
Abstract
Determining the effects of antimicrobial therapies on airway microbiology at a population-level is essential. Such analysis allows, for example, surveillance of antibiotic-induced changes in pathogen prevalence, the emergence and spread of antibiotic resistance, and the transmission of multi-resistant organisms. However, current analytical strategies for understanding these processes are limited. Culture- and PCR-based assays for specific microbes require the a priori selection of targets, while antibiotic sensitivity testing typically provides no insight into either the molecular basis of resistance, or the carriage of resistance determinants by the wider commensal microbiota. Shotgun metagenomic sequencing provides an alternative approach that allows the microbial composition of clinical samples to be described in detail, including the prevalence of resistance genes and virulence traits. While highly informative, the application of metagenomics to large patient cohorts can be prohibitively expensive. Using sputum samples from a randomised placebo-controlled trial of erythromycin in adults with bronchiectasis, we describe a novel, cost-effective strategy for screening patient cohorts for changes in resistance gene prevalence. By combining metagenomic screening of pooled DNA extracts with validatory quantitative PCR-based analysis of candidate markers in individual samples, we identify population-level changes in the relative abundance of specific macrolide resistance genes. This approach has the potential to provide an important adjunct to current analytical strategies, particularly within the context of antimicrobial clinical trials.
Similar content being viewed by others

Introduction
As in all clinical disciplines, the management of patients with chronic respiratory diseases is subject to a process of ongoing refinement, including through the development of novel antimicrobial drugs and treatment strategies. Understanding the impact of antimicrobial treatments for individual recipients allows the personalisation of clinical management. However, determining the effects of treatments at a population level is also crucial, providing a means to predict shifts in the prevalence of respiratory pathogens, or the emergence of antimicrobial resistance, within large patient groups.
The impact that evolving treatment strategies can have on airway microbiology can be seen, for example, in changes in the cystic fibrosis (CF) airway microbiota during recent decades. Within this context, the use of anti-Pseudomonal treatments, including parenteral therapies and fluoroquinolones, have been implicated in the emergence of Stenotrophomonas maltophilia as an airway pathogen [1, 2]. Likewise, increasingly intensive antibiotic use appears to be a contributory factor in the increasing prevalence of non-tuberculous mycobacteria [3, 4]. The impact of antibiotic use is also reflected in the increasing frequency of multi-drug resistant organisms in the airways of patients with chronic respiratory disease, with an estimated 25–45% of adult CF patients chronically infected with multi-drug resistant bacteria [5]. For example, CF-derived methicillin-resistant Staphylococcus aureus (MRSA) isolates increasingly show resistance to newer therapies, including linezolid [6, 7], ceftaroline [8] and tigecycline [6], presumably as a result of frequent and prolonged exposures [9].
Despite the importance of understanding the impact of antimicrobial exposure on the airway microbiome in those with respiratory disease, characterising this process remains challenging. Assessments of antibiotic-associated changes in microbiology are typically limited to a small group of predefined pathogens or resistance genes. The standard analytical approaches employed in clinical antibiotic trials fail to assess major aspects of antibiotic resistance, including shifts in the composition of the wider airway microbiota, and the carriage of transmissible resistance determinants by populations of commensal microbes. The absence of suitable strategies to determine antibiotic impact has resulted in significant gaps in our understanding of how widely employed therapies affect the complex microbiota of the respiratory tract.
Shotgun metagenomic sequencing is an emerging technology that allows highly detailed characterisation of airway microbiota through the analysis of total microbial DNA from clinical samples, including determination of the prevalence of virulence factors and resistance determinants [10]. While metagenomic approaches have been shown to be highly effective in describing changes in the microbiome across a wide range of clinical contexts [11], the cost of its employment within population-scale studies is commonly prohibitive.
We describe a novel, cost-effective, strategy to inform the use of assays for specific resistance genes or microbial taxa, based on deep metagenomic screening of pooled study cohort DNA. We illustrate the application of this approach through the analysis of samples from a previously reported randomised controlled trial of long-term low dose macrolide therapy in adults with bronchiectasis.
Methods
The BLESS randomised placebo-controlled trial assessed the effect of 12 months of low dose erythromycin therapy (twice-daily erythromycin ethylsuccinate; 400 mg) on exacerbation rates in adults with non-CF bronchiectasis [12]. The analysis reported here was based on paired baseline and week 48 sputum samples from 32 members of the treatment group, and subsequent analysis between treatment group and placebo group subjects (n = 32, and n = 31, respectively). Patient baseline characteristics are described in Additional file 1: Table S1.
Sputum DNA extracts were pooled according to time-point and subject to microbial DNA enrichment (NEBNext® Microbiome DNA Enrichment Kit). DNA fragmentation was performed with TruSeq Nano DNA Library Prep Kit (Illumina), prior to 150 bp paired-end metagenomic shotgun sequencing using the Illumina HiSeq system at the South Australian Health and Medical Research Institute, Adelaide. Reads have been uploaded to the Sequence Read Archive (SRA) under BioProject ID: 397083.
Sequences were quality filtered using Trimmomatic v0.32 [13] and human-derived reads removed using BBMap v35.40 (comparing reads to the NCBI human reference genome release GRCh38) [14]. Contigs were de novo assembled using IDBA-UD v1.1.1 [15], followed by identification of open-reading frames using MetaGeneMark v3.26 [16]. A non-redundant gene catalogue was constructed using CDHIT v4.6 [17] and resistome composition annotated by BLASTP search to the Comprehensive Antibiotic Resistance Database (CARD) v1.1.7 [18]. Quantification of gene hits was determined by SOAP v2.20 [19] and normalised to counts per million reads.
Specific resistance genes that were identified as associated with erythromycin treatment through metagenomics were subsequently quantified in DNA extracts from individual sputum samples by qPCR. Previously published assays were used for ermA [20], ermB [21], ermC [22], 16S [23], and smpB [24] genes. Primers for quantification of hmrM were designed within this study (see Additional file 1). For analysis of qPCR results, Wilcoxon rank tests were performed on fold change normalised to 16S copy number to compare erythromycin paired samples to placebo control paired samples (n = 31 pairs).
Results
A schematic of the pooled-template metagenomic sequencing strategy, and subsequent qPCR-based validation, is shown in Fig. 1. Following removal of low-quality reads and human DNA (approximately 90% of total read depth), a mean sample read depth of 12,866,780 was achieved. Approximately half a million reads has been previously found to be sufficient for sputum metagenome analysis in individual samples [25]. Mapping of sequence reads to the CARD database resulted in the detection of a total of 102 resistance-associated genetic determinants. The distribution of normalised reads that mapped to the CARD database in pre- and post-trial pooled samples is shown in Fig. 2. Detected genes represented a range of resistance mechanisms, including antibiotic inactivating enzymes, efflux pumps, and effector site protection proteins, and conferred resistance to a number of antibiotic classes, including aminoglycosides, beta-lactams, glycopeptides, and tetracyclines.

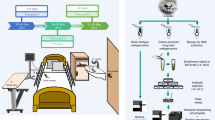
Principle of pooled-template metagenomic sequencing. Sample DNA extracts from a population of interest are pooled together according to a pre-specified variable of interest (such as treatment or time-point). Metagenomic sequencing is then performed on pooled samples and regions that discriminate between populations are determined. Targeted assays (such as qPCR) are then performed on individual samples for gene specific enumeration

Resistome of pooled-template sputum before and after erythromycin therapy. Square root-transformed counts per million total reads (CPM) of major antibiotic resistance genes identified by CARD database. Change in CPM where red indicates higher in samples post erythromycin. Resistance genes grouped by function as defined by CARD where: brown = aminoglycoside resistance genes, red = beta-lactam resistance genes, orange = efflux pump resistance genes, yellow = glycopeptide resistance genes, green = tetracycline resistance genes, blue = other resistance genes
A substantial proportion of the genes identified through resistome analysis were chromosomally-encoded, non-transmissible, resistance determinants. Changes in the level of carriage of these genes during the trial therefore reflected shifts in the relative abundance of the species in whose genomes they are encoded, rather than resistance gene acquisition or loss. For example, the multidrug efflux pump gene, hmrM, appeared to increase in response to erythromycin therapy. This gene is chromosomally-encoded by Haemophilus influenzae however, and subsequent qPCR analysis revealed hmrM levels to be correlated with H. influenzae levels (r = 0.74, p < 0.001, Fig. 3). The observed increase in prevalence of hmrM is therefore likely to simply reflect an increase in the relative abundance in H. influenzae in the assessed patient group (a median increase of 1.4 × 103 copies was observed between pre- and post-erythromycin samples). This phenomenon could explain apparent changes in the group-level abundance of other chromosomally-encoded resistance genes, such as an observed decrease in the relative abundance of patA, a chromosomally-encoded fluoroquinolone resistance gene carried by Streptococcus pneumoniae [26], and aph(3′)-IIb, a chromosomally-encoded aminoglycoside resistance gene carried by Pseudomonas aeruginosa [27].

Correlation between hmrM and H. influenzae copy number. hmrM (normalised to total bacteria) against H. influenzae copy number (determined by comparing to known standard curve). Significance determined by Spearman’s rank order correlation
Many of the other resistance genes identified through pooled-template metagenomic sequencing were, however, encoded on mobile genetic elements, and have been shown previously to be transmissible between bacterial species. These include a number of transmissible genetic elements that confer resistance to macrolide antibiotics. For example, qPCR-based validation analysis, revealed a significant increase in the relative abundance of the plasmid-encoded erythromycin resistance methylase gene, ermB, in subjects who received erythromycin (p = 0.007), but not in those who received placebo (p = 0.073, Fig. 4). The ermB gene can be carried by a number of respiratory pathogens, including S. pneumoniae, S. aureus, and H. influenzae [28,29,30], and confers substantial resistance to all macrolide drugs. In contrast, other transmissible macrolide-resistance determinants were shown by follow-up qPCR analysis to not contribute substantially to the post-trial resistome. For example, ermA, a resistance gene found in staphylococci [29], was present in only four subjects (two in the treatment group and two in the control group). The ermC resistance determinant, which is also found in staphylococci [29], was detected more frequently (68% of subjects receiving placebo and 81% of subjects receiving erythromycin), however, ermC levels did not change significantly over the course of the trial. The rates of carriage of ermA and ermC are consistent with those reported in S. aureus clinical isolates more widely [29, 31].

Changes in ermB levels in erythromycin and placebo groups. Paired sample analysis of ermB (normalised to total bacteria). Significance determined by Wilcoxon signed-rank test
Discussion and conclusion
We describe a cost effective approach that can be used to guide the assessment of changes in antibiotic resistance gene carriage, which might represent a useful adjunct to conventional approaches that are based on a priori target selection. As an illustration, the BLESS randomised placebo-controlled trial that preceded this study included an assessment of whether erythromycin therapy resulted in an increased relative abundance of macrolide resistant oropharyngeal streptococci using culture-based proportional sensitivity testing [12]. While this narrow analysis identified a significant increase in the proportion of macrolide-resistant streptococci, neither the level of transmissible resistance gene carriage in non-streptococcal species, nor the molecular basis of resistance, were determined. Our use of pooled metagenomic sequencing revealed a number of resistance determinants for follow-up analysis where targeted qPCR assays were subsequently applied to DNA extracts from individual samples. This validation step confirmed significant increases in the abundance of, for example, the transmissible macrolide resistance gene, ermB, in patients receiving erythromycin.
By pooling sample DNA at the pre-sequencing, rather the post-sequencing, library-construction stage (as performed in standard metagenomic sequencing approaches), we calculate the cost of our analysis to be approximately 15% of that required to analyse all of the samples individually (although precise costs will be influenced by sample number, processing methodologies, and desired sequencing depth). However, despite this substantial reduction in expense, it is important to be aware of some of the limitations that are inherent in this approach. For example, variations in bacterial load between samples from different patients mean that pooling DNA based on total concentration could result in the contribution of individual samples to meta-microbiome characteristics being unequal. In addition, the non-normal distribution of microbiome traits within a population could lead to the identification of traits as potential inter-group discriminators based on their particularly high abundance in a small number of individuals (although the impact of this effect is likely to decrease with increasing cohort size).
A limitation of all metagenomic sequencing is the challenge to differentiate between changes in the carriage of resistance determinants due to direct selective pressure versus changes in resistance gene carriage, because of shifts in the relative abundance of the bacterial populations that encode them. Due to such limitations, the approach that we describe should be used as an additional means to identify markers for further analysis, rather than as a means to characterise antibiotic associated effects on airway microbiology in itself.
As an illustration of the potential of the pooled-template metagenomic analysis, we examined shifts in the airway resistome. This application targeted the global health concern of monitoring of antibiotic resistance. Patients with chronic lung diseases have an increased exposure to antibiotics, with the emergence of resistance correlating closely with consumption [32]. The resistome associated with the airway microbiota in these patients is likely to be a substantial contributor to the emergence and expansion of populations of multi-resistant organisms [33] and their potential transmission to individuals within the wider community. However, despite its application to the assessment of the airway resistome here, pooled-template metagenomic analysis can be applied equally to assessment of species distribution [34], or to identify changes in community level carriage of pathogenicity traits (for example, through alignment to virulence factor genetic databases). By aligning regions that encode antibiotic binding sites, it may also be possible to determine the relative abundance of resistance-conferring single nucleotide polymorphisms (SNPs). Obtaining such information could provide important clinical insight. For example, while de novo mutations in the 23S rRNA are the principal cause of macrolide resistance in non-tuberculous mycobacteria [35], relatively little is known currently about the dynamics of their emergence during macrolide therapy.
The costs of metagenomic sequencing, and the associated costs of high performance computing required for bioinformatic analysis, are likely to continue to fall. However, by providing a low-cost means to perform unbiased surveys of large patient cohorts, strategies such as the one described here represent a useful means of identifying potentially important discriminatory microbiome features for follow-up analysis.
References
Burns JL, Van Dalfsen JM, Shawar RM, Otto KL, Garber RL, Quan JM, et al. Effect of chronic intermittent administration of inhaled tobramycin on respiratory microbial flora in patients with cystic fibrosis. J Infect Dis. 1999;179(5):1190–6.
Denton M, Todd NJ, Littlewood JM. Role of anti-pseudomonal antibiotics in the emergence of Stenotrophomonas maltophilia in cystic fibrosis patients. Eur J Clin Microbiol Infect Dis. 1996;15(5):402–5.
Bar-On O, Mussaffi H, Mei-Zahav M, Prais D, Steuer G, Stafler P, et al. Increasing nontuberculous mycobacteria infection in cystic fibrosis. J Cyst Fibros. 2015;14(1):53–62.
Catherinot E, Roux AL, Vibet MA, Bellis G, Ravilly S, Lemonnier L, et al. Mycobacterium avium and Mycobacterium abscessus complex target distinct cystic fibrosis patient subpopulations. J Cyst Fibros. 2013;12(1):74–80.
Lechtzin N, John M, Irizarry R, Merlo C, Diette GB, Boyle MP. Outcomes of adults with cystic fibrosis infected with antibiotic-resistant Pseudomonas aeruginosa. Respiration. 2006;73(1):27–33.
Champion EA, Miller MB, Popowitch EB, Hobbs MM, Saiman L, Muhlebach MS, et al. Antimicrobial susceptibility and molecular typing of MRSA in cystic fibrosis. Pediatr Pulmonol. 2014;49(3):230–7.
Hill RL, Kearns AM, Nash J, North SE, Pike R, Newson T, et al. Linezolid-resistant ST36 methicillin-resistant Staphylococcus aureus associated with prolonged linezolid treatment in two paediatric cystic fibrosis patients. J Antimicrob Chemother. 2010;65(3):442–5.
Long SW, Olsen RJ, Mehta SC, Palzkill T, Cernoch PL, Perez KK, et al. PBP2a mutations causing high-level Ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob Agents Chemother. 2014;58(11):6668–74.
Parkins MD, Floto RA. Emerging bacterial pathogens and changing concepts of bacterial pathogenesis in cystic fibrosis. J Cyst Fibros. 2015;14(3):293–304.
Rogers GB, Shaw D, Marsh RL, Carroll MP, Serisier DJ, Bruce KD. Respiratory microbiota: addressing clinical questions, informing clinical practice. Thorax. 2015;70(1):74–81.
Wang J, Jia H. Metagenome-wide association studies: fine-mining the microbiome. Nat Rev Microbiol. 2016;14(8):508–22.
Serisier DJ, Martin ML, McGuckin MA, Lourie R, Chen AC, Brain B, et al. Effect of long-term, low-dose erythromycin on pulmonary exacerbations among patients with non-cystic fibrosis bronchiectasis: the BLESS randomized controlled trial. JAMA. 2013;309(12):1260–7.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Bushnell B. BBMap short read aligner. Berkeley: University of California; 2016. URL http://sourceforge.net/projects/bbmap
Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28(11):1420–8.
Besemer J, Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005;33(Web Server):W451–4.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–2.
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–73.
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–7.
Jung J, Yoon EJ, Choi EC, Choi SS. Development of TaqMan probe-based real-time PCR method for erm(a),erm(B), and erm(C), rapid detection of macrolide-lincosamide-streptogramin B resistance genes, from clinical isolates. J Microbiol Biotechnol. 2009;19(11):1464–9.
Zhang L, Kinkelaar D, Huang Y, Li Y, Li X, Wang HH. Acquired antibiotic resistance: are we born with it? Appl Environ Microb. 2011;77(20):7134–41.
Martineau F, Picard FJ, Grenier L, Roy PH, Ouellette M, Bergeron MG. Multiplex PCR assays for the detection of clinically relevant antibiotic resistance genes in staphylococci isolated from patients infected after cardiac surgery. The ESPRIT Trial. J Antimicrob Chemother. 2000;46(4):527–34.
Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148(1):257–66.
Reddington K, Schwenk S, Tuite N, Platt G, Davar D, Coughlan H. Comparison of established diagnostic methodologies and a novel bacterial smpB real-time PCR assay for specific detection of Haemophilus influenzae isolates associated with respiratory tract infections. J Clin Microbiol. 2015;53(9):2854–60.
Moran, Losada P, Chouvarine P, Dorda M, Hedtfeld S, Mielke S, Schulz A, et al. The cystic fibrosis lower airways microbial metagenome. ERJ Open Res. 2016;2(2).
El Garch F, Lismond A, Piddock LJ, Courvalin P, Tulkens PM, Van Bambeke F. Fluoroquinolones induce the expression of patA and patB, which encode ABC efflux pumps in Streptococcus pneumoniae. J Antimicrob Chemother. 2010;65(10):2076–82.
Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406(6799):959–64.
Kresken M, Henrichfreise B, Bagel S, Brauers J, Wiedemann B. High prevalence of the ermB gene among erythromycin-resistant Streptococcus pneumoniae isolates in Germany during the winter of 2000-2001 and in vitro activity of Telithromycin. Antimicrob Agents Chemother. 2004;48(8):3193–5.
Ghanbari F, Ghajavand H, Havaei R, Jami MS, Khademi F, Heydari L, et al. Distribution of erm genes among Staphylococcus aureus isolates with inducible resistance to clindamycin in Isfahan, Iran. Ad Biomed Res. 2016;5:62.
Tristram S, Jacobs MR, Appelbaum PC. Antimicrobial resistance in Haemophilus influenzae. Clin Microbiol Rev. 2007;20(2):368–89.
Aktas Z, Aridogan A, Kayacan CB, Aydin D. Resistance to macrolide, lincosamide and streptogramin antibiotics in staphylococci isolated in Istanbul. Turkey J Microbiol. 2007;45(4):286–90.
Goossens H. Antibiotic consumption and link to resistance. Clin Microbiol Infect. 2009;15(Suppl 3):12–5.
Sherrard LJ, Tunney MM, Elborn JS. Antimicrobial resistance in the respiratory microbiota of people with cystic fibrosis. Lancet. 2014;384(9944):703–13.
Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015;12(10):902–3.
Brown-Elliott BA, Nash KA, Wallace RJ Jr. Antimicrobial susceptibility testing, drug resistance mechanisms, and therapy of infections with nontuberculous mycobacteria. Clin Microbiol Rev. 2012;25(3):545–82.
Funding
Collection of data was internally funded by the South Australian Health and Medical Research Institute and supported by a project grant (PG1104000) from the Australian National Health and Medical Research Council (NHMRC). Publication costs of this article will be funded by Novamedia and Italian Respiratory Society (IRS).
About this supplement
This article has been published as part of Multidisciplinary Respiratory Medicine Volume 13 Supplement 1, 2018: Bronchiectasis: still an orphan disease?. The full contents of the supplement are available online at https://mrmjournal.biomedcentral.com/articles/supplements/volume-13-supplement-1.
Author information
Authors and Affiliations
Contributions
SLT and LEXL performed data collection; SLT, LEXL and FMM performed analyses; SLT, LEXL, LDB, JMC, and GBR, interpreted the data; SLT, SW, and GBR wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Mater Health Service human research ethics committee (ACTRN12609000578202), and all participants provided written, informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
hmrM primers and patient characteristics. (DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Taylor, S.L., Leong, L.E.X., Mobegi, F.M. et al. Understanding the impact of antibiotic therapies on the respiratory tract resistome: a novel pooled-template metagenomic sequencing strategy. Multidiscip Respir Med 13 (Suppl 1), 30 (2018). https://doi.org/10.1186/s40248-018-0140-9
Published:
DOI: https://doi.org/10.1186/s40248-018-0140-9