatidylethanolamine (PE) from porcine brain (#840022C, Avanti Polar Lipids, Alabaster, AL) was supplied as 25 mg/ml stock in chloroform. Immediately before use, an aliquot of PE was lyophilized in a glass tube under a stream of compressed nitrogen, and then re-suspended at 10 mM in 0.05% Triton-X by sonication in a water bath until clear. PolyA (#P9403, Sigma) was dissolved at 5 mg/ml in 10 mM Tris, pH 8.0, 1 mM EDTA buffer, and stored frozen in aliquots.
Protein misfolding cyclic amplification (PMCA)
QSonica S-3000, S-4000 or Q700 sonicators (Newtown, CT) equipped with a microplate horn were used for PMCA reactions. Samples in 0.2 ml thin-wall PCR tubes (Fisher #14230205) were placed in a floating rack inside the horn filled with 300 ml water and covered with foil. To maintain 37 °C temperature, two coils of rubber tubing attached to a circulating water bath were installed inside the horn. Alternatively, the horn and its enclosure were put inside 37 °C incubator.
rPrPresPE was produced in 20 mM Tris, pH 7.5, 135 mM NaCl, 5 mM EDTA and 0.15% Triton X-100 supplemented with 5 μg/ml rPrP and 2.5 mM PE. For the first round of PMCA, 90 μl aliquots of reaction mixture were supplemented with 10 μl of diluted scrapie brain homogenates as indicated (see Brain Material). PMCA sonication program consisted of 5 s sonication pulses at 150–200 W applied every 10 min during a 24 h period. For each subsequent round, 30 μl of the reaction from the previous round were added to 60 μl of fresh substrate. Each PMCA reaction was carried out in the presence of two 3/32” Teflon beads (McMaster-Carr). To analyze production of PK-resistant PrP material in PMCA, the samples were digested with 10 μg/ml PK at 37 °C for 1 h. The digestion was terminated by addition of SDS-sample buffer and heating the samples for 10 min in a boiling water bath.
rPrPresPE + PolyA was produced in the presence of 2.5 mM PE and 20 μg/ml polyA using the same conditions as for the reactions with PE as a sole cofactor, except the Teflon beads were omitted, and for each subsequent round, 20 μl of the reaction mixture from the previous rounds were added to 80 μl of fresh substrate. The ingredients were mixed in the following order. A master mix consistent of water, Tris, NaCl, EDTA and Triton X-100 was prepared first. Then rPrP was mixed with PE and incubated for 10 min at room temperature. After that, the polyA was added, followed by additional 5 min incubation. Finally, the master mix and rPrP mix were combined and aliquoted. The 90 μl aliquots for the first round were supplemented with 10 μl of seeds and subjected to sonication immediately. The 80 μl aliquots for the subsequent rounds were frozen at − 20 °C. Completed rounds were used to seed the following rounds on the same day. The leftovers were kept frozen before the subsequent analysis. PK-digestion was performed with 10 μg/ml PK at 37 °C for 1 h and terminated by addition of SDS-sample buffer and heating the samples for 10 min in a boiling water bath.
To establish amplification efficiency in a standard PMCA, 10% normal brain homogenate (NBH) from healthy hamsters was prepared as described previously [44] and used as a substrate [26]. To produce desialylated substrate (dsNBH), 10% NBH was treated with Arthrobacter ureafaciens sialidase (cat # N3786, Sigma-Aldrich, St. Louis, MO) as described before [33]. For the first round, 90 μl of NBH or dsNBH were supplemented with 10 μl of scrapie brain homogenates serially diluted in PBS. The standard sonication program consisted of 20 s sonication pulses at 150–200 W applied every 20 min during a 24 h period. For each subsequent round, 10 μl of the reaction mixtures from the previous round were added to 90 μl of fresh substrate. Each PMCA reaction was carried out in the presence of two 3/32” Teflon beads (McMaster-Carr). To analyze production of PK-resistant PrP material in PMCA, 10 μl of sample were supplemented with 5 μl of SDS and 5 μl of PK to a final concentration of SDS and PK of 0.25% and 50 μg/ml, respectively, followed by incubation at 37 °C for 1 h. The digestion was terminated by addition of SDS-sample buffer and heating the samples for 10 min in a boiling water bath.
Formation of PrPresPolyA was achieved by serial PMCA of 5 μg/ml rPrP in the buffer conditions similar to previously described [18]: 20 mM MOPS, pH 7.5, 150 mM NaCl, 500 mM Imidasole, 0.5% Triton X-100, 50 mM EDTA with the addition of 20 μg/ml PolyA. No seeds were added to these reactions. PMCA sonication program consisted of 30 s sonication pulses at 150–200 W applied every 30 min during a 24 h period. For each subsequent round, 10 μl of the reaction from the previous round were added to 90 μl of fresh substrate. To analyze production of PK-resistant PrP material in PMCA, the samples were digested with 20 μg/ml PK at 37 °C for 1 h. For PK-digestion pattern comparison, R-fibrils (F2M) produced from rPrP as described previously [41] were digested with 1:5000 PK:rPrP ratio at 37 °C for 1 h. The digestion of all samples was terminated by addition of SDS-sample buffer and heating the samples for 10 min in a boiling water bath.
Bioassay
Each animal received 50 μl (S. hamsters) or 20 ul (Tg7 mice) of inoculum intracerebrally, under 2% O2/4 minimum alveolar concentration (MAC) isoflurane anesthesia. After inoculation, animals were observed daily for disease using a ‘blind’ scoring protocol. Non-habituating startle response to sound and/or touch was considered the first clinical sign and was marked as an onset of the disease when consistently observed during consecutive scoring sessions. The animals were euthanized at a terminal stage when unable to rear and having troubles to right themselves after flipping to their backs.
PrPSc detection by Western blot
An aliquot of 10% BH was mixed with an equal volume of 4% sarcosyl in PBS, supplemented with 50 mM Tris, pH 7.5, and digested with 20 μg/ml PK (New England BioLabs) for 30 min at 37 °C with 1000 rpm shaking using a DELFIA plate shaker (Wallac) placed in 37 °C incubator. PK digestion was stopped by adding SDS sample buffer and heating the samples for 10 min in a boiling water bath. Samples were loaded onto NuPAGE 12% Bis-Tris gels, transferred to PVDF membrane, and probed with 3F4 or SAF-84 antibody [45].
Analysis of conformational stability and proteinase K resistance
Ten percent brain homogenate was diluted 10 times into PMCA conversion buffer, then supplemented with an equal volume of GdnHCl solution in PBS to a final concentration of GdnHCl ranging from 0.4 to 4 M and incubated at room temperature for 1 h. Next, nine volumes of 2% sarkosyl in PBS were added to all samples followed by 1 h incubation at room temperature, and then the samples were treated with 20 μg/mL PK for 1 h at 37 °C with shaking. The digestion was stopped with 2 mM PMSF, and the proteins were precipitated in four volumes of ice-cold acetone, incubated overnight at − 20 °C, and subsequently centrifuged for 30 min at 16000 x g. Pellets were dried for 30 min, resuspended in 1 × SDS-sample buffer, loaded into NuPAGE 12% bisTris gels, then transferred to PVDF membrane, and stained with 3F4 antibody.
Histopathological study
Histopathological studies were performed on three animals per group. Formalin fixed brain halves were divided at the midline. Right hemisphere was frozen, and left hemisphere was fixed in 10% neutral buffered formalin solution. Brains were treated for 1 h with 96% formic acid prior to embedding in paraffin to deactivate prion infectivity. Paraffin embedded brains were sliced into 4 μm sections and processed for hematoxylin-eosin stain as well as for immunohistochemistry for PrP using the mouse monoclonal anti-PrP antibody 3F4 (1:1000, Covance, Berkeley, CA, USA), or rabbit anti-glial fibrillary acidic protein (GFAP; 1:500, Novus, Littleton, CO, USA), or rabbit anti-ionized calcium-binding adapter molecule 1 (Iba1; 1:500, Wako, Richmond, VA, USA). Horse radish peroxidase-labeled goat anti-mouse and anti-rabbit antibody (KPL, Milford, MA) were used as secondary antibody. For detection of disease-associated PrP, we applied a pretreatment of 30 min hydrated autoclaving at 121 °C followed by 5 min in 96% formic acid. Detection was performed using DAB Quanto chromogen and substrate (VWR, Radnor, PA).
Procedure for purification of scrapie material
Extraction of PrPSc (in the form of PrP27–30) from a 263K scrapie hamster and from three hamsters (i, ii, and iii) from the second passage of SSLOWPE + PolyA for FTIR microspectroscopic analysis was performed as described by Daus et al. [16] with the following modifications: hemispheres of mid-saggitally split hamster brains (approximately 0.5 g) were each homogenized in adjusted volumes of homogenization buffer for the preparation of 10% (w/v) brain tissue homogenates. From each donor animal, two aliquots of 1 mL of 10% (w/v) brain tissue homogenate were subjected to the extraction procedure. This yielded two final pellets of highly purified PrPSc per donor animal, each corresponding to 0.1 g of brain tissue. For infrared spectroscopic analysis final PrPSc pellets were washed in double-distilled water as described [16] and resuspended in 10 μL of double-distilled H2O. 1 μL aliquots of these PrPSc suspensions were transferred for drying onto a CaF2 window of 1 mm thickness (Korth Kristalle GmbH, Altenförde, Germany).
Infrared microspectroscopy (IR-MSP)
IR-MSP analysis of highly purified PrPSc extracts were carried out as previously described [16]. Briefly, mid-IR spectra were acquired in transmission mode using an IFS 28/B FT-IR spectrometer from Bruker (Bruker Optics GmbH, Ettlingen Germany) that was linked to an IRscope II infrared microscope (Bruker). IR microspectra were recorded with a spatial resolution of approximately 80 μm. Nominal spectral resolution was 4 cm− 1, and the zero filling factor was 4. For each background and for each sample spectrum, 512 individual interferograms were averaged, zero-filled and apodized using a Blackman-Harris 3-term apodization function. For each examined PrPSc extract from one 263K scrapie hamster and from hamsters i, ii, and iii infrared spectra were recorded at three different positions in PrPSc sample spots dried on CaF2 windows. Data acquisition and spectral preprocessing was carried out by utilizing Bruker’s instrument software OPUS v. 5.5. Second derivative spectra were obtained by means of a 9-smoothing point Savitzki-Golay derivative filter. Spectra from the 263K scrapie hamster and the three hamsters i, ii, and iii were vector normalized in the wave number region between 1610 and 1700 cm− 1.
Results
PolyA as a sole cofactor is not sufficient for assisting conversion of hamster rPrP into PrPSc
Previous studies revealed that RNA molecules including synthetic, homopolymeric nucleic acids such as polyA assisted conversion of PrPC into self-propagating, PK-resistant, PrPSc-like states in sPMCA [17, 20]. Moreover, RNAs were found to facilitate conversion of hamster PrPC, but not mouse or vole PrPC, into PrPSc [19]. These results emphasized species-specific differences in biochemical environment important for conversion. Taking previous data into consideration, we decided to test whether PolyA is sufficient for assisting conversion of hamster rPrP into authentic PrPSc in vitro. To answer this question, non-seeded sPMCA reactions that utilized hamster rPrP as a substrate were carried out in the presence or absence of synthetic polyA. In the presence of polyA, PK-resistant products appeared between 3rd and 5th sPMCA round, whereas no products were detected in the reactions conducted in the absence of polyA (Additional file 1: Figure S1A). The PMCA-derived, PK-resistant products (rPrPresPolyA) consisted of expected peptide of ~ 16 kDa and two shorter peptides of 10 kDa and 8 kDa, which were all detectable by SAF-84 antibody. Once formed, rPrPresPolyA was able to propagate in sPMCA with rPrP as a substrate, albeit with some variations in yield (Additional file 1: Figure S1A).
For testing whether rPrPresPolyA is infections, Syrian hamsters and transgenic mice that overexpress hamster PrPC on an ablated background (tg7) were inoculated with PMCA-derived rPrPresPolyA material. Hamsters did not develop any clinical signs of the disease and were euthanized at 661 days postinoculation. No PK-resistant material was found in brains of hamsters by Western blots (Additional file 1: Figure S1B). Despite expression of PrPC at a level of 3.5-fold higher than that in a hamster [34], tg7 mice did not develop any clinical signs of the disease for up to 524 days postinoculation and were euthanized. However, Western blot analysis of tg7 mice revealed PK-resistant products that were detectable by SAF-84 and consisted of three bands with molecular weight of 23, 16 and 10 kDa. Such PK-digested pattern suggests that upon inoculation of rPrPresPolyA, tg7 mice produced PrPres state different from authentic PrPSc, but similar to the atypical PrPres described in our previous studies [44, 46, 48, 49]. Serial transmission of rPrPresPolyA in tg7 mice displayed dynamics similar to those previously observed for the serial transmission of atypical PrPres [44, 46, 48, 49]. In a 2nd passage, tg7 mice did not develop clinical disease, yet again three PK-resistant bands of 23, 16 and 10 kDa were observed using SAF-84 antibody (Additional file 1: Figure S1B) documenting a self-replicating nature of this state. In addition, small amounts of PrPSc were detectable by SAF-84 and 3F4 antibodies (Additional file 1: Figure S1B). In summary, animals inoculated with rPrPresPolyA material did not develop clinical disease nor did they produce PrPSc in a first passage arguing that rPrPresPolyA preparation does not contain authentic PrPSc.
Both PE and polyA are required for efficient conversion of hamster rPrP into PrPSc in vitro
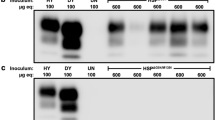
While the experiments on polyA had been carried out, cellular lipids and, specifically, PE were shown to be essential for converting rPrP into infectious PrPSc in vitro, albeit with a loss of strain identity [21, 22, 63, 64]. Therefore, next we assessed the effect of PE on converting hamster rPrP. sPMCA reactions with rPrP as a substrate were seeded by hamster strains Hyper (HY), Drowsy (DY) or synthetic strain SSLOW. Small amounts of PK-resistant product (referred to as rPrPresPE) with molecular weight ~ 16 kDa, expected for recombinant PrPSc, were detected in reactions seeded with SSLOW, but not with HY, DY or non-seeded reactions (Fig. 1a). For testing whether PE also facilitates alternative misfolding pathway leading to atypical PrPres, sPMCA reactions were seeded with brain-derived atypical PrPres, yet no PK-resistant bands were observed (Fig. 1a). Among the strains used for seeding, only reactions seeded with SSLOW showed positive results in the presence of PE (Additional file 1: Figure S2). It is unlikely that this difference could be attributed to the strain-specific differences in the efficiency of amplification in sPMCA, because HY was found to display significantly higher amplification efficiency than SSLOW in conventional sPMCA [24, 27]. Nevertheless, in the presence of PE alone, SSLOW-seeded rPrPresPE propagated with low efficiency and only at low dilution factor between serial PMCA rounds. Next, we tested whether supplementing both polyA and PE will improve the yield and the efficiency of amplification. Serial PMCA reactions were seeded with brain-derived SSLOW or PMCA-derived rPrPresPE and conducted in the presence of a mixture of PE and polyA or PE alone. In both rPrPresPE- and SSLOW-seeded reactions, stable amplification of the 16 kDa PK-resistant product was observed only in the presence of a mixture of PE and polyA (will be referred to as rPrPresPE + PolyA), but not PE alone (Fig. 1b, c). rPrPresPE + PolyA could be detected by 3F4 antibodies, arguing that the central PrP region that is missing in atypical PrPres was present in rPrPresPE + PolyA. No lower molecular weight bands characteristic for atypical PrPres were detected in PK-digested rPrPresPE + PolyA upon immunoblotting with SAF-84 antibody, suggesting that rPrPresPE + PolyA conformation is different from rPrPresPolyA.

Attempts to produce Ha-rPrPSc with assistance of PE and PolyA. a Duplicate sPMCA reactions were seeded with 103-fold diluted brain-derived Hyper (HY), Drowsy (DY) or SSLOW, or atypical PrPres produced in vitro, then subjected to four sPMCA rounds in the presence of PE with 3-fold dilutions between rounds and analyzed by Western blot. Products of 4th sPMCA rounds are shown. Small amounts of PK-resistant material (rPrPresPE) were detected in the reactions seeded with SSLOW (indicated by arrow). b sPMCA reactions were seeded with PMCA-derived rPrPresPE or 103-fold diluted SSLOW brain material, then subjected to four serial rounds in the presence of PE alone or a mixture of PE and polyA with 5-fold dilutions between rounds and analyzed by Western blots. c Serial amplification of rPrPresPE + PolyA in PMCA. sPMCA reactions were seeded with 103-fold diluted SSLOW brain material, then subjected to 18 serial rounds in the presence of PE and polyA with 5-fold dilutions between rounds and analyzed by Western blots. SAF-84 antibody was used to verify the absence of low molecular weight bands upon PK-digestion (middle panel)
rPrPresPE + PolyA is transmissible
To test whether rPrPresPE + PolyA is infectious, sPMCA reaction with hamster rPrP was seeded by 103-diluted SSLOW brain material, subjected to 18 rounds of sPMCA (Fig. 1c), then ten-fold diluted sPMCA-derived products were inoculated in Syrian hamsters. The final inoculum contained 1.3 × 1016-fold diluted SSLOW brain material; this dilution is approximately 107-fold higher than the limiting dilution of SSLOW as determined by bioassay [47]. Three out of four hamsters inoculated with rPrPresPE + PolyA developed first clinical signs of disease by 517 days postinoculation showing non-habituating startle response to sound and an agitated, fidgeting behavior. With disease progression, three animals with clinical signs showed increasing difficulty righting themselves when rolled onto their back. Their hair became dry and detached in clumps. Three clinical animals were euthanized along with one non-clinical animal at 622 days postinoculation (Table 1). All four animals showed PrPSc on Western blots detectable by 3F4 and SAF-84 with the lowest amount observed in the sub-clinical animal (Fig. 2a). In three clinical animals the amounts of PrPSc were similar to those in terminal SSLOW-inoculated animals (Fig. 2a). Histopathological analysis of sick animals revealed accumulation of PrPSc in forms of diffused synaptic deposits or small aggregates in multiple brain regions including subpial areas, deep layers of cortex, thalamus, hippocampus, cerebellum and subventricular regions (Fig. 3). Notably, hippocampus showed intense deposition of PrPSc in the stratum-lacunosum region (Fig. 3f, h), a pattern of deposition reminiscent of SSLOW [30, 43, 45]. Hematoxylin and eosine staining revealed moderate vacuolation in several brain areas including thalamus (Fig. 3d). PrPSc material from animals inoculated with rPrPresPE + PolyA will be referred to as SSLOWPE + PolyA.

Serial transmission and characterization of the strain produced upon inoculation of rPrPresPE + PolyA. a Western blots of brain materials from animals inoculated with PMCA-derived rPrPresPE + PolyA and sacrificed at 622 days post-inoculation. One and ten percent brain homogenates from SSLOW-inoculated animals are provided as a reference. b Comparison of amplification efficiency of PrPSc in PMCA and dsPMCA that utilizes desialylated substrate. PMCA or dsPMCA reactions were seeded with SSLOWPE + PolyA, SSLOW or 263K brain materials serially diluted to up to 1010-fold as indicated, subjected to one amplification round and analyzed by Western blot. In a manner similar to SSLOW, amplification efficiency of SSLOWPE + PolyA increased drastically in dsPMCA conditions relative to that of PMCA conditions. c Western blots of brain materials of the first and second passages of SSLOWPE + PolyA. Arrows indicate animals that were used for the serial transmission. 3F4 antibody was used. d Kaplan-Meier survival curves for animals inoculated with SSLOW (circles) or SSLOWPE + PolyA (2nd passage, two individual groups, squares and triangles)

Histopathological analysis of animals from the first passage of rPrPresPE + PolyA. PrPSc was found in form of diffused synaptic deposition and small aggregates in subpial areas (a), deep layers of cortex (b), thalamus (c), cerebellum (e), hippocampus (f), and subventricular regions (g). Stratum-lacunosum (s-l) of hippocampus has characteristically intense deposition of PrPSc (f, h) with a pattern reminiscent of SSLOW [30, 43, 45]. Hematoxylin and eosine staining revealed moderate vacuolation in several brain areas including thalamus (d). Scale bars = 200 μm (a-d, g, h) or 300 μm (e, f)
For examining strain-specific biochemical properties of SSLOWPE + PolyA, we analyzed amplification efficiency of brain-derived PrPSc using normal PMCA and dsPMCA, in which desialylated substrate is used. Our previous studies revealed that desialylation of PrPC increases the amplification rate in a strain-specific manner. For instance, in dsPMCA the amplification rate of SSLOW PrPSc increased by several orders of magnitudes relative to its amplification rate in PMCA [33]. PMCA and dsPMCA reactions were seeded with serially-diluted brain-derived SSLOWPE + PolyA, SSLOW or 263K and amplified in one round. The amplification rate of 263K increased only by 10-fold in dsPMCA relative to that of PMCA, whereas the amplification rates of SSLOWPE + PolyA and SSLOW increased by approximately 103–104 folds. This experiment illustrates that SSLOWPE + PolyA material has amplification dynamics similar to that of SSLOW.
For assessing transmissibility of SSLOWPE + PolyA, 10% SSLOWPE + PolyA brain homogenates from two animals were inoculated into two new hamster groups. Animals of both groups developed first clinical signs of the disease between 274 and 335 days postinoculation (Table 1). The clinical disease progressed slowly and all animals approached terminal stage by 420–440 days postinoculation (Table 1, Fig. 2c, d). For the second passage of SSLOWPE + Poly, the incubation time to the first clinical signs, the duration of the clinical diseases, the timing of terminal stage of the disease and set of clinical signs were highly reminiscent to SSLOW (Fig. 2d) [45]. Non-habituating startle response to sound and touch and an agitated, fidgeting behavior were the first signs of the disease. As the disease progressed the animals had increasing difficulty righting themselves when rolled onto their back. The hair became dry and detached in clumps. Most animals appeared overweight, pear shaped with enlarged abdomens and hind quarters. At the time of euthanasia the animals had become less active and unable to rear. Histopathological examination revealed strong PrPSc deposition in multiple brain areas including deep layers of cortex, thalamus, hippocampus and cerebellum (Fig. 4a, e, f, g). Similar to SSLOW, SSLOWPE + Poly was characterized by strong perivascular deposition of PrPSc (Fig. 4g). Vacuolation and neuroinflammation of microglia and astrocytes were observed throughout the brain, but particularly strong in the areas of intense PrPSc accumulation (Fig. 4b, c, d).

Histopathological analysis of animals from the second passage of rPrPresPE + PolyA. PrPSc deposition in hippocampus (a), deep layers of cortex (e), thalamus (f), and cerebellum (g) as shown by 3F4 staining. Vacuolation and reactive gliosis were observed in the areas of PrPSc accumulation, as shown for hippocampus using staining with hematoxylin and eosine (c), or staining of microglia with anti-Iba1 (b) or astrocytes with anti-GFAP (d). Scale bars = 300 μm (a-d) or 200 μm (e-g)
SSLOWPE + PolyA displays SSLOW-specific structural and neuropathological features
The question of considerable interest is whether propagation of SSLOW in vitro with an assistance of PE and polyA preserved strain-specific disease phenotype. For addressing this question, first we examined patterns of PrPSc deposition peculiar to SSLOW. SSLOW is characterized by intense accumulation of diffuse PrPSc in the stratum lacunosum-moleculare of the hippocampus, which is accompanied by a marked astrocytic inflammation in the same area [30]. Analysis of SSLOWPE + Poly revealed intense deposition of PrPSc and astrocytic gliosis in stratum lacunosum-moleculare typical for SSLOW (Fig. 4a, d). In a manner similar to SSLOW, SSLOWPE + Poly-infected animals also displayed other SSLOW-specific neuropathological features including diffuse PrPSc in deeper layers of cortex, intense narrow PrPSc staining in subpial area and PrPSc plaques in subependymal areas (Fig. 5a-g). Overall, the region-specific intensity of PrPSc deposition was very similar in animals infected with SSLOWPE + Poly and SSLOW, where thalamus and subependymal areas displaying the strongest deposition, and cerebellum the weakest (Figs. 4 and 5) [43, 45].

Comparison of neuropathological features of SSLOWPE + PolyA and SSLOW. a, b Both SSLOWPE + PolyA and SSLOW shows similar patterns of PrPSc accumulation in cortex including deposition in subpial area (black arrowhead), strong deposition in deeper layers of cortex (white arrowhead), and plaques in subependymal areas (arrow). c Cortex of 263K-infected animals displays different pattern of PrPSc deposition (d – g). Subependymal plaques (d, e) and subpial deposition of PrPSc (f, g) in SSLOWPE + PolyA (d, f) and SSLOW (f, g) animals. Scale bars = 300 μm (a-c) or 200 μm (d-g)
Second, PrPSc was purified in the form of PrP27–30 from SSLOWPE + Poly-infected animals and its structural features were examined using infrared microspectroscopy (Fig. 6). Infrared spectroscopy and microspectroscopy have been established as a powerful analytical tool for the detection of structural differences between PrPSc from various native prion strains or PMCA parent and progeny seeds [13, 16, 31, 55, 61]. SSLOWPE + Poly and SSLOW showed very similar if not identical IR spectra that were characterized by a major peak at 1626–1627 cm− 1 that indicates the presence of β-sheet secondary structure elements, a small peak at 1696 cm− 1 that also reports on β-sheet structures and a moderate peak at 1658–1659 cm− 1 which is typically assigned to an α-helical conformation and/or disordered structures [3, 4] (Fig. 6a, b). The shape of IR spectra displayed by SSLOWPE + Poly and SSLOW was markedly different from that of 263K (Fig. 6c) [16, 57].

Comparison of the secondary structure of PrPSc materials by infrared microspectroscopy. a Second derivative IR microspectra obtained from PrPSc material purified in the form of PrP27–30 from three individual SSLOW-infected hamsters (these spectral data originate from the study [31]). b Second derivative IR microspectra obtained from PrPSc material purified in the form of PrP27–30 from three individual hamsters (i, ii, and iii) from the second passage of SSLOWPE + PolyA. Technical replicate spectra were acquired for each hamster at three different positions in PrPSc sample spots dried on CaF2 windows. c Second derivative IR microspectra obtained from PrPSc material purified in the form of PrP27–30 from one 263K scrapie hamster brain. Technical replicate spectra were acquired at three different positions in PrPSc sample spots dried on a CaF2 window. Similar spectra of highly purified PrPSc from 263K scrapie hamster brain were previously published elsewhere [16, 57]
Third, we examined conformational stability of PrPSc in GdnHCl-induced denaturation experiment. The conformational stability was found to be similar for four groups examined: SSLOWPE + Poly from the 1st and 2nd passages, SSLOW and 263K (Additional file 1: Figure S3A). This result is consistent with the previous study that documented similar stability of SSLOW and 263K [43]. However, the band pattern was different between 263K and the three members of SSLOW group (SSLOWPE + Poly from the 1st and 2nd passages, SSLOW) (Additional file 1: Figure S3A). Because electrophoretic mobility of PK is similar to the band corresponding to diglycosylated PrP isoform, the area on the membrane where PK is present has reduced binding of anti-prion antibody, which creates a blind spot. Since PK-related blind sport serves as an internal reference for each lane of SDS-PAGE, this approach was found to be reliable for comparing minor differences between electrophoretic mobility of prion strains, as documented in our previous studies [32]. We found that the PK-related blind spot hides the upper half of diglycosylated PrP band for SSLOW group, while it cuts though the middle of the band for 263K, illustrating slightly higher electrophoretic mobility of the members of SSLOW group relative to that of 263K (Additional file 1: Figure S3A). Direct comparison of electrophoretic mobility confirmed that SSLOWPE + Poly and SSLOW displayed slightly higher mobility in comparison to 263K (Additional file 1: Figure S3B).
Discussion
In the recent decade, enormous progress has been made in generating highly infectious prions in vitro using rPrP as a substrate. Several experimental protocols for converting rPrP into PrPSc have been developed that highlight importance of lipids and/or polyanionic molecules for assisting rPrP conversion in vitro [21,22,23, 35, 63, 67]. These studies established that rPrP that lacks posttranslational modification is able to support replication of highly infectious PrPSc, yet it remains unclear whether prion replication in rPrP can preserve strain identity. In previous studies, seeding of rPrP by brain-derived PrPSc gave rise to new prion strains with new disease phenotypes documenting loss of a strain identity upon replication in rPrP substrate [21,22,23, 35]. Remarkably, loss of prion strain identity upon replication in rPrP was mirrored by the studies conducted in transgenic mice with deficient posttranslational modifications of PrPC [1, 40]. Transmission of mouse-adapted prion strain RML or mCWD to transgenic mice expressing PrPC devoid of GPI anchor and deficient in N-linked glycosylation led to formation of novel prion strains, which maintained their novel properties upon transmission to wild type mice [1, 40]. In a similar fashion, passaging of prion strains through transgenic mice expressing PrPC devoid of just N-linked glycans resulted in changes of strain-specific infectious properties upon passaging back to wild type host [11].
In the current study, replication of hamster strain SSLOW was achieved in vitro using rPrP as a substrate with assistance of the mixture of polyA and PE. The disease phenotype generated upon transmission of rPrPresPE + PolyA in hamsters was strikingly similar to the original SSLOW diseases phenotype. In the second passage of SSLOWPE + Poly, the incubation time to the first clinical signs, the duration of the disease progression, the incubation time to terminal stage and the set of clinical signs were highly reminiscent to the characteristics of prion infection by SSLOW (Fig. 2d). Moreover, neuropathological analysis demonstrated remarkable resemblance between animals affected by SSLOWPE + Poly and those affected by SSLOW with respect to brain regions affected by PrPSc and PrPSc deposition patterns (Figs. 4 and 5). Analysis of purified PrPSc using infrared microspectroscopy indicated that SSLOWPE + PolyA and SSLOW had very similar, if not identical, secondary structures (Fig. 6). IR-MSP is very sensitive technique which can be used to detect even very small structural changes [13, 16, 31, 55, 61]. While the fact that we cannot detect any spectroscopic differences does not fundamentally exclude conformational differences, yet our IR-MSP results indicate that conformational differences, if there are any, must be very small. In addition to similar secondary structures, SSLOWPE + PolyA and SSLOW PrPSc showed very similar amplification dynamics in PMCAs that employed normal and desialylated substrate (Fig. 2b). Finally, SSLOWPE + PolyA and SSLOW PrPSc displayed similar electrophoretic mobility, which was slightly faster in comparison to that of 263K (Additional file 1: Figure S3). The current study is the first to demonstrate the proof of principle that rPrP is capable of preserving strain identity of brain-derived PrPSc.
In previous studies, the majority of work on generating infectious recombinant prions has been conducted using mouse rPrP [21, 22, 63, 67]. The current study is the first to document that successful propagation of a hamster strain could be achieved in vitro using hamster rPrP. Propagating of hamster strains in vitro using rPrP or unglycosylated PrPC was found to be very challenging. All hamster strains, whether of natural or synthetic origin, are predominantly diglycosylated [2, 27]. In fact, previous studies showed that diglycosylated PrPC molecules were required for propagating hamster Sc237 strain in PMCA [50]. Surprisingly, while unglycosylated mouse PrPC were required for replicating mouse prions, unglycosylated hamster PrPC molecules inhibited replication of hamster prions [50]. In vivo, N-linked glycans might play a role in facilitating the assembly of hamster PrPSc or stabilizing PrP molecules within hamster PrPSc [50]. The current work provides a proof of principle that faithful replication of hamster prion strain that typically relies on diglycosylated PrPC molecules could be achieved in the absence of N-linked glycan, but with assistance of two cofactors.
It is not clear whether the results presented in the current study represent a rare exception or general rule. We do not know whether other hamster-adapted strains might have more stringent requirements for propagation using rPrP as a substrate including not only a different set of cofactors, but also PMCA amplification conditions (dilution between rounds, sonication time and power). While failure of DY to utilize rPrP substrate in the current study could be attributed to its very low rate of replication, as assessed in conventional PMCA reactions [2], this is not the case for HY. In fact, with PrPC as a substrate the replication rate of HY was found to be faster than that of SSLOW [27]. One possibility behind faithful replication of SSLOW in rPrP substrate could be attributed to its synthetic origin, as it was generated via serial transmission of rPrP amyloid fibril prepared in vitro [43]. However, such possibility, should be considered with great caution, because structure of rPrP fibrils that gave rise to SSLOW were fundamentally different from that of authentic PrPSc including SSLOW PrPSc, which emerged in hamster upon serial passaging [51, 66]. In fact, four serial passages in hamsters were required to stabilize SSLOW-specific disease phenotype and PrPSc properties [42, 45, 48, 49]. While SSLOW is a strain of synthetic origin, animals infected with SSLOW display all key neuropathological and biochemical characteristics of transmissible spongiform encephalopathies including chronic neuroinflammation and neurodegeneration, spongiform vacuolation, deposition of bona fide PrPSc, efficient transmission of the disease between animals, high infectivity titer and efficient amplification of SSLOW PrPSc in PMCA [30, 38, 45, 47]. In a manner similar to PMCA amplification of hamster strains of natural origin, amplification of SSLOW in PMCA displayed dependence on RNAs, the sialylation status of N-linked glycans, and also showed a species barrier upon amplification in mouse substrate [26, 27, 33].
Previous studies documented that RNA molecules including polyA facilitate in vitro replication of hamster prion strains in PMCAs that employ brain-derived hamster PrPC as a substrate [18, 20]. Notably, the effect of RNAs on stimulating prion replication was found to be species- and strain-dependent [19, 27, 54]. RNAs had strong stimulating effects on replication of hamster strains, yet their effect on mouse strains was considerably less pronounced and strain-dependent [19, 27, 54]. The current study demonstrated that polyA can facilitate misfolding of hamster rPrP (Additional file 1: Figure S1). However, in the presence of polyA as a sole cofactor, rPrP misfolded into PK-resistant, self-replicating state, which was different from PrPSc (Additional file 1: Figure S1). These data suggest that polyA and, perhaps, other RNAs indeed promote PrP misfolding, however without imposing strict constraints with respect to misfolding pathways. Interestingly, our previous studies demonstrated that serial replication of hamster strain 263K in PMCA conducted in RNA-depleted brain homogenates resulted in self-replicating PrP states that failed to produce prion disease in hamsters [25, 32] .
In previous studies, PE was found to be sufficient as a sole cofactor for propagating mouse prion strains in vitro and generating highly infectious recombinant PrPSc from rPrP [21, 22]. In the present work, we were not able to efficiently propagate SSLOW using rPrP as a substrate in the presence of PE as a sole cofactor (Fig. 1b). The current results are consistent with previous observations that replication of hamster strains exhibit stronger dependency on polyanions than mouse strains [19]. Our work also suggests that faithful replication of prion strains from different species using rPrP might requires different sets of cofactors.
The current study demonstrates that rPrP can support replication of brain-derived PrPSc preserving its stain identity despite lack of posttranslational modifications. In contrast to rPrP, PrPC that serves as a replication substrate in a brain is posttranslationally modified with GPI anchor and N-linked glycans [58, 59, 62]. Previously, we proposed that in PrPC, posttranslational modifications might limit the diversity of misfolding pathways that are otherwise accessible to rPrP [6, 10, 36]. Consistent with this view, previous studies documented changes in strain-specific disease phenotype and physical properties of PrPSc upon passaging of prion stains in transgenic mice expressing PrPC devoid of GPI anchor and/or N-linked glycans [1, 11]. In the absence of posttranslational modifications and cofactors, rPrP alone displays a broad spectrum of misfolding pathways [7, 36, 41]. What is the mechanism behind PE-assisted conversion of rPrP into PrPSc? Our previous studies that employed steady-state spectroscopic techniques failed to find any evidence of direct physical interactions between PE and rPrP [56]. Bearing this in mind, one could propose that interactions between PE and rPrP are very weak and/or transient (PE-rPrP complexes exists for very short time periods). If this is the case, only a tiny fraction of rPrP could be found in a state bound to PE at any given time, the fraction that could be presumably an intermediate toward PrPSc. According to this mechanism, PE could promote misfolding of rPrP directly, along the pathway that leads to PrPSc. Alternatively, PE might assist rPrP conversion into infectious states indirectly, i.e. by binding and neutralizing intermediates toward alternative, non-infectious amyloid states. This mechanism proposes that PE might limit the diversity of misfolding pathways. If this is the case, one would expect that PE would promote replication of other hamster strains, which was not supported by current observations. A third possibility is that PE is involved transiently at the stage of interaction of rPrP with PrPSc seeds. Whether such transient interactions depend on strain-specific properties of PrPSc seeds remains to be established. Regardless of the specific mechanism, PE was found to be essential for propagating SSLOW-specific features using rPrP.
Incomplete attack rate and prolonged incubation time to disease observed in the first passage of SSLOWPE + PolyA argues that sPMCA-derived rPrPresPE + PolyA material had low specific prion infectivity (Table 1). A drop in specific prion infectivity could be due to accumulation of alternative, non-infectious, self-replicating states that replicate faster than SSLOW PrPSc in sPMCA with rPrP. In addition, such drop could also be due to conformational changes and/or changes in size of SSLOW PrPSc particles during sPMCA. Notably, the diminished specific prion infectivity in sPMCA is not specific to sPMCA that employs rPrP as a substrate, as it was previously documented for conventional sPMCAs conducted with PrPC as a substrate. In fact, previous studies established that replication of hamster strains including 263K and SSLOW in sPMCA reactions consisting of multiple rounds reduced prion infectivity [31, 37]. In our previous study, hamsters inoculated with sPMCA-derived SSLOW subjected to 24 rounds of PMCA in normal brain homogenates did not develop clinical diseases for at least 621 days postinoculation, but showed PrPSc accumulation in their brains and spleens [31]. Nevertheless, the fact that SSLOWPE + PolyA animals from the 2nd passage showed incubation time to diseases, disease phenotype and structural PrPSc features typical for SSLOW argued that at least a fraction of sPMCA-derived rPrPresPE + PolyA material preserved authentic properties of SSLOW upon replication in rPrP.
Conclusions
The current study is the first to demonstrate that faithful replication of a prion strain that preserves strain-specific identity could be achieved in vitrousing recombinant prion protein despite lack of posttranslational modifications. Faithful replication required two cofactors to be present in the reaction mixture with recombinant prion protein: poly A and phosphatidylethanolamine.
Change history
24 September 2018
Figure 6 of the original publication [1] contained an error in the Wavenumber in panels B and C. The wavenumbers 1616 (Cm-1) in panels B and C should have been 1516 (cm-1). The updated figure has been published in this correction article; the original article has been updated.
Abbreviations
- GPI:
-
Glycosylphosphatidylinositol
- IR-MSP:
-
Infrared microspectroscopy
- PE:
-
Phosphatidylethanolamine
- PrPC :
-
Normal, cellular isoform of the prion protein
- PrPSc :
-
Infectious, disease-associated, pathogenic form of the prion protein;
- rPrP:
-
recombinant prion protein
- sPMCA:
-
serial protein misfolding cyclic amplification
References
Aguilar-Calvo P, Xiao X, Bett C, Erana H, Soldau K, Castilla J, Nilsson KPR, Surewicz WK, Sigurdson CJ (2017) Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci Rep 7:43295. https://doi.org/10.1038/srep43295
Ayers JL, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC (2011) The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog 7:e1001317
Barth A (2007) Infrared spectroscopy of proteins. Biochim Biophys Acta 1767:1073–1101
Barth A, Zscherp C (2002) What vibrations tell us about proteins. Q Rev Biophys 35:369–430
Baskakov IV, Breydo L (2007) Converting the prion protein: what makes the protein infectious. Biochim Biophys Acta (Molecular Basis of Disease) 1772:692–703
Baskakov IV, Katorcha E (2016) Multifaceted role of sialylation in prion diseases. Front Neurosci 10:358. https://doi.org/10.3389/fnins.2016.00358
Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE (2002) Pathway complexity of prion protein assembly into amyloid. J BiolChem 277:21140–21148
Baskakov IV, Legname G, Prusiner SB, Cohen FE (2001) Folding of prion protein to its native à-helical conformation is under kinetic control. JBiolChem 276:19687–19690
Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV (2005) In vitro conversion of full length mammalian prion protein produces amyloid form with physical property of PrPSc. JMolBiol 346:645–659
Breydo L, Sun Y, Makarava N, Lee CI, Novitskaia V, Bocharova OV, Kao JPY, Baskakov IV (2007) Nonpolar substitution at the C-terminus of the prion protein, a mimic of the glycosylphosphatidylinositol anchor, partially impairs amyloid fibril formation. Biochemistry 46:852–861
Cancellotti E, Mahal SP, Somerville R, Diack A, Brown D, Piccardo P, Weissmann C, Manson JC (2013) Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J 32:756–769
Castilla J, Morales R, Saa P, Barria M, Gambetti P, Soto C (2008) Cell-free propagation of prion strains. EMBO J 27:2557–2566
Caughey B, Raymond GJ, Bessen RA (1998) Strain-dependent differences in b-sheet conformations of abnormal prion protein. JBiolChem 273:32230–32235
Cohen FE, Prusiner SB (1998) Pathologic conformations of prion proteins. Annu Rev Biochem 67:793–819
Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen HO, Lemus A, Cohen FE, DeArmond SJ, Prusiner SB (2010) Protease-sensitive synthetic prions. PLoS Pathog 6:e1000736
Daus ML, Wagenfuhr K, Thomzig A, Boerner S, Hermann P, Hermelink A, Beekes M, Lasch P (2013) Infrared microspectroscopy detects protein Misfolding cyclic amplification (PMCA)-induced conformational alterations in hamster scrapie progeny seeds. J Biol Chem 288:35068–35080
Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S (2005) Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic Polyanions. JBiolChem 280:26873–26879
Deleault NR, Harris BT, Rees JR, Supattapone S (2007) Formation of native prions from minimal components in vitro. ProcAcadNatlSciUSA 104:9741–9746
Deleault NR, Kascsak R, Geoghegan JC, Supattapone S (2010) Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49:3928–3934
Deleault NR, Lucassen RW, Supattapone S (2003) RNA molecules stimulate prion protein conversion. Nature 425:717–720
Deleault NR, Piro JR, Walsh DJ, Wang F, Ma J, Geoghegan JC, Supattapone S (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Acad Natl Sci U S A 109:8546–8551
Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S (2012) Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. ProcAcadNatlSciUSA 109:E1938–E1946
Fernández-Borges N, Di Bari MA, Erana H, Sánchez-Martín M, Pirisinu L, Parra B, Elezgarai SR, Vanni I, Lopez-Moreno R, Vaccari G, Charco JM, Gil D, Harratchi C, D'Agostino C, Agrimi U, Mayoral T, Requena JR, Nonno R, Castilla J (2018) Cofactors influence the biological properties of infectious recombinant prions. Acta Neuropathol 135:179–199
Gonzalez-Montalban N, Baskakov IV (2012) Assessment of strain-specific PrPSc elongation rates revealed a transformation of PrPSc properties during protein Misfolding cyclic amplification. PLoS One 7:e41210
Gonzalez-Montalban N, Lee YJ, Makarava N, Savtchenko R, Baskakov IV (2013) Changes in prion replication environemnt cause prion strain mutation. FASEB J 27:3702–3710
Gonzalez-Montalban N, Makarava N, Ostapchenko VG, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov IV (2011) Highly efficient protein Misfolding cyclic amplification. PLoS Pathog 7:e1001277
Gonzalez-Montalban N, Makarava N, Savtchenko R, Baskakov IV (2011) Relationship between conformational stability and amplification efficiency of prions. Biochemistry 50:7933–7940
Green KM, Castilla J, Seward TS, Napier DL, Jewell JE, Soto C, Telling GC (2008) Accelerated high Fidelity prion amplification within and across prion species barriers. PLoS Pathog 4:e1000139
Groveman BR, Raymond GJ, Campbell KJ, Race B, Raymond LD, Hughson AG, Orru CD, Kraus A, Phillips K, Caughey B (2017) Role of the central lysine cluster and scrapie templating in the transmissibility of synthetic prion protein aggregates. PLoS Pathog 13:e1006623. https://doi.org/10.1371/journal.ppat.1006623
Jeffrey M, McGovern G, Makarava N, Gonzalez L, Kim YS, Rohwer RG, Baskakov IV (2014) Pathology of SSLOW, a transmissible and fatal synthetic prion protein disorder, and comparison with naturally occurring classical transmissible spongoform encephalopathies. Neuropath Appl Neurobiol 40:296–310
Katorcha E, Daus ML, Gonzalez-Montalban N, Makarava N, Lasch P, Beekes M, Baskakov IV (2016) Reversible off and on switching of prion infectivity via removing and reinstalling prion sialylation. Sci Rep 6:33119. https://doi.org/10.1038/srep33119
Katorcha E, Gonzalez-Montalban N, Makarava N, Kovacs GG, Baskakov IV (2018) Prion replication environment defines the fate of prion strain adaptation. PLoS Pathog 14:e10007093
Katorcha E, Makarava N, Savtchenko R, D’Azzo A, Baskakov IV (2014) Sialylation of prion protein controls the rate of prion amplification, the cross-species barrier, the ratio of PrPSc glycoform and prion infectivity. PLOS Pathog 10:e1004366. https://doi.org/10.1371/journal.ppat.1004366
Kercher L, Favara C, Chan CC, Race R, Chesebro B (2004) Differences in scrapie-induced pathology of the retina and brain in transgenic mice that express hamster prion protein in neurons, astrocytes, or multiple cell types. Am J Pathol 165:2055–2067
Kim C, Xiao X, Chen S, Haldiman T, Smirnovas V, Kofskey D, Warren M, Surewicz K, Maurer NR, Kong Q, Surewicz W, Safar JG (2018) Artificial strain of human prions created in vitro. Nat Commun 9:e2166
Klimova N, Makarava N, Baskakov IV (2015) The diversity and relationship of prion protein self-replicating states. Virus Res 207:113–119
Klingeborn M, Race B, Meade-White KD, Chesebro B (2011) Lower specific infectivity of protease-resistant prion protein generated in cell-free reactions. Proc Acad Natl Sci U S A 108:E1244–E1253
Kovacs GG, Makarava N, Savtchenko R, Baskakov IV (2013) Atypical and classical forms of the disease-associated state of the prion protein exhibit distinct neuronal tropism, deposition patterns, and lesion profiles. Am J Pathol 183:1539–1547
Legname G, Baskakov IV, Nguyen HOB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB (2004) Synthetic mammalian prions. Science 305:673–676
Mahal SP, Jablonski J, Suponitsky-Kroyter I, Orlschlegel AM, Herva ME, Oldstone M, Weissmann C (2012) Propagating of RML prions in mice expressing PrP devoid of GPI anchor leads to formation of a novel, stable prion strain. PLoS Pathog 8:e1002746
Makarava N, Baskakov IV (2008) The same primary structure of the prion protein yields two distinct self-propagating states. J Biol Chem 283:15988–15996
Makarava N, Baskakov IV (2013) The evolution of transmissible prions: the role of deformed templating. PLoS Pathog 9:e1003759
Makarava N, Kovacs GG, Bocharova OV, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV (2010) Recombinant prion protein induces a new transmissible prion disease in wild type animals. Acta Neuropathol 119:177–187
Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV (2011) Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog 7:e1002419
Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV (2012) Stabilization of a prion strain of synthetic origin requires multiple serial passages. J Biol Chem 287:30205–30214
Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Ostapchenko VG, Budka H, Rohwer RG, Baskakov IV (2012) A new mechanism for transmissible prion diseases. J Neurosci 32:7345–7355
Makarava N, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov IV (2012) Fast and ultrasensitive method for quantitating prion infectivity titer. Nat Commun 3:741
Makarava N, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov IV (2016) New molecular insight into mechanism of evolution of mammalian synthetic prions. Am J Pathol 186:1006–1014
Makarava N, Savtchenko R, Baskakov IV (2015) Two alternative pathways for generating transmissible prion disease de novo. Acta Neuropathologica Communications 3:69. https://doi.org/10.1186/s40478-015-0248-5
Nishina K, Deleault NR, Mahal S, Baskakov I, Luhrs T, Riek R, Supattapone S (2006) The stoichiometry of host PrPC Glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry 45:14129–14139
Ostapchenko VG, Sawaya MR, Makarava N, Savtchenko R, Nilsson KP, Eisenberg D, Baskakov IV (2010) Two amyloid states of the prion protein display significantly different folding patterns. J Mol Biol 400:908–921
Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144
Prusiner SB, Hsiao KK, Bredesen DE, DeArmond SJ (1989) Prion disease. In: Vinken PJ, Bruyn GW, Klawans HL (eds) Handbook of Clinical Neurology, Vol. 12 (56): Viral Disease. Elsevier Science Publishers, Amsterdam, pp 543–580
Saa P, Sferrazza GF, Ottenberg G, Oelschlegel AM, Dorsey K, Lasmezas CI (2012) Strain-specific role of RNAs in prion replication. J Virol 86:10494–10504
Spassov S, Beekes M, Naumann D (2006) Structural differences between TSEs strains investigated by FT-IR spectroscopy. Biochim Biophys Acta 1760:1138–1149
Srivastava S, Baskakov IV (2015) Contrasting Effects of Two Lipid Cofactors of Prion Replication on the Conformation of the Prion Protein. PLoS One 10:e0130283. https://doi.org/10.1371/journal.pone.0130283
Srivastava S, Katorcha E, Daus ML, Lasch P, Beekes M, Baskakov IV (2017) Sialylation controls prion fate in vivo. J Biol Chem 292:2359–2368. https://doi.org/10.1074/jbc.M116.768010
Stahl N, Baldwin MA, Hecker R, Pan KM, Burlingame AL, Prusiner SB (1992) Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry 31:5043–5053
Stahl N, Borchelt DR, Hsiao K, Prusiner SB (1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51:229–240
Supattapone S (2014) Synthesis of high titer infectious prions with cofactor molecules. J Biol Chem 289:19850–19854
Thomzig A, Spassov S, Friedrich M, Naumann D, Beekes M (2004) Discriminating scrapie and bovine spongiform encephalopathy isolates by infrared spectroscopy of pathological prion protein. J Biol Chem 279:33854
Turk E, Teplow DB, Hood LE, Prusiner SB (1988) Purification and properties of the cellular and scrapie hamster prion proteins. Eur J Biochem 176:21–30
Wang F, Wang X, Yuan CG, Ma J (2010) Generating a prion bacterially expressed recombinant prion protein. Science 327:1132–1135
Wang X, McGovern G, Zhang Y, Wang F, Zha L, Jeffrey M, Ma J (2015) Intraperitoneal infection of wild-type mice with synthetically generated mammalian prion. PLoS Pathog 11:e1004958
Wilham JM, Orru CD, Bessen RA, Atarashi R, Sano K, Race B, Meade-White KD, Taubner LM, Timmes A, Caughey B (2010) Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog 6:e1001217
Wille H, Bian W, McDonald M, Kendall A, Colby DW, Bloch L, Ollesh J, Borovinskiy AL, Cohen FE, Prusiner SB, Stubbs G (2009) Natural and synthetic prion structure from X-ray fiber diffraction. Proc Acad Natl Sci USA 106:16990–16995
Zhang Z, Zhang Y, Wang F, Wang X, Xu Y, Yang H, Yu G, Yuan C, Ma J (2013) De novo generation of infectious prions with bacterially expressed recombinant prion protein. FASEB J 27:4768–4775
Funding
Financial support for this study was provided by National Institute of Health Grants R01 NS045585 and R01 AI128925 to IVB.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information file.
Author information
Authors and Affiliations
Contributions
NM and IVB conceived the idea and designed the experiments; NM, RS and MB performed experiments; NM, PL, MB and IVB analyzed the data; IBV wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal protocol was approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore (Assurance Number A32000–01; Permit Number: 0215002).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
Figure 6 of the original publication contained erroneous wavenumbers. Details of the update are available in the correction article. The original article has been corrected.
Additional file
Additional file 1:
Supplementary Materials. (PDF 937 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Makarava, N., Savtchenko, R., Lasch, P. et al. Preserving prion strain identity upon replication of prions in vitro using recombinant prion protein. acta neuropathol commun 6, 92 (2018). https://doi.org/10.1186/s40478-018-0597-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-018-0597-y