
Abstract
Background
The recently updated European LeukemiaNet risk stratification guidelines combine cytogenetic abnormalities and genetic mutations to provide the means to triage patients with acute myeloid leukemia for optimal therapies. Despite the identification of many prognostic factors, relatively few have made their way into clinical practice.
Methods
In order to assess and improve the performance of the European LeukemiaNet guidelines, we developed novel prognostic models using the biomarkers from the guidelines, age, performance status and select transcript biomarkers. The models were developed separately for mononuclear cells and viable leukemic blasts from previously untreated acute myeloid leukemia patients (discovery cohort, N = 185) who received intensive chemotherapy. Models were validated in an independent set of similarly treated patients (validation cohort, N = 166).
Results
Models using European LeukemiaNet guidelines were significantly associated with clinical outcomes and, therefore, utilized as a baseline for comparisons. Models incorporating age and expression of select transcripts with biomarkers from European LeukemiaNet guidelines demonstrated higher area under the curve and C-statistics but did not show a substantial improvement in performance in the validation cohort. Subset analyses demonstrated that models using only the European LeukemiaNet guidelines were a better fit for younger patients (age < 55) than for older patients. Models integrating age and European LeukemiaNet guidelines visually showed more separation between risk groups in older patients. Models excluding results for ASXL1, CEBPA, RUNX1 and TP53, demonstrated that these mutations provide a limited overall contribution to risk stratification across the entire population, given the low frequency of mutations and confounding risk factors.
Conclusions
While European LeukemiaNet guidelines remain a critical tool for triaging patients with acute myeloid leukemia, the findings illustrate the need for additional prognostic factors, including age, to improve risk stratification.
Similar content being viewed by others
Background
The recently revised European LeukemiaNet (ELN-2017) recommendations for diagnosis and management of adult patients with acute myeloid leukemia (AML) are broadly accepted by physicians as a gold standard and provide guidelines to stratify patients into three outcome groups: favorable, intermediate, and adverse based on cytogenetics and mutation status of ASXL1, CEBPA, FLT3, NPM1, RUNX1, and TP53 [1]. This stratification scheme provides a simple, yet powerful means to triage patients for appropriate therapies. Two previous studies have validated the performance of ELN-2017 guidelines in patients ≤65 years old with AML [2, 3] and one study has evaluated the use of non-coding RNA expression to improve the prognostic significance of the ELN-2017 risk classification [4]. However, the majority of AML patients are older than the previously studied patients [5], and clinical prognostic factors such as age and performance status (PS) were not examined in the previous studies, nor are these factors included in the ELN-2017 guidelines [1, 6,7,8]. Similarly, prognostic guidelines, including ELN-2017, do not provide recommendations or guidance about the optimal material for clinical testing. Prognostic biomarkers have generally been identified, optimized, and validated using cryopreserved samples comprised of heterogeneous populations of mononuclear cells (MNCs). The inter-sample variability in the percentage and viability of leukemic blasts may impact continuous biomarkers like FLT3-ITD allelic ratio (AR) or transcript expression [6]. Thus, studies are needed to determine if examining a more homogenous population of malignant cells may improve the precision of risk stratification guidelines, and these studies, including those examining the current ELN-2017 guidelines, need to be extended to older patients [5, 9,10,11,12].
Therefore, we developed novel prognostic models using ELN-2017 risk stratification guidelines (ELN2017), clinical factors such as age and PS, and expression of select transcripts reported to be associated with prognosis [6, 13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. The models provided continuous risk scores that were used to define risk stratification thresholds. The models were developed separately for unsorted MNCs and highly enriched viable leukemic blasts (VLBs) to examine the potential prognostic benefit of testing a more homogenous population of malignant cells, representing the largest systematic evaluation of paired MNCs and VLBs from patients with AML to date. These risk models were then validated in an independent population of patients. Given that clinical assays for ASXL1, CEBPA, RUNX1 and TP53 are not available at every institution, we also examined the performance of models without the mutation status of these four genes (ELN2017-MOD). The results demonstrated the utility of the ELN-2017 guidelines for younger patients with AML and caution for applying the same risk strata to older patients. Integration of the selected expression biomarkers into models did not markedly improve the model performance. In addition, the findings highlight the need for new prognostic biomarkers and risk stratification approaches for older adults with AML.
Methods
Patient materials
A review of SWOG Cancer Research Network leukemia repository inventory identified 383 out of 1042 previously untreated AML patients 1) with pretreatment bone marrow or peripheral blood samples containing ≥3 cryopreserved vials and 2) who were enrolled onto trials to receive cytarabine- and daunorubicin-based induction chemotherapy and consolidation with curative intent. These patients were enrolled onto protocols SWOG-9031, SWOG-9333, S0106 and S0112 and treated as previously described [28,29,30,31]. The included patients were assigned into two cohorts by simple randomization: a discovery cohort (n = 190) and a validation cohort (n = 193) by the SWOG Statistical Center [32]. Specimen handling and cryopreservation were consistent across the studies per previously described standard operating procedures [6]. All participants provided written informed consent to participate in correlative research in compliance with the Declaration of Helsinki, and all studies were conducted with the approval of Fred Hutch Institutional Review Board.
Thawing, FACS preparation, analyses, and nucleic acid extraction
Cryopreserved samples were thawed as previously described [6]. A portion of unsorted MNCs was lysed, while the remainder of the sample underwent fluorescence-activated cell sorting (FACS) to isolate VLBs using forward-by-side scatter, DAPI staining and fluorescently-labeled antibodies to CD45, CD34 and CD117 as previously described [6]. RNA and DNA from unsorted MNCs and VLBs were extracted and quantified as previously described [6]. Of the randomly selected samples, 185/190 (97%) and 166/193 (86%) yielded sufficient materials in each cohort for downstream analyses.
Identification of genomic mutation
Internal tandem duplication in FLT3 (FLT3-ITD) and mutations in NPM1 were assayed via fragment analyses as previously described [7, 33, 34]. If the wild type FLT3 was not detected in a sample with an ITD, an allelic ratio (AR) of 20 was assigned [35]. MNCs from the discovery cohort were sequenced for mutations in ASXL1, RUNX1 and TP53 using Wafergen technology by the British Columbia Cancer Agency (BCCA) per standard protocols (Additional File, Tables S1A and B). VLBs from the discovery and validation cohorts were sequenced using TruSight™ Myeloid Sequencing Panel (Illumina) as previously described [36]. Neither BCCA nor TruSight™ platforms provided optimal coverage for CEBPA, therefore, targeted MiSeq CEBPA assays were developed (Additional File, Table S1C). Paired-end short reads were first aligned to the human genome GRCh37/hg19 reference assembly using Burrows-Wheeler Aligner, BWA, v0.7.12 [37]. The resulting alignment data were further processed based on the best practice of Genome Analysis Toolkit (GATK, v3.5 https://www.broadinstitute.org/gatk/). The overview of sequence alignment statistics was computed for each sample using Samtools (v1.0 http://samtools.sourceforge.net), and the sequence coverage was computed for each sample using GATK DepthOfCoverage. Variants were called per sample using GATK HaplotypeCaller in GVCF mode, then jointly as a cohort using GenotypeGVCFs. The resulting collection of variants, in the form of a VCF file, were annotated using Annovar, version 2016Feb01 [38]. The following exclusion filters were applied: synonymous variants, low quality (Qual score < 150), variant read depth < 65 [39], variant allele frequency (VAF) < 10%, and non-exonic loci outside of splice sites. For in-frame missense amino acid substitutions, additional filters excluded changes with a FATHMM_score < 0.7 [40] and/or if ExAC_ALL score was > 0.0001, unless the missense amino acid change was defined as likely pathogenic or pathogenic by ClinVar and described as somatic in the COSMIC databases (https://cancer.sanger.ac.uk) [41]. Sequencing depth for each read loci were calculated to determine average percent coverage. For quality control (QC), loci with > 20% of samples displaying < 65 read coverage were removed from downstream analyses. Three loci failed QC, but the excluded loci displayed a very low abundance of previously reported somatic mutations confirmed to be present in hematopoietic malignancies in Cosmic Database (Additional File, Table S2).
Expression of transcript biomarkers
Taqman gene expression assays for BAALC, CEBPA, CCNA1, CD34, ERG1, EVI1, FLT3, GATA2, IL3RA, JAG1, KIT, MN1, RUNX1, and WT1 were used to quantify gene expression as previously described [6, 22]. The fold change (FC) for each transcript was computed using the comparative Ct method with Beta-glucuronidase (GUSB) normalization to pooled non-malignant bone marrow calibrator, except for WT1 FC, which used LAMA-84 cell line as a calibrator [6]. The FC was censored at maximum cycle threshold of 45 for samples without evidence of expression by qRT/PCR. Transcript expression was examined in experimental duplicates, with the geometric mean of the duplicates used for downstream analyses. If either of the replicates was censored, their mean was also marked as censored. If a duplicate was not available, we used the FC and censoring of that single expression value. Censored values were assigned the minimum expression observed for that gene, divided by the square root of two [42].
Statistical analyses
Cytogenetic and mutation risk classification was based on the ELN-2017 guidelines [1]. Complete remission (CR) required the following: > 20% marrow cellularity with maturation of all cell lines, < 5% blasts, no Auer rods, absolute neutrophil count (ANC) ≥1500/μL, platelets > 100,000/μL, no peripheral blasts, and no extramedullary disease. Study S0106 required ANC ≥1000/μL and did not have any marrow cellularity or peripheral blasts requirements. Overall survival (OS) was measured from the date of study registration to the date of death by any cause, with patients last known to be alive censored at the date of last contact. Relapse-free survival (RFS) was measured from date of CR to date of death or relapse, with patients last known to be alive and without report of relapse censored at the date of last contact. Transplant data were not collected on these trials and, thus, are not available for incorporation into the modeling algorithms. Disease characteristics, patient demographics, and clinical responses were compared between the pool of patients who were selected to be analyzed herein (N = 351) versus patients enrolled on the four trials who were not analyzed (N = 691) using Chi-squared, Fisher’s exact, or Wilcoxon rank-sum tests as appropriate. The same analyses were used to compare the discovery (N = 185) and validation (N = 166) cohorts.
Differences in mutation status, gene expression, and FLT3-ITD AR in paired MNC and VLB samples were assessed using McNemar’s test or the Wilcoxon signed rank test, as appropriate. OS and RFS were estimated using the Kaplan-Meier method and compared across groups using log-rank tests. RFS and OS models used Cox proportional hazards regression; CR models used logistic regression. Model building in the discovery cohort was composed of the following steps, done separately for each outcome and for each type of material (unsorted MNCs and VLBs). 1) Univariate models were fit for each of the following baseline variables: age (quantitative), performance status (0–1 vs. 2–3), AML onset (secondary vs de novo), clinical trial, immunophenotype (IP) and ELN-2017 risk group. 2) Multivariable models were fit with covariates with p-value < 0.10 from step 1 for each of the expression variables. These adjusted expression p-values were ranked, and the 5 expression variables with the smallest p-values were selected for additional modeling. If ELN-2017 risk group or IP were included in the multivariable models, interactions with expression variables were also evaluated, and interactions with p-values less than 0.15 were selected for additional modeling; if more than 5 interactions had p-values less than 0.15, the 5 with the smallest p-values were selected for additional modeling. 3) A multivariable model including selected baseline variables, selected expression variables, and selected interaction variables was built using backwards selection based on the Aikike Information Criterion (AIC). Area under the Receiver Operating Characteristic curve (AUC) and C-statistics were estimating using 5-fold cross validation of the entire (3-step) model building process. The locked parameter values from step 3 were fit to the validation cohort and AUC and C-statistics were calculated. We note that AUC and C-statistic values of 0.50 indicate prediction equivalent to a coin flip (random prediction), and values of 1.00 indicate perfect prediction. Analyses were performed using SAS version 9.4 (SAS Institute, Cary NC) and R version 3.4.3 [43].
Results
Characteristics of patient population
Patients who were included in this study displayed higher WBC, blast percentage, and ANC (P < 0.0001 for all) compared to patients enrolled on these trials who were not included in this study. In addition, there was a significant difference in cytogenetic profiles (P = 0.0031), FAB class (P < 0.0001), and proportions across clinical trials (P = 0.0129, Additional File, Table S3). These differences between included and not included patients likely reflect reported biases for patients within repositories having a higher burden of disease at diagnosis and depletion of specimens from older trials [6]. The differences between trial representation likely reflect the higher abundance of samples from the more recent trials. However, there were no significant differences between the included and not included patients with respect to CR rates (60% vs. 58% P = 0.52), RFS (5-year RFS 32% vs. 33%; P = 0.52) or OS (5-year OS 30% vs. 32%; P = 0.62, Additional File, Table S3 and Fig. S1). Comparing the discovery and validation patients, the two cohorts displayed some differences in clinical characteristics despite randomization (e.g., WBC, P = 0.0188; cytogenetics, P = 0.0296; cytogenetics risk group, P = 0.028 and distribution across clinical trials, P = 0.0209; Additional File, Table S4), however there were no significant differences in clinical outcomes between the discovery and validation cohorts (CR 57% vs. 63% P = 0.31; 5-yr RFS 30% vs. 34%; P = 0.54; or 5-yr OS 30% vs. 31%; P = 0.82, Additional File, Table S4 and Fig. S2).
Characterization of mutations and transcript expression
Mutation analyses focused on genes utilized for ELN-2017 risk stratification. FLT3-ITD and NPM1 mutations were examined in all specimens with available material (i.e., MNCs and VLBs). There was 100% concordance for NPM1 mutations in MNCs and VLBs. One FLT3-ITD was observed in the MNCs but not VLBs (99.7% concordant). FLT3-ITD and NPM1 mutations were detected in 109 (31%) and 125 (36%) patients, respectively. The distribution and mutation frequencies of NPM1 and FLT3-ITD, as well as FLT3-ITD AR, were not significantly different between discovery and validation cohorts in either population of cells (Additional File, Table S5 and Fig. S3). Excluding the patient with discordant FLT3-ITD results, FLT3-ITD AR was significantly higher in VLBs than the MNCs (AR ranges 0.03–20 and 0.04–13.2, respectively, P < 0.0001). Given that the ELN-2017 guidelines utilize FLT3-ITD AR of 0.5 for risk stratification, we examined the impact that testing the FLT3-ITD AR in VLBs had on ELN-2017 classification. In the MNCs, percentages of patients with low and high FLT3-ITD ARs were 34 and 66%, respectively, while percentages for low and high AR in VLBs were 23 and 77%. Examining FLT3 in VLBs resulted in a different AR classification for 19 patients, with 15 patients changing from low AR in MNCs to high AR in VLBs and 4 patients changing from high AR in MNCs to low AR in VLBs.
ASXL1, CEBPA, RUNX1, and TP53 mutations were examined in both MNC and VLB populations for the discovery cohort. Similar to the results for NPM1 and FLT3, there was a 99.4% concordance in mutations between MNCs and VLBs, with only one patient displaying a discrepancy for an ASXL1 mutation. Therefore, mutation analyses for ASXL1, CEBPA, RUNX1, and TP53 were examined in only VLBs for the validation cohort. Overall, the frequencies of mutations in the examined patients were as follows: ASXL1 (N = 35, 10%), CEBPA (N = 20, 6%), RUNX1 (N = 40, 11%), and TP53 (N = 26, 7%). The frequency of ASXL1 mutations was modestly higher in the discovery cohort (13% discovery vs. 7% validation, P = 0.044); other mutations displayed similar frequencies in both groups of patients (Additional File, Table S5 and Fig. S3).
Building upon the results examining transcript biomarkers in the discovery cohort [6], analyses examined transcript expression as a continuous variable for 13 genes, which had been previously reported to be potential prognostic biomarkers: BAALC, CCNA1, CEBPA, ERG1, EVI1, FLT3, GATA2, IL3RA, JAG1, KIT, MN1, RUNX1, and WT1 [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. In the case of EVI1, transcript expression was not detectable and thus censored in 69% of VLBs and 70% of MNCs. Given the dichotomous nature of EVI1 expression, we also examined the prognostic significance of EVI1 expression as a binary variable (expressed vs. not expressed). In the discovery cohort, univariate analyses showed a significant increase in expression in VLBs relative to MNCs for BAALC (P < 0.0001), CCNA1 (P = 0.005), ERG1 (P < 0.0001), EVI1 (P = 0.001), FLT3 (P = 0.024), MN1 (P < 0.0001), RUNX1 (P = 0.001) and WT1 (P < 0.0001), while none of the transcripts were expressed at significantly lower levels in VLBs than MNCs (Additional File, Table S6).
Prognostic significance of biomarkers in univariate analyses
Univariate analyses examined the prognostic significance of FLT3-ITD AR, NPM1 mutation, and transcript expression in MNCs and VLBs in the discovery cohort. Increasing FLT3-ITD AR in MNCs was associated with worse OS (Table 1). NPM1 mutations were not associated with clinical outcome in univariate analyses (Table 1). The prognostic significance for some transcripts varied depending upon tested cell type (Table 1, Additional File, Table S7). Overall, increased expression of CCNA1, ERG1, EVI1, FLT3, IL3RA, KIT and MN1 was significantly associated with adverse risk for one or more clinical outcomes in one or both cell populations (Table 1), while expression of BAALC, CEBPA, GATA2, JAG1, RUNX1 and WT1 were not significantly associated with clinical outcomes in either MNCs or VLBs (Additional File, Table S7). Univariate analyses also evaluated the prognostic significance of age, cytogenetics, PS, secondary AML status, and ELN risk groups in the discovery cohort. As expected, increasing age, adverse cytogenetics, poor PS, and secondary AML status were significantly associated with poor clinical outcomes (Table 2). Favorable ELN-2017 risk was significantly associated with improved CR, whether examining MNCs or VLBs (OR = 3.11, P = 0.024 and OR = 3.69, P = 0.014, respectively), while adverse and unknown ELN-2017 risks were not significantly associated with CR (Table 2). Favorable ELN-2017 risk was also significantly associated with improved OS in VLBs (MNCs, HR = 0.58, P = 0.060 and VLBs, HR = 0.38, P = 0.001, Table 2). Adverse ELN-2017 risk was associated with reduced OS in MNCs (HR = 1.66, P = 0.050) but not in VLBs (HR = 1.10, P = 0.720). In keeping with the CR and OS analyses, favorable ELN-2017 was significantly associated with improved RFS in both MNCs and VLBs (HR = 0.47, P = 0.027 and HR = 0.37, P = 0.008, respectively, Table 2).
Performance of novel risk models utilizing ELN and other prognostic factors
Multivariable models for CR, OS, and RFS were developed separately for each cell population using age, ELN-2017 risk group, PS, AML onset, immunophenotype, clinical trial, transcript biomarker and expression as possible covariates (Additional File, Models Details). In the discovery cohort, the models with the best performance were obtained when clinical variables and expression biomarkers were integrated; however, when applied to an independent population of patients in the validation cohort, the performances of integrated models for most outcomes were not superior to AGE + ELN2017 models (Table 3). If a model is generalizable to a broad population, AUCs or C-statistics will be nearly equivalent in the two cohorts. Generalizability of the developed integrated models was inconsistent across CR, OS and RFS outcomes.
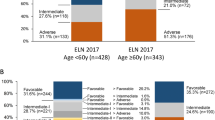
The ELN2017 model divides patients into 4 groups: favorable, intermediate, adverse, and unknown. Figure 1 shows OS by ELN2017 risk in MNCs and VLBs from the validation cohort. Since previous studies demonstrated a worse prognosis for intermediate risk patients over the age of 55 [8, 44], the ELN2017 models were also applied to younger (age < 55) and older (age ≥ 55) patients. ELN2017 models were a better fit for the younger patients, whether using data derived from MNCs (Fig. 1) or VLBs (Fig. 2). To visualize the AGE + ELN2017 model for OS, the continuous risk score from the AGE + ELN2017 model in the discovery data was divided into quartiles to parallel the ELN2017 model, and boundaries of these quartiles were applied to the validation data (Figs. 3 and 4). Though these plots are intended to be exploratory, the quartiles defined by the AGE + ELN2017 models visually show more separation between curves than do the ELN2017 risk groups in MNCs and in VLBs (Figs. 3a and 4a vs Figs. 1a and 2a). The c-statistics for the AGE + ELN2017 models are also slightly higher than the c-statistics for the ELN2017 models. There were no patients younger than 55 in the quartiles representing the poorest outcomes (3rd and 4th quartiles in MNCs and 4th quartile in VLBs) and no patients older than 55 in the 1st quartile, representing the best outcomes. This is due to the fact that older age was associated with poorer outcomes in the multivariable models controlling for ELN risk and age, and these models were used to derive the quartiles in the figure.

Performance of ELN2017 model in Mononuclear cells. Overall Survival probability over time by ELN2017 risk group in MNCs from the validation cohort (n = 166). C-statistics are for the ELN2017 model fit to the validation cohort for all patients (age 18.5–88.8, a), patients younger than 55 years old (N = 86, b), and patients 55 years and older (N = 80, c). The total number of patients who were at risk of death (alive and uncensored) are shown for each year of follow-up

Performance of ELN2017 model in Viable Leukemic Blasts. Overall Survival probability over time by ELN2017 risk group in VLBs from the validation cohort (n = 166). C-statistics are for the ELN2017 model fit to the validation cohort for all patients (age 18.5–88.8, a), patients younger than 55 years old (N = 86, b), and patients 55 years and older (N = 80, c). The total number of patients who were at risk of death (alive and uncensored) are shown for each year of follow-up

Performance of AGE + ELN2017 Model in Mononuclear Cells. Overall Survival probability over time as predicted by the AGE + ELN2017 models developed using the discovery cohort in MNCs. The continuous risk score from the AGE + ELN2017 model in the discovery cohort was divided into quartiles and the boundaries of these quartiles were used to define a four-level categorical variable. A model was fit using this categorical variable in the validation cohort for all patients (N = 166, age 18.5–88.8, a), patients younger than 55 years old (N = 86, b), and patients 55 years and older (N = 80, c). There were no patients younger than 55 in 3rd and 4th quartiles (b) or patients older than 55 in 1st quartile (c). The total number of patients who were at risk of death (alive and uncensored) are shown for each year of follow-up

Performance of AGE + ELN2017 Model in Viable Leukemic Blasts. Overall Survival probability over time as predicted by the AGE + ELN2017 models developed using the discovery cohort in MNCs. The continuous risk score from the AGE + ELN2017 model in the discovery cohort was divided into quartiles and the boundaries of these quartiles were used to defined a four-level categorical variable. A model was fit using this categorical variable in the validation cohort for all patients (N = 166, age 18.5–88.8, a), patients younger than 55 years old (N = 86, b), and patients 55 years and older (N = 80, c). There were no patients younger than 55 in 4th quartiles (b) or patients older than 55 in 1st quartile (c). The total number of patients who were at risk of death (alive and uncensored) are shown for each year of follow-up
Evaluation of simplified ELN-2017 and AGE + ELN2017 models
To investigate the impact of ASXL1, CEBPA, RUNX1 and TP53 mutations on risk stratification, we evaluated the performance of modified models that did not include mutation data for these 4 genes without age (ELN2017-MOD) and with age (AGE + ELN2017-MOD). Exclusion of mutation status of these four genes resulted in an overall reassignment of risk groups for 46 patients in MNCs and 44 patients in VLBs of the 351 patients (Additional File, Table S8). Both models were developed using the discovery data from the MNCs and VLBs. In the validation cohort, the AUCs and C-statistics were similar between the ELN2017 and ELN2017-MOD models, allowing comparable population risk prediction at the community sites that may not have access to genomic mutation screening. Furthermore, the AGE + ELN2017-MOD models had almost the exact same performance characteristics as the AGE + ELN2017 models (Table 4).
Discussion
Risk stratification of AML patients enables physicians to triage patients for optimal therapy. Many prognostic factors have been identified, but relatively few have made their way into clinical practice. The revised ELN-2017 guidelines combine cytogenetic abnormalities and genetic mutations to stratify patients with AML into favorable, intermediate, and adverse risk groups [1]. We examined the predictive accuracy of risk models developed using the ELN-2017 guidelines with and without incorporating additional prognostic factors, as well as how these models performed in VLBs and MNCs. ELN2017 predictive models were a better fit for younger patients. Models utilizing the expression results from selected transcript biomarkers did not provide substantial improvement over the ELN2017 models, regardless of whether transcripts were tested in MNCs or VLBs. In addition, we examined the potential contribution of mutations in ASXL1, CEBPA, RUNX1 and TP53, given that clinical testing for these genes may not be readily available in many settings. The ELN2017-MOD demonstrated that while these mutations may provide additional benefit for risk stratification of individual patients, their contribution to the predictive accuracy of the ELN2017 models was limited in our cohorts of AML patients. Thus, ELN-2017 guidelines remain a critical tool for risk stratifying AML patients, but the findings illustrate the need for additional prognostic factors to improve risk stratification, especially in older adults with AML. Moreover, observations suggest that risk stratification models may need to be developed separately for older patients.
Previous studies have examined the performance of ELN-2017 guidelines in patients with AML [2,3,4, 10]. Similar to our observation in patients younger than 55, authors confirmed the prognostic significance of the ELN-2017 guidelines, with patients stratified as favorable, intermediate, and adverse having a 5-year OS of approximately 60, 40, and 20%, respectively [2,3,4]. Our results are consistent with the previous report that ELN-2017 guidelines are not as informative for older adults, supporting the need for additional studies for this population [10]. Age remains one of the most robust prognostic factors for patients with AML. As demonstrated in Table 3, AGE models performed comparably to ELN2017 models, with the AGE + ELN2017 models displaying the best performances. Given that a majority of patients diagnosed with AML are over the age of 65 [45], current risk stratification guidelines for patients with AML need to be adjusted for age or recalibrated for older patients. Similarly, the findings demonstrate the need for more accurate risk stratification models for older adults with AML. Such models will likely require the inclusion of novel prognostic biomarkers.
Although age-related comorbidities and differences in therapy play a role in the poor outcomes for older adults with AML, these factors cannot fully explain the higher relapse rates for these patients [7, 8]. Older adults with AML frequently harbor mutations in genes associated with the spliceosome, methylation and chromatin remodeling, which are commonly identified in patients with MDS or secondary AML [10, 12, 46,47,48,49]. This age-related mutational profile, as well as unknown molecular factors, may contribute to the resistant biology that leads to higher relapse rates and an adverse prognosis for older adults with AML. The integration of age into prognostic models partially compensated for some of the age-related adverse biology. This approach, however, cannot fully account for the intra- and inter-patient heterogeneity in AML blasts from older adults, and as such, remains a relatively imprecise surrogate for the biological factors causing resistance in older patients. Investigations into the biology governing resistance in older adults with AML are warranted to elucidate the molecular factors responsible for the poor outcomes.
The ELN recently integrated mutations in ASXL1, RUNX1, and TP53 into their guidelines. In addition, the ELN-2017 guidelines now require double CEBPA mutations for patients to be deemed favorable risk. These changes require either a part of or the entire reading frame of genes to be sequenced. Such sequencing technology is either not available or may be cost-prohibitive in many areas. To better understand the prognostic benefit of these changes, we evaluated the performance of a modified model (ELN2017-MOD), which excluded the mutation data for ASXL1, RUNX1, TP53 and CEBPA. The ELN2017-MOD had a similar performance to the ELN2017 model. Inclusion of age into the model (AGE + ELN2017-MOD) demonstrated an improved performance over the ELN2017-MOD model. While a small number of patients changed risk group between the ELN2017 and ELN2017-MOD models, the incremental improvement does not negate the potential individual prognostic value of these additional mutations.
The studies also examined the prognostic impact of testing biomarkers in a more homogenous cell population (i.e., VLBs). The concordance in dichotomous mutation calls was almost 100% between MNCs and VLBs. Assays employed to detect mutation (fragment analyses PCR for FLT3-ITD and targeted deep sequencing for ASXL1, CEBPA, NPM1, RUNX1 and TP53) paralleled those currently used in clinical testing. However, the sequencing depth of the experiments were not intended to detect very low mutation loads, and as such, sequencing at higher depths may have yielded different results. Unlike dichotomous results, the FLT3-ITD AR was higher in VLBs than MNCs, resulting in a shift of the risk classification for 19 patients. Nevertheless, these differences in risk classification did not markedly impact the prognostic significance of the biomarker by itself or when incorporated into models. The transcript biomarkers were primarily selected based on their reported promise as prognostic biomarkers, and some previously validated transcript biomarkers, such as those involving leukemia stem cell signatures, were not examined [50,51,52,53]. Similarly, we assayed expression of select transcripts via q-RT/PCR due to the focused nature of the studies and global transcription profiling was not performed. Although expression of the examined transcripts in VLBs did not markedly improve the predictive accuracy of the models, the analyses confirmed that expression of the transcript biomarkers significantly differs between MNCs and VLBs, with most transcripts being expressed at higher levels in the VLBs. Therefore, it remains unclear whether examining VLBs may or may not provide a mechanism to identify novel prognostic biomarkers or improve the prognostic performance of other transcript biomarkers. Studies are currently underway to examine these questions using a more comprehensive approach, which includes global RNA sequencing of the MNCs and VLBs.
Although the current report represents the largest analysis of paired MNCs and VLBs from AML patients, the number and source (i.e., BM vs. PB) of samples may limit the ability to detect significant differences between models utilizing MNCs vs. VLBs. The number of examined patients was limited by the availability of specimens with adequate vials and the resources. Nevertheless, the data suggest that prognostic biomarkers (e.g., FLT3-ITD AR) yield different results depending upon the examined material (i.e., MNCs vs. VLBs) and highlight the need to identify additional biomarkers to improve current risk stratification guidelines. Unfortunately, large numbers of paired diagnostic BM and PB samples are not readily available for correlative studies to evaluate the impact of specimen source. However, some comparisons between MNCs from paired BM and PB have been performed by our group and others. While some report potential functional differences [54], others found subtle differences between the two tissue sources [55, 56]. Our previous examination of transcript and mutation biomarkers in paired BM and PB samples did not find any significant differences between unsorted MNCs from the PB versus BM with respect to the immunophenotype of leukemic blasts, mutation detection in FLT3 and NPM1 genes, relative quantities of mutations (allelic ratio of FLT3-ITD and NPM1 insertions), or the expression of majority of specific transcripts reported in this paper [6]. These additional biomarker studies will likely require investigations into previously untapped molecular components driving the biology of AML such as the proteome. As a means to improve the homogeneity of treatment, the study examined only those patients who received intensive chemotherapy with curative intent as part of SWOG trials. Despite randomization, the more recent trials were better represented in evaluated populations than older trials, however, the treatment regimens were comparable among the four trials from which the patients were drawn. Thus, the results may not be generalizable to patients receiving therapy outside of evaluated clinical trials, low-intensity regimens (e.g., azacytidine), or targeted agents (e.g., midostaurin). Nonetheless, some recent biomarker studies suggest that previously recognized prognostic factors remain highly informative and predictive for responses to more “targeted’ agents [57,58,59], and as such, there likely remains some role for the identification of prognostic biomarkers that are applicable across a variety of therapies.
Conclusions
In summary, this study represents the largest systematic evaluation of prognostic biomarkers in paired MNC and VLB from patients with AML. Overall, the ELN-2017 guidelines risk stratified younger adults with AML more accurately than older adults with AML. In addition, models developed utilizing ELN-2017 guidelines and other selected biomarkers did not substantially improve risk stratification. Similarly, the performance of these models was not significantly impacted by the source of material examined, (i.e., MNC vs. VLB).
Availability of data and materials
The datasets used and/or analyses used in the current study are available from the corresponding author on reasonable request. Clinical data pertaining to individual patients are available from SWOG upon request and execution of a data use agreement per SWOG and NCI policy.
Abbreviations
- Adv.:
-
Adverse
- AIC:
-
Aikike Information Criterion
- AML:
-
Acute myeloid leukemia
- ANC:
-
Absolute neutrophil count
- AR:
-
Allelic ratio
- AUC:
-
Area under the Receiver Operating Characteristic curve
- BCCA:
-
British Columbia Cancer Agency
- BWA:
-
Burrows-Wheeler Aligner
- Cont.:
-
Continuous
- CR:
-
Complete remission
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DN:
-
De novo
- DNA:
-
Deoxyribonucleic acid
- ELN:
-
European LeukemiaNet
- FAB:
-
French-American-British
- FACS:
-
Fluorescence-activated cell sorting
- Fav.:
-
Favorable
- FC:
-
Fold Change
- GATK:
-
Genome Analysis Toolkit
- Interm.:
-
Intermediate
- IP:
-
Immunophenotype
- IQR:
-
Interquartile range
- ITD:
-
Internal tandem duplication
- MNCs:
-
Mononuclear Cells
- MOD:
-
Modified
- OS:
-
Overall survival
- PS:
-
Performance status
- QC:
-
Quality control
- RFS:
-
Relapse-free survival
- RNA:
-
Ribonucleic acid
- Ukn.:
-
Unknown
- Unfav.:
-
Unfavorable
- VAF:
-
Variant allele frequency
- VLBs:
-
Viable leukemic blasts
References
Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47.
Boddu PC, Kadia TM, Garcia-Manero G, Cortes J, Alfayez M, Borthakur G, et al. Validation of the 2017 European LeukemiaNet classification for acute myeloid leukemia with NPM1 and FLT3-internal tandem duplication genotypes. Cancer. 2019;125(7):1091–100.
Harada Y, Nagata Y, Kihara R, Ishikawa Y, Asou N, Ohtake S, et al. Prognostic analysis according to the 2017 ELN risk stratification by genetics in adult acute myeloid leukemia patients treated in the Japan adult leukemia study group (JALSG) AML201 study. Leuk Res. 2018;66:20–7.
Tsai CH, Yao CY, Tien FM, Tang JL, Kuo YY, Chiu YC, et al. Incorporation of long non-coding RNA expression profile in the 2017 ELN risk classification can improve prognostic prediction of acute myeloid leukemia patients. EBioMedicine. 2019;40:240–50.
Lazarevic VL, Bredberg A, Lorenz F, Ohlander E, Antunovic P, Cammenga J, et al. Acute myeloid leukemia in very old patients. Haematologica. 2018;103(12):e578–e80.
Pogosova-Agadjanyan EL, Moseley A, Othus M, Appelbaum FR, Chauncey TR, Chen IL, et al. Impact of specimen heterogeneity on biomarkers in repository samples from patients with acute myeloid leukemia: a SWOG report. Biopreservation biobanking. 2018;16(1):42–52.
Ostronoff F, Othus M, Lazenby M, Estey E, Appelbaum FR, Evans A, et al. Prognostic significance of NPM1 mutations in the absence of FLT3-internal tandem duplication in older patients with acute myeloid leukemia: a SWOG and UK National Cancer Research Institute/Medical Research Council report. J Clin Oncol. 2015;33(10):1157–64.
Appelbaum FR, Gundacker H, Head DR, Slovak ML, Willman CL, Godwin JE, et al. Age and acute myeloid leukemia. Blood. 2006;107(9):3481–5.
Hernandez-Boluda JC, Sierra J, Esteve J, Nomdedeu B, Montserrat E. Treatment of elderly patients with AML: results of an individualized approach. Haematologica. 1998;83(1):34–9.
Eisfeld AK, Kohlschmidt J, Mrozek K, Blachly JS, Walker CJ, Nicolet D, et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years or older who respond favorably to standard chemotherapy: an analysis of Alliance studies. Leukemia. 2018;32(6):1338–48.
Heiblig M, Labussiere-Wallet H, Nicolini FE, Michallet M, Hayette S, Sujobert P, et al. Prognostic Value of Genetic Alterations in Elderly Patients with Acute Myeloid Leukemia: A Single Institution Experience. Cancers (Basel). 2019;11(4):570. https://doi.org/10.3390/cancers11040570.
Tsai CH, Hou HA, Tang JL, Liu CY, Lin CC, Chou WC, et al. Genetic alterations and their clinical implications in older patients with acute myeloid leukemia. Leukemia. 2016;30(7):1485–92.
Schwind S, Marcucci G, Maharry K, Radmacher MD, Mrozek K, Holland KB, et al. BAALC and ERG expression levels are associated with outcome and distinct gene and microRNA expression profiles in older patients with de novo cytogenetically normal acute myeloid leukemia: a Cancer and leukemia group B study. Blood. 2010;116(25):5660–9.
Langer C, Radmacher MD, Ruppert AS, Whitman SP, Paschka P, Mrozek K, et al. High BAALC expression associates with other molecular prognostic markers, poor outcome, and a distinct gene-expression signature in cytogenetically normal patients younger than 60 years with acute myeloid leukemia: a Cancer and leukemia group B (CALGB) study. Blood. 2008;111(11):5371–9.
Baldus CD, Thiede C, Soucek S, Bloomfield CD, Thiel E, Ehninger G. BAALC expression and FLT3 internal tandem duplication mutations in acute myeloid leukemia patients with normal cytogenetics: prognostic implications. J Clin Oncol. 2006;24(5):790–7.
Miglino M, Colombo N, Pica G, Grasso R, Clavio M, Bergamaschi M, et al. WT1 overexpression at diagnosis may predict favorable outcome in patients with de novo non-M3 acute myeloid leukemia. Leuk Lymphoma. 2011;52(10):1961–9.
Rockova V, Abbas S, Wouters BJ, Erpelinck CA, Beverloo HB, Delwel R, et al. Risk stratification of intermediate-risk acute myeloid leukemia: integrative analysis of a multitude of gene mutation and gene expression markers. Blood. 2011;118(4):1069–76.
Testa U, Riccioni R, Diverio D, Rossini A, Lo Coco F, Peschle C. Interleukin-3 receptor in acute leukemia. Leukemia. 2004;18(2):219–26.
Nakamaki T, Hamano Y, Hisatake J, Yokoyama A, Kawakami K, Tomoyasu S, et al. Elevated levels of cyclin A1 and a (A2) mRNA in acute myeloid leukaemia are associated with increased survival. Br J Haematol. 2003;123(1):72–80.
Becker H, Marcucci G, Maharry K, Radmacher MD, Mrozek K, Margeson D, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and leukemia group B study. J Clin Oncol. 2010;28(4):596–604.
Groschel S, Schlenk RF, Engelmann J, Rockova V, Teleanu V, Kuhn MW, et al. Deregulated expression of EVI1 defines a poor prognostic subset of MLL-rearranged acute myeloid leukemias: a study of the German-Austrian acute myeloid leukemia study group and the Dutch-Belgian-Swiss HOVON/SAKK cooperative group. J Clin Oncol. 2013;31(1):95–103.
Stirewalt DL, Meshinchi S, Kopecky KJ, Fan W, Pogosova-Agadjanyan EL, Engel JH, et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer. 2008;47(1):8–20 PMID:17910043.
Reuss-Borst MA, Buhring HJ, Schmidt H, Muller CA. AML: immunophenotypic heterogeneity and prognostic significance of c- kit expression. Leukemia. 1994;8(2):258–63.
Schwind S, Marcucci G, Kohlschmidt J, Radmacher MD, Mrozek K, Maharry K, et al. Low expression of MN1 associates with better treatment response in older patients with de novo cytogenetically normal acute myeloid leukemia. Blood. 2011;118(15):4188–98.
Fu L, Fu H, Tian L, Xu K, Hu K, Wang J, et al. High expression of RUNX1 is associated with poorer outcomes in cytogenetically normal acute myeloid leukemia. Oncotarget. 2016;7(13):15828–39.
Niavarani A, Herold T, Reyal Y, Sauerland MC, Buchner T, Hiddemann W, et al. A 4-gene expression score associated with high levels of Wilms Tumor-1 (WT1) expression is an adverse prognostic factor in acute myeloid leukaemia. Br J Haematol. 2016;172(3):401–11.
Ochsenreither S, Majeti R, Schmitt T, Stirewalt D, Keilholz U, Loeb KR, et al. Cyclin-A1 represents a new immunogenic targetable antigen expressed in acute myeloid leukemia stem cells with characteristics of a cancer-testis antigen. Blood. 2012;119(23):5492–501.
Anderson JE, Kopecky KJ, Willman CL, Head D, O'Donnell MR, Luthardt FW, et al. Outcome after induction chemotherapy for older patients with acute myeloid leukemia is not improved with mitoxantrone and etoposide compared to cytarabine and daunorubicin: a southwest oncology group study. Blood. 2002;100(12):3869–76.
Godwin JE, Kopecky KJ, Head DR, Willman CL, Leith CP, Hynes HE, et al. A double-blind placebo-controlled trial of granulocyte colony-stimulating factor in elderly patients with previously untreated acute myeloid leukemia: a southwest oncology group study (9031). Blood. 1998;91(10):3607–15.
Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121(24):4854–60.
Chauncey TR, Gundacker H, Shadman M, List AF, Dakhil SR, Erba HP, et al. Sequential phase II southwest oncology group studies (S0112 and S0301) of daunorubicin and cytarabine by continuous infusion, without and with ciclosporin, in older patients with previously untreated acute myeloid leukaemia. Br J Haematol. 2010;148(1):48–58.
Suresh K. An overview of randomization techniques: an unbiased assessment of outcome in clinical research. J Hum Reprod Sci. 2011;4(1):8–11.
Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. 2006;108(12):3654–61.
Stirewalt DL, Kopecky KJ, Meshinchi S, Appelbaum FR, Slovak ML, Willman CL, et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97(11):3589–95 PMID:11369655.
Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–35.
Au CH, Wa A, Ho DN, Chan TL, Ma ES. Clinical evaluation of panel testing by next-generation sequencing (NGS) for gene mutations in myeloid neoplasms. Diagn Pathol. 2016;11:11.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Goode DL, Hunter SM, Doyle MA, Ma T, Rowley SM, Choong D, et al. A simple consensus approach improves somatic mutation prediction accuracy. Genome Med. 2013;5(9):90.
Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31(10):1536–43.
Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–D83.
Hornung RW, Reed LD. Estimation of average concentration in the presence of nondetectable values. Appl Occup Environ Hyg. 1990;5(1):46–51.
Team RC. A language and environment for statistical computing: R Foundation for statistical computing; 2017 Available from: www.R-project.org.
Appelbaum FR. What is the impact of hematopoietic cell transplantation (HCT) for older adults with acute myeloid leukemia (AML)? Best Pract Res Clin Haematol. 2008;21(4):667–75.
Cancer Stat Facts: Leukemia - Acute Myeloid Leukemia (AML) [Internet]. NIH. 2019. Available from: https://seer.cancer.gov/statfacts/html/amyl.html. Accessed 16 May 2019.
Eisfeld AK, Mrozek K, Kohlschmidt J, Nicolet D, Orwick S, Walker CJ, et al. The mutational oncoprint of recurrent cytogenetic abnormalities in adult patients with de novo acute myeloid leukemia. Leukemia. 2017;31(10):2211–8.
Hou HA, Liu CY, Kuo YY, Chou WC, Tsai CH, Lin CC, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget. 2016;7(8):9084–101.
Cao XX, Cai H, Mao YY, Wu Q, Zhang L, Zhou DB, et al. Next-generation sequencing-based genetic landscape and its clinical implications for Chinese acute myeloid leukemia patients. Cancer Cell Int. 2018;18:215.
Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Gorlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686–98.
Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–93.
Metzeler KH, Maharry K, Kohlschmidt J, Volinia S, Mrozek K, Becker H, et al. A stem cell-like gene expression signature associates with inferior outcomes and a distinct microRNA expression profile in adults with primary cytogenetically normal acute myeloid leukemia. Leukemia. 2013;27(10):2023-31. https://doi.org/10.1038/leu.2013.181.
Ng SW, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433–7.
Duployez N, Marceau-Renaut A, Villenet C, Petit A, Rousseau A, Ng SWK, et al. The stem cell-associated gene expression signature allows risk stratification in pediatric acute myeloid leukemia. Leukemia. 2019;33(2):348–57.
Sellar RS, Fraser L, Khwaja A, Gale RE, Marafioti T, Akarca A, et al. Cell cycle status in AML blast cells from peripheral blood, bone marrow aspirates and trephines and implications for biological studies and treatment. Br J Haematol. 2016;174(2):275–9.
Hutter G, Letsch A, Nowak D, Poland J, Sinha P, Thiel E, et al. High correlation of the proteome patterns in bone marrow and peripheral blood blast cells in patients with acute myeloid leukemia. J Transl Med. 2009;7:7.
Tong WG, Sandhu VK, Wood BL, Hendrie PC, Becker PS, Pagel JM, et al. Correlation between peripheral blood and bone marrow regarding FLT3-ITD and NPM1 mutational status in patients with acute myeloid leukemia. Haematologica. 2015;100(3):e97–8.
Fournier E, Duployez N, Ducourneau B, Raffoux E, Turlure P, Caillot D, et al. Mutational profile and benefit of gemtuzumab ozogamicin in acute myeloid leukemia patients treated in the ALFA0701 trial. Blood. 2019.
DiNardo CD, Tiong IS, Quaglieri A, MacRaild S, Loghavi S, Brown FC, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020.
Dohner K, Thiede C, Jahn N, Panina E, Gambietz A, Larson RA, et al. Impact of NPM1/FLT3-ITD genotypes defined by the 2017 European LeukemiaNet in patients with acute myeloid leukemia. Blood. 2020;135(5):371–80.
Acknowledgements
The authors wish to gratefully acknowledge the important contributions of the late Dr. Stephen H. Petersdorf to SWOG and to study S0106.
Trial registration
N/A
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers CA114563, CA160872, CA180819, CA180828, CA180861, CA180888, CA190661 and CA196175. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Concept and design: ELPA, AM, MO, SM, ES and DLS. Financial support: SM and DLS; Provision of study materials or patients: FRA, TRC, IMLC, HPE, JEG, AFL, JPR, CLW, and DLS; Collection and assembly the data: ELPA, AM, MO, IMLC, MF, KJK, MH, BEW, ES, JN, CLW, BLW, QZ, SM and DLS; Data analysis and interpretation: ELPA, AM, MO, FRA, TRC, IMLC, HPE, JEG, ICJ, MF, MH, KJK, AFL, JN, JPR, ES, BEW, CLW, BLW, QZ, SM, and DLS; Manuscript writing: ELPA, AM, MO, FRA, TRC, IMLC, HPE, JEG, ICJ, MF, MH, KJK, AFL, JN, JPR, ES, BEW, CLW, BLW, QZ, SM, and DLS; Final approval of the manuscript: ELPA, AM, MO, FRA, TRC, IMLC, HPE, JEG, ICJ, MF, MH, KJK, AFL, JN, JPR, ES, BEW, CLW, BLW, QZ, SM, and DLS.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participants provided written informed consent to participate in correlative research in compliance with the Declaration of Helsinki. All studies were conducted with the approval of Fred Hutch Institution Review Board.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Targeted Sequencing Details. Table S1A. Target regions for Wafergen Sequencing. Table S1B Primers and Amplicon Details for Wafergen Sequencing. Table S1C. Primers for CEBPA targeted MiSeq Assay. Table S2. Loci that Failed Quality Control. Table S3. Characteristics of selected and unselected SWOG patients. Fig. S1. Comparison of Performance Characteristics of selected and unselected SWOG patients. Table S4. Characteristics of SWOG patients in the discovery and validation cohorts. Fig. S2. Comparison of Performance Characteristics of SWOG patients in the discovery and validation cohorts. Table S5. Mutation distribution in discovery and validation cohorts. Fig. S3. Mutation Frequency (OncoPrint). Table S6. Expression fold change differences between MNCs and VLBs in discovery cohort. Table S7. Univariate analyses results, non-significant findings Models Details. Table S8. ELN2017 versus ELN2017-MOD risk assignment in MNCs and VLBs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pogosova-Agadjanyan, E.L., Moseley, A., Othus, M. et al. AML risk stratification models utilizing ELN-2017 guidelines and additional prognostic factors: a SWOG report. Biomark Res 8, 29 (2020). https://doi.org/10.1186/s40364-020-00208-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-020-00208-1