
Abstract
In recent years, many studies have investigated the correlations between Parkinson’s disease (PD) and vitamin D status, but the conclusion remains elusive. The present review focuses on the associations between PD and serum vitamin D levels by reviewing studies on the associations of PD with serum vitamin D levels and vitamin D receptor (VDR) gene polymorphisms from PubMed, Web of Science, Cochrane Library, and Embase databases. We found that PD patients have lower vitamin D levels than healthy controls and that the vitamin D concentrations are negatively correlated with PD risk and severity. Furthermore, higher vitamin D concentrations are linked to better cognitive function and mood in PD patients. Findings on the relationship between VDR gene polymorphisms and the risk of PD are inconsistent, but the FokI (C/T) polymorphism is significantly linked with PD. The occurrence of FokI (C/T) gene polymorphism may influence the risk, severity, and cognitive ability of PD patients, while also possibly influencing the effect of Vitamin D3 supplementation in PD patients. In view of the neuroprotective effects of vitamin D and the close association between vitamin D and dopaminergic neurotransmission, interventional prospective studies on vitamin D supplementation in PD patients should be conducted in the future.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is the most common type of parkinsonism, a syndrome characterized by bradykinesia, postural instability, rigidity, and resting tremor [1]. The pathophysiological cause of PD is the progressive loss of dopaminergic (DA) neurons in the substantia nigra (SN) of the midbrain [2,3,4] and the formation of Lewy bodies, which are neuronal inclusions mainly consisting of α-synuclein protein aggregations [5,6,7]. In high-income countries, the annual incidence of PD is 14 per 100,000 in the general population, and rises to 160 per 100,000 in the population aged 65 years or older [8]. A systematic review estimated that there were 6.1 million PD patients worldwide in the year 2016, a significant increase from 2.5 million in 1990, with further projected increases in the future. Moreover, the increase cannot entirely be explained by the growth of number of older people [9]. The etiology of PD remains unknown and is presumably multifactorial [10]. The exact mechanism of neurodegeneration in PD is not yet fully elucidated, but it likely involves a series of events including interactions between genetic and environmental factors, oxidative stress, mitochondrial dysfunction, inflammation, immune regulation, and others [11,12,13,14,15,16,17,18,19,20,21,22]. Due to the unclear etiology, no medications have been proven to cure PD [1]. Therefore, there is a critical demand for new and targeted drugs that focus on protecting DA neurons from degeneration in PD.
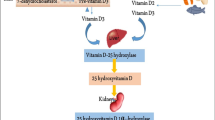
Vitamin D obtained via sun exposure or through the diet is converted by 25-hydroxylase mainly located in the liver into 25-hydroxyvitamin D (25(OH)D), the major circulating form of vitamin D. The 25(OH)D can be used as a serum marker to measure vitamin D levels in PD patients, however, it is biologically inactive and must be transformed into the active form 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) by 25-hydroxyvitamin D-1α-hydroxylase (CYP27B1) in the kidney. Increased concentrations of 1,25(OH)2D3 can raise the expression of 25-hydroxyvitamin D-24-hydroxylase (CYP24A1) to catabolize 1,25(OH)2D3 into calcitroic acid [23,24,25]. The biological functions of 1,25(OH)2D3 are mediated by the vitamin D receptor (VDR), a member of the nuclear receptor superfamily of transcription factors [25, 26]. Upon ligand binding, the VDR interacts with the retinoid X receptor (RXR) to form a heterodimer, which then binds to vitamin D response elements (VDREs) in target genes to promote their expression [27, 28]. It has been predicted that 1,25(OH)2D3 regulates more than 200 genes, influencing a variety of cellular processes (Fig. 1) [29]. There is ample evidence from in vitro and animal studies that vitamin D plays an important role in cell proliferation and differentiation, neurotrophic regulation and neuroprotection, neurotransmission, immune regulation, and neuroplasticity [30,31,32,33]. Studies have confirmed the presence of vitamin D metabolites, their metabolizing enzymes CYP27B1 and CYP24A1, as well as VDR in the human brain. This indicates that the human brain can regulate 1,25(OH)2D3 levels, and vitamin D may play a key role in the maintenance of normal nervous system function [34, 35]. Moreover, VDR and CYP27B1 expression is most abundant in the substantia nigra (SN; a brain region rich in dopaminergic neurons) according to immunofluorescence [36]. Studies have also found that the earliest time of VDR expression in the midbrain is on embryonic day 12 (E12), which coincides with the time of development of a majority of dopaminergic neurons in the SN region [37, 38]. Vitamin D is a fat-soluble hormone that can pass the blood-brain barrier, which supports its significance in the central nervous system [39].

Vitamin D metabolism. DBP, Vitamin D-binding protein; RXR, retinoid X receptor; VDREs, vitamin D response elements; VDR, vitamin D receptor; 1-OHase, 25-hydroxyvitamin D-1α- hydroxylase; 24-OHase, 25-hydroxyvitamin D-24-hydroxylase
Considering the neuroprotective effect of vitamin D in the human brain, researchers have proposed a ‘two-hit hypotheses’ to explain how low vitamin D levels make the nervous system more susceptible to secondary harmful effects, which may aggravate the development of diseases such as PD and cerebrovascular disease [40,41,42]. Notably, many studies have indicated that vitamin D metabolism may be directly or indirectly related to the pathogenesis of PD [30, 31, 33, 39, 42,43,44]. Further, vitamin D can act as a neuroprotective agent to provide partial protection for DA neurons [45]. Accordingly, inadequate vitamin D status may play a significant role in PD, resulting in a progressive loss of DA neurons in the human brain [46]. However, experimental data from the Asymptomatic Parkinson Associated Vitamin D Intake Risk Syndrome cohort were not consistent with the hypothesis that chronically inadequate levels of vitamin D threaten the integrity of the DA system, leading to the pathogenesis of PD [47]. The conundrum of the connection between serum vitamin D levels and PD therefore remains unsolved. In this paper, we review the serum vitamin D concentrations in PD patients, the relationships of serum vitamin D concentrations and VDR gene polymorphisms with PD risk, the relationship between serum vitamin D concentrations and clinical manifestations of PD patients, as well as the preventive and therapeutic roles of vitamin D in PD.
Main text
PD patients often have low serum vitamin D concentrations
Vitamin D insufficiency is prevalent in the elderly worldwide [48], and it is also a common health problem in neurodegenerative diseases such as PD and Alzheimer’s disease (AD). Recently, it has been reported that vitamin D insufficiency is more common among PD patients than healthy controls [49,50,51]. If the insufficiency of vitamin D is a consequence of neurodegenerative disease, the incidence of vitamin D insufficiency in AD and PD patients should be similar, but a study revealed that vitamin D insufficiency in PD patients was more pronounced than that in AD patients and controls (55% versus 41% and 36%, respectively) [52]. The 1,25(OH)2D3 levels were normal in all PD patients, whereas the serum levels of 25(OH)D were insufficient (< 20 ng/mL) in 49% of patients in a prospective cohort study [53]. This can be explained by the fact that circulating 25(OH)D levels are 1000 times higher than 1,25(OH)2D3, and that the 25(OH)D can be converted into 1,25(OH)2D3 by 1a-OHase [29, 53].
PD patients experience mobility problems more frequently, and the typical course of PD is longer than that of AD. Both factors may decrease sunlight exposure, thus reducing the cutaneous synthesis of vitamin D. Many studies have reported that the more severe the motor symptoms, the lower the 25(OH)D concentrations in PD patients [53,54,55,56,57]. The reduced mobility and sunlight deprivation may be responsible for the higher incidence of vitamin D deficiency in PD patients. However, compared with controls, there are significantly lower levels of 25(OH)D in PD patients with sufficient sunlight exposure [51]. This may be because that as some vitamin D should be from the diet [29], the gastrointestinal dysfunction in PD patients may result in chronically inadequate vitamin D intake [58, 59]. Interestingly, a longitudinal cohort study discovered that the 25(OH)D concentrations were slightly increased over the study period, which means that these patients did not have digestive dysfunction. The study also reported that there was a high incidence of vitamin D insufficiency in subjects with early PD who did not require symptomatic therapy [60].
However, another study showed that compared with controls, the PD patients had slightly yet not significantly lower serum vitamin D concentrations [61]. There may be two factors that affect the results. First, the case-control study was conducted in the Faroe Islands at a high latitude and with harsh climate and frequent cloud cover, which may have decreased sunlight exposure. Second, Faroese food is not rich in vitamin D, which resulted in the common vitamin D insufficiency in the Faroe Islands.
The relationship between vitamin D deficiency and PD risk
Lower vitamin D levels may be a result of PD due to the limited mobility and digestive symptoms of PD patients. However, several studies suggested that vitamin D deficiency may be associated with the etiology of PD [46, 54]. A study showed that the prevalence of vitamin D deficiency was higher among patients with PD, even if they had normal ambulation and gastrointestinal functions [60]. Newmark and colleagues concluded that chronic vitamin D deficiency is likely to be linked to the pathogenesis or progression of PD rather than only being a consequence of the disease [46, 54]. This hypothesis was also supported by a 29-year prospective study in Finland, which confirmed that those who had higher vitamin D concentrations were less likely to develop PD. When comparing probands in the highest and the lowest quartiles of vitamin D levels, the relative risk of PD was 0.33 for the highest quartile (95% confidence interval, 0.14–0.80) [62]. In line with these findings, a large case-control sample study revealed a negative correlation between PD risk and the level of 25(OH)D, and additionally showed an inverse correlation between 25(OH)D2 and PD [59]. Moreover, Danish and Chinese case-control studies both suggested that outdoor work can reduce the risk of PD in later life [63,64,65]. One possible protective mechanism of outdoor work is to increase sunlight exposure, which contributes to vitamin D3 synthesis in the skin. Interestingly, a nationwide ecological study in France showed that increasing sunlight exposure can reduce the risk of PD in the young population [66]. However, a population-based prospective study with 17 years of follow-up and a Mendelian randomization study did not explore the association between 25(OH)D concentrations and the prevalence of PD [67, 68].
The relationship between VDR gene polymorphisms and PD risk
The VDR is the key mediator of the functions of vitamin D. A transcriptome-wide scan indicated that the VDR gene expression is increased in the blood cells of early-stage PD patients [69]. Consequently, it is reasonable that the VDR polymorphic variants might also have an effect on the pathogenesis of PD. In recent years, the polymorphisms of BsmI (rs1544410), FokI (rs10735810), ApaI (rs7975232), and TaqI (rs731236) have been most widely studied in research on the correlation between VDR gene variants and PD, but the results were inconsistent [70,71,72]. A polymerase chain reaction-based restriction analysis of VDR gene polymorphisms in Korea indicated that the BsmI (B/b) polymorphism is a candidate allele influencing the pathogenesis of PD. The study further showed that the bb genotype was more common in the group with predominant postural instability and gait disorders than in the tremor-predominant group and the healthy controls [73]. In addition, Hungarian, Japanese and Chinese studies suggested that the FokI (C/T) polymorphism located in exon 2 in the 5′ coding region of the gene was significantly linked with PD, and the C allele can increase the risk of PD [74,75,76,77].
The most significant start codon polymorphism of the VDR gene is the functional FokI polymorphism, which results in different translation initiation sites, one producing a long version of the VDR protein (the T-allele) and the other producing a protein shortened by three amino acids (the C-allele) [70]. In spite of the small difference, the functional characteristics of the two forms of VDR (C-VDR and T-VDR) are significantly different. Compared with T-VDR, the C-VDR has a better capacity for intestinal calcium absorption [70, 78]. Therefore, the C allele may forecast higher vitamin D levels and reduce the risk of PD. However, research findings have suggested that the C allele is a risk factor for PD rather than being a protective factor [74,75,76,77]. Suzuki et al. revealed that there was a stronger association of the FokI CC genotype with milder forms of PD (odds ratio, 0.32; 95% confidence interval, 0.16–0.66) [53]. Moreover, the Parkinson Environment Gene study, a population-based case-control study of PD in the Central Valley of California, showed that FokI polymorphism was linked to cognitive decline in PD [79].
In 2015, a study in California with higher ultraviolet radiation levels than in previous studies of VDR gene polymorphisms showed that the major allele TaqI TT genotype and the ApaI GG genotype are associated with decreased risk of PD [80]. However, some studies did not find any association between the VDR genotypes (BsmI, FokI, ApaI, and TaqI loci) and PD risk [61, 81]. The different results may be explained by a number of reasons. First, the effect of VDR gene polymorphism on PD risk may be related to the vitamin D levels. Second, these studies involved different ethnicities, environmental factors, gene-gene and gene-environment interactions, or small sample sizes. Therefore, future studies should shift to the interactions of vitamin D levels and VDR gene polymorphisms in PD, and take into account the environmental factors.
The relationship between vitamin D level and clinical manifestations of PD patients
There is accumulating evidence that the PD patients have an increased prevalence of osteoporosis and osteopenia [82,83,84], and PD is recognized as a cause of secondary osteoporosis [85]. In a study conducted in Korea, researchers found that 6542 (18.3%) of 35,663 PD patients experienced osteoporosis, and that fractures occurred most commonly within 6 months after PD onset and decreased after 3 years from PD diagnosis [86]. Other studies have shown that bone loss and fractures in PD patients are multifactorial [87,88,89], with causes including vitamin D deficiency [90]. As PD patients experience more bone loss, more falls and more fractures (particularly at the hip) [91] than controls, osteoporosis should be screened and treated early [51, 82, 84, 92,93,94], particularly for older female patients within 3 years of PD diagnosis [86]. A meta-analysis of randomized controlled trials found that daily supplementation of 700 IU to 1000 IU of vitamin D could reduce the risk of falls by 19%, and that this advantage may not be dependent on additional calcium supplementation [95].
Many recent studies have confirmed the significant negative correlation between the severity of PD evaluated using the Hoehn & Yahr scale or Unified Parkinson’s Disease Rating Scale (UPDRS) and the circulating serum 25(OH)D levels [53,54,55,56,57, 96]. Prospective observational studies have also found a negative association between the vitamin D status at baseline and severity of PD motor symptoms during disease progression [41, 97]. Therefore, supplementation of vitamin D may delay the worsening of symptoms in PD patients. A cross-sectional, observational study supported the relationship between postural balance and serum vitamin D levels. Further analysis showed that among balance measures, vitamin D levels were associated with an automatic posture response to backwards translation, particularly with response strength and weight-bearing asymmetry [98].
PD patients often ignore nonmotor symptoms, which may, however, have been present for years before diagnosis. A large population-based sample of French older people found a strong relationship of lower 25(OH)D concentrations with cognitive decline, as well as increased risk of dementia and AD over 12 years of follow-up [99]. Likewise, in a sample of PD patients without cognitive impairment, higher vitamin D levels were associated with better cognition and mood [100]. The impact of vitamin D on cognition can partially be explained by its effect on amyloid beta (Aβ) [101], which has been shown to deposit in PD as well, probably leading or contributing to cognitive decline [102]. Interestingly, vitamin D has been reported to affect the Aβ-producing enzymes BACE1 and γ-secretase to reduce Aβ anabolism and elevate Aβ catabolism. Furthermore, vitamin D3 could reduce the cytotoxicity of Aβ peptide by ameliorating the decrease of the sphingosine-1-phosphate/ceramide ratio caused by Aβ [103]. A randomized double-blind trial found that vitamin D supplementation can effectively reduce the levels of Aβ, amyloid precursor protein (APP), BACE1, APP mRNA, and BACE1 mRNA [104]. Vitamin D and its receptors are important components of neuronal amyloid processing pathways [105]. Mayne et al. found that vitamin D deficiency may affect synaptic plasticity, leading to a decline of cognition [31]. Studies have shown that vitamin D signaling can affect the expression of L-type voltage-gated calcium channels, which are involved in neurotransmitter release, neuronal excitability change, learning and memory, etc. [31, 106]. Treatment of aging rodents with high-dose vitamin D3 could prevent cognitive decline and enhance hippocampal synaptic excitability [107].
Many studies have shown that brain regions involved in regulating olfactory function are closely related to cognitive decline, and the severity of olfactory disorder in PD patients may precede dementia [108,109,110]. In 2018, Kim et al. firstly demonstrated that the 25(OH)D3 levels were correlated with the severity of olfactory dysfunction in PD [111]. According to the Braak model of neuropathological staging of PD, the early stages 1 and 2 start from the medulla and the olfactory bulb [112], supporting a relationship between vitamin D and early PD. In addition to being associated with dementia and olfactory function in PD patients, serum 25(OH)D3 concentrations can also affect the gastric emptying time [113] and orthostatic hypotension [114].
Preventive and therapeutic effects of vitamin D in PD
The pathophysiology of PD is affected by 1,25(OH)2D3 via genomic (Table 1) and non-genomic routes (rapid vitamin D-dependent membrane-associated effects) [120]. 1,25(OH)2D3 can increase or decrease the expression of a number of genes, thereby affecting intracellular signaling pathways. Recent pieces of evidence suggest that there is an inverse correlation between vitamin D concentrations and PD risk.
1,25(OH)2D3 affects PD by genomic actions mediated by VDR
Neuroprotective effects of vitamin D
Glial cell-derived neurotrophic factors (GDNFs) facilitate neuronal regrowth and protect dopaminergic nerve terminals, which make them a very promising candidate for neuro-restoration therapy of PD [121, 122]. GDNF binds to the GDNF family receptor alpha 1 (GFRa1) and then associates with the proto-oncogene tyrosine-protein kinase receptor Ret (C-Ret). This complex enables GDNF to exert intracellular signaling in DA neurons [115]. However, GDNF cannot pass the blood-brain barrier, and injecting GDNF into the CNS has many negative effects [121]. As a fat-soluble vitamin, 1,25(OH)2D3 can pass the blood-brain barrier, which consolidates the importance of this hormone in PD [39]. Upon VDR binding, 1,25(OH)2D3 directly upregulates the transcription of genes targeted by C-Ret and GDNF. There is a positive feedback between GDNF and C-Ret, and both can suppress GFRa1 production [115]. Depletion of 1,25(OH)2D3 results in decreased expression of GDNF, Nurr1 and p57kip2 [123, 124], which may alter the differentiation and maturation of DA neurons in the developing rat brain [116, 117]. Nurr1 is also crucial for the expression of C-Ret [115], which in turn triggers Src-family kinases and tyrosine kinase, activating several downstream signaling cascades, including the phosphoinositide 3-kinase (PI3K) pathway, the phospholipase Cγ (PLC-γ) pathway, and the p42/p44 mitogen-activated protein kinase (MAPK) pathway [125, 126]. The activation of the MAPK pathway requires a basal activity of the PI3K pathway [126]. The activation of these pathways may promote the survival and differentiation of midbrain DA neurons (Fig. 2). In conclusion, 1,25(OH)2D3 exerts its neuroprotective effects by increasing the expression of GDNF gene and then activating several downstream intracellular signaling cascades.

1,25(OH)2D3 exerts neuroprotective effects via GDNF. CAT, catalase; C-Ret, proto-oncogene tyrosine-protein kinase receptor Ret; DA, dopaminergic; GFR α1, GDNF family receptor alpha 1; GPx, glutathione peroxidase; MAPK pathway, p42/p44 mitogen-activated protein kinase pathway; PLC-γ pathway, phospholipase Cγ pathway; PI3K pathway, phosphoinositide 3-kinase pathway; ROS, reactive oxygen species; RXR, retinoid X receptor; SOD, superoxide dismutase; VDR, vitamin D receptor; VDREs, vitamin D response elements. The arrows indicate signaling components that are either enhanced (red arrows) or reduced (green arrows)
In addition, 1,25(OH)2D3 is also an antioxidant, which may further contribute to its neuroprotective effects. Studies have demonstrated that 1,25(OH)2D3 increases the expression of GDNF, a powerful antioxidant that can reduce reactive oxygen species (ROS). GDNF markedly increases the levels of superoxide dismutase, glutathione peroxidase and catalase in the striatum (Fig. 2) [127]. In addition to the upregulation of GDNF expression, 1,25(OH)2D3 can also exert its antioxidant effect through genomic and/or nongenomic activation. Under inflammatory stimulation, microglial cells can produce 1,25(OH)2D3 in situ, where it potentiates the mRNA expression of gamma-glutamyl transferase (γ-GT) and γ-GT activity induced by proinflammatory stimuli. γ-GT mediates the import of glutathione (GSH) into the cell, after which the intracellular GSH reduces the production of reactive nitrogen species and hydrogen peroxide [128]. In addition, 1,25(OH)2D3 also increases the expression of the nuclear factor erythroid 2-related factor 2 (Nrf2). When ROS rise, they bind to antioxidant response elements (AREs) in the nucleus, enhancing the expression of antioxidant genes, detoxifying enzymes and various signaling components. By increasing the expression of Fos and JUN, Nrf2 also increases the expression of both VDR and RXR [129]. Moreover, 1,25(OH)2D3 can directly inhibit lipid peroxidation as a membrane antioxidant, which protects the membranes of normal cells from ROS-induced oxidative damage [130, 131]. Therefore, 1,25(OH)2D3 contributes to the enhancement of antioxidative systems by increasing the expression of GDNF, γ-GT and Nrf2 (Fig. 3).

1,25(OH)2D3 also exerts neuroprotective effects through genomic and/or non-genomic activation. ARE, antioxidant response element; GFR α1, GDNF family receptor alpha 1; γ-GT, gamma-glutamyl transferase; GSH, glutathione; GSNOH, S-nitrosoglutathione; RXR, retinoid X receptor; ROS, reactive oxygen species; PMCA, plasma membrane Ca2+ ATP-ase; Nrf2, nuclear factor erythroid 2-related factor 2; VDR, vitamin D receptor; VDREs, vitamin D response elements. The arrows indicate signaling components that are either enhanced (red arrows) or reduced (green arrows)
It has also been reported that 1,25(OH)2D3 has anti-inflammatory properties. It can attenuate pro-inflammatory and upregulate anti-inflammatory processes [132]. In the 6-OHDA-induced PD model, pre- or post-treatment with 1,25(OH)2D3 reduced tissue immunopositivity for TNF-α, partially restored tyrosine hydroxylase (TH) immunoreactivity, and prevented the decrease of VDR immunoreactivity in the lesioned striatum [133, 134]. Cell culture studies revealed that the increased intracellular free calcium can induce the aggregation of α-synuclein, and proved that the increase of intracellular calcium and oxidative stress can act cooperatively to promote α-synuclein aggregation [135,136,137,138]. By reducing the expression of L-type Ca2+ channels and increasing the expression of the plasma membrane Ca2+ ATP-ase, NCX1, anti-apoptotic factor Bcl-2 and buffering protein calbindin D28k, 1,25(OH)2D3 can maintain the low cytosolic Ca2+ concentrations and thereby protect against Ca2+-induced oxidative damage in SN dopaminergic neurons [106, 129]. High concentrations of divalent metal ions exhibit toxic effects that may cause an elevation of ROS levels and mitochondrial dysfunction, and even induce neuronal cell death. Notably, 1,25(OH)2D3 can maintain zinc, iron and manganese homeostasis by regulating the expression of related genes. It can transactivate SLC30A10 to increase the expression of zinc and manganese transporter ZnT10. It can also decrease the expression of SLC39A2, which encodes the ZIP (SLC39) protein implicated in zinc, iron and/or manganese transport. The ZnT (SLC30) protein reduces the cytoplasmic concentrations of metal ions, while ZIP (SLC39) transporters lead to an increase (Fig. 3) [118]. In short, 1,25(OH)2D3 can maintain the homeostasis of calcium, zinc, iron and manganese by regulating the expression of some genes, thereby reducing oxidative stress and mitochondrial damage.
Vitamin D is closely associated with dopaminergic neurotransmission
Studies have shown that 1,25(OH)2D3 and VDR are directly involved in regulating the expression of genes in dopaminergic neurons [139]. Many studies have found that VDR protein levels and TH expression are enhanced in the brains of rats following 1,25(OH)2D administration [119, 140]. Notably, TH is the rate-limiting enzyme of dopamine synthesis. It has been reported that a likely mediator of the regulation of TH expression by vitamin D is N-cadherin [141]. In line with these findings, researchers found that pre- or post-treatments with 1,25(OH)2D3 restored the decreased DA content, and increased the expression of TH and dopamine transporter in 6-OHDA-lesioned rats according to striatal neurochemical and immunohistochemical assays [133]. GDNF can act directly on DA neurons to enhance their activity and increase DA release [127]. In a word, 1,25(OH)2D3 may participate in dopaminergic neurotransmission via TH expression regulation and the direct effect of GDNF on DA neurons, which mediates the relationship between vitamin D concentrations and the severity of PD.
1,25(OH)2D3 affects PD by rapid vitamin D-dependent membrane-associated effects
Protein disulfide isomerase 3 (PDIA3), also known as the endoplasmic reticulum stress protein 57 (ERp57), acts as another 1,25(OH)2D3 membrane receptor [142]. Compared to the kidney and liver, PDIA3 is highly expressed in all types of brain cells and can be considered as the main VDR in the brain [143]. It is a multifunctional protein that can not only control the quality of protein processing, but also maintain Ca2+ homeostasis and regulate cellular stress responses [144, 145]. In the PD model induced by 6-OHDA, the level of PDIA3 protein in the striatum is increased, which may be a cellular response to oxidative stress. In this case, PDIA3 may act as a chaperone to prevent the misfolding and aggregation of α-synuclein [144]. After generation of ROS by 6-OHDA, protein oxidation occurs first, and early in the protein oxidation process, the PDIA3 rapidly forms juxtanuclear aggresome-like structures (ERp57/DNA) in dopaminergic cells, which may induce downstream sequelae such as the unfolded protein response, cell stress, and apoptosis [146, 147]. ERp57 has an affinity for Ref-1, which has a synergistic effect and jointly regulates the gene expression mediated by redox-sensitive transcription factors and the adaptive responses of cells to oxidative damage [147]. As a result, it is likely that vitamin D functions in the PD through PDIA3.
Vitamin D supplementation in PD patients
The most important studies on vitamin D supplementation in PD patients are shown in Table 2. An interventional trial supported the role of vitamin D in postural balance of PD patients and suggested that daily supplementation of vitamin D could improve the balance of younger PD patients [148]. Other studies have also confirmed that supplemental 25(OH)D has beneficial effects on strength and balance in older adults [149]. Therefore, there is a debate on whether vitamin D supplementation can specifically delay the progression of motor symptoms in PD patients, or only lead to a non-specific improvement in muscle strength and balance. However, the complex automatic postural response not only requires muscle function, but also involves the spinal cord, midbrain/brainstem, and cerebellum/basal ganglia/cerebral cortex [150, 151]. In addition, vitamin D3 supplementation has an age-dependent effect on PD [148]. Another randomized controlled trial of vitamin D supplementation found that vitamin D3 supplementation may retard the progression of PD for a short period in patients with FokI CT and TT genotypes [41]. Therefore, the extensive roles of vitamin D in the skeletal muscle and neural systems suggest that vitamin D can affect the symptoms of PD.
Conclusions and future directions
In summary, the most consistent view at present is that the concentration of vitamin D is low in PD patients. Higher vitamin D concentrations are linked to reduced risk and severity of PD, as well as better cognition and mood of the patients. Furthermore, the VDR gene phenotypes may influence the risk and severity of PD, as well as the effect of vitamin D supplementation in PD patients. Although there are limited data on the effectiveness of vitamin D3 supplementation in PD patients, related studies have highlighted the effectiveness of vitamin D3 supplementation in preventing osteoporotic fractures in the aging population and retarding the progression of PD for a short period.
A recent study has found that vitamin D has the potential to be used as a biomarker for PD [152], inspiring great interest in the relationship between PD and vitamin D. Vitamin D can improve protein homeostasis and slow down the aging process [153], but vitamin D insufficiency is prevalent worldwide [48]. Moreover, vitamin D supplementation is readily available, affordable and safe. The earliest detectable side-effects of vitamin D supplementation are hypercalciuria and hypercalcemia, which are only a concern when 25(OH)D levels exceed 88 ng/mL (220 nmol/L) [154, 155]. Vitamin D supplementation in PD patients at a dose of 1200 IU/day for 12 months [41] or 10,000 IU/day for 16 weeks [148] did not lead to obvious adverse events such as hypercalcemia (Table 2). Therefore, vitamin D supplementation in PD patients seems to be promising, although the dose of vitamin D that may cause toxicity remains unclear. Despite the limited long-term safety data, in 2010, the Institute of Medicine (IOM) defined a safe upper limit dosage for vitamin D of 4000 IU/day, although practitioners should keep in mind the intake of other dietary supplements [156]. The possibility of neuroprotection is the most exciting aspect of vitamin D therapy in PD. Considering the neuroprotective effects of vitamin D and the role of vitamin D in dopaminergic neurotransmission, interventional prospective studies on vitamin D supplementation in PD patients should be conducted in the future.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- ARE:
-
Antioxidant response element
- Aβ:
-
Amyloid beta
- APP:
-
Amyloid precursor protein
- C-Ret:
-
Proto-oncogene tyrosine-protein kinase receptor Ret
- DA:
-
Dopaminergic
- ERp57:
-
Endoplasmic reticulum stress protein 57
- γ-GT:
-
Gamma-glutamyl transferase
- GSH:
-
Glutathione
- GDNF:
-
Glia-derived neurotrophic factors
- GFRa1:
-
GDNF family receptor alpha 1
- MAPK pathway:
-
p42/p44 mitogen-activated protein kinase pathway
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- PD:
-
Parkinson’s disease
- PLC-γ pathway:
-
Phospholipase Cγ pathway
- PMCA:
-
Plasma membrane Ca2+ ATP-ase
- PI3K pathway:
-
Phosphoinositide 3-kinase pathway
- PDIA3:
-
Protein disulfide isomerase 3
- ROS:
-
Reactive oxygen species
- RXR:
-
Retinoid X receptor
- SN:
-
Substantia nigra
- TH:
-
Tyrosine hydroxylase
- UPDRS:
-
Unified Parkinson’s Disease Rating Stage
- VDR:
-
Vitamin D receptor
- VDREs:
-
Vitamin D response elements
- 25(OH)D:
-
25-hydroxyvitamin D
- 1,25(OH)2D3 :
-
1,25-dihydroxyvitamin D3
- 1-OHase:
-
25-hydroxyvitamin D-1α-hydroxylase
- 24-OHase:
-
25-hydroxyvitamin D-24-hydroxylase
References
Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA. 2020;323:548–60.
Benazzouz A, Mamad O, Abedi P, Bouali-Benazzouz R, Chetrit J. Involvement of dopamine loss in extrastriatal basal ganglia nuclei in the pathophysiology of Parkinson's disease. Front Aging Neurosci. 2014;6:87.
Pakkenberg B, Moller A, Gundersen HJ, Mouritzen Dam A, Pakkenberg H. The absolute number of nerve cells in substantia nigra in normal subjects and in patients with Parkinson’s disease estimated with an unbiased stereological method. J Neurol Neurosurg Psychiatry. 1991;54:30–3.
Hornykiewicz O. The discovery of dopamine deficiency in the parkinsonian brain. J Neural Transm Suppl. 2006;9:20015.
Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson's disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology. 2007;27:494–506.
Shoji TMM, Imai Y, Inoue H, Kawarabayashi T, Matsubara E, Sasaki YHA, et al. Pael-R is accumulated in Lewy bodies of Parkinson's disease. Ann Neurol. 2004;55:439–42.
Bodner RA, Outeiro TF, Altmann S, Maxwell MM, Cho SH, Hyman BT, et al. Pharmacological promotion of inclusion formation: a therapeutic approach for Huntington’s and Parkinson’s diseases. Proc Natl Acad Sci U S A. 2006;103:4246–51.
Ascherio A, Schwarzschild MA. The epidemiology of Parkinson's disease: risk factors and prevention. Lancet Neurol. 2016;15:1257–72.
Dorsey ER, Elbaz A, Nichols E, Abd-Allah F, Abdelalim A, Adsuar JC, et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2018;17:939–53.
Logroscino G. The role of early life environmental risk factors in Parkinson disease: what is the evidence? Environ Health Perspect. 2005;113:1234–8.
Feany MB. New genetic insights into Parkinson's disease. N Engl J Med. 2004;351:1937–40.
Meredith GE, Halliday GM, Totterdell S. A critical review of the development and importance of proteinaceous aggregates in animal models of Parkinson's disease: new insights into Lewy body formation. Parkinsonism Relat Disord. 2004;10:191–202.
Cory-Slechta DA, Thiruchelvam M, Barlow BK, Richfield EK. Developmental pesticide models of the Parkinson disease phenotype. Environ Health Perspect. 2005;113:1263–70.
Ahlskog JE. Challenging conventional wisdom: the etiologic role of dopamine oxidative stress in Parkinson’s disease. Mov Disord. 2005;20:271–82.
Greenamyre JT, Hastings TG. Parkinson’s--divergent causes, convergent mechanisms. Science. 2004;304:1120–2.
Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49.
Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, et al. Lysosomal impairment in Parkinson’s disease. Mov Disord. 2013;28:725–32.
Bové J, Prou D, Perier C, Przedborski S. Toxin-induced models of Parkinson’s disease. NeuroRx. 2005;2:484–94.
Lim KL, Zhang C. Molecular events underlying Parkinson’s disease – an interwoven tapestry. Front Neurol. 2013;4:33.
Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009;119:650–60.
Selvaraj S, Sun Y, Watt JA, Wang S, Lei S, Birnbaumer L, et al. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J Clin Invest. 2012;122:1354–67.
Maiti P, Manna J, Dunbar GL. Current understanding of the molecular mechanisms in Parkinson's disease: targets for potential treatments. Transl Neurodegener. 2017;6:28.
Tuckey RC, Cheng CYS, Slominski AT. The serum vitamin D metabolome: what we know and what is still to discover. J Steroid Biochem Mol Biol. 2019;186:4–21.
Christakos S, Ajibade DV, Dhawan P, Fechner AJ, Mady LJ. Vitamin D: metabolism. Endocrinol Metab Clin N Am. 2010;39:243–53.
Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. 2016;96:365–408.
DeLuca HF. Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr. 2004;80:1689S–96S.
Rachez C, Freedman LP. Mechanisms of gene regulation by vitamin D3 receptor: a network of coactivator interactions. Gene. 2000;246:9–21.
Takeyama K, Kato S. The vitamin D3 1alpha-hydroxylase gene and its regulation by active vitamin D3. Biosci Biotechnol Biochem. 2011;75:208–13.
Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–81.
Almeras L, Eyles D, Benech P, Laffite D, Villard C, Patatian A, et al. Developmental vitamin D deficiency alters brain protein expression in the adult rat: implications for neuropsychiatric disorders. Proteomics. 2007;7:769–80.
Mayne PE, Burne THJ. Vitamin D in synaptic plasticity, cognitive function, and neuropsychiatric illness. Trends Neurosci. 2019;42:293–306.
Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol. 2014;21:319–29.
Groves NJ, McGrath JJ, Burne TH. Vitamin D as a neurosteroid affecting the developing and adult brain. Annu Rev Nutr. 2014;34:117–41.
Harms LR, Burne TH, Eyles DW, McGrath JJ. Vitamin D and the brain. Best Pract Res Clin Endocrinol Metab. 2011;25:657–69.
Eyles D, Burne T, McGrath J. Vitamin D in fetal brain development. Semin Cell Dev Biol. 2011;22:629–36.
Eyles DW, Smith S, Kinobe R, Hewison M, McGrath JJ. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29:21–30.
Gates MA, Torres EM, White A, Fricker-Gates RA, Dunnett SB. Re-examining the ontogeny of substantia nigra dopamine neurons. Eur J Neurosci. 2006;23:1384–90.
Veenstra TD, Prufer K, Koenigsberger C, Brimijoin SW, Grande JP, Kumar R. 1,25-Dihydroxyvitamin D3 receptors in the central nervous system of the rat embryo. Brain Res. 1998;804:193–205.
DeLuca GC, Kimball SM, Kolasinski J, Ramagopalan SV, Ebers GC. Review: the role of vitamin D in nervous system health and disease. Neuropathol Appl Neurobiol. 2013;39:458–84.
Balden R, Selvamani A, Sohrabji F. Vitamin D deficiency exacerbates experimental stroke injury and dysregulates ischemia-induced inflammation in adult rats. Endocrinology. 2012;153:2420–35.
Suzuki M, Yoshioka M, Hashimoto M, Murakami M, Noya M, Takahashi D, et al. Randomized, double-blind, placebo-controlled trial of vitamin D supplementation in Parkinson disease. Am J Clin Nutr. 2013;97:1004–13.
Cui X, Gooch H, Groves NJ, Sah P, Burne TH, Eyles DW, et al. Vitamin D and the brain: key questions for future research. J Steroid Biochem Mol Biol. 2015;148:305–9.
Eyles DW, Feron F, Cui X, Kesby JP, Harms LH, Ko P, et al. Developmental vitamin D deficiency causes abnormal brain development. Psychoneuroendocrinology. 2009;34(Suppl 1):S247–57.
Garcion E, Wion-Barbot N, Montero-Menei CN, Berger FO, Wion D. New clues about vitamin D functions in the nervous system. Trends Endocrinol Metab. 2002;13:100–5.
Smith MP, Fletcher-Turner A, Yurek DM, Cass WA. Calcitriol protection against dopamine loss induced by intracerebroventricular administration of 6-hydroxydopamine. Neurochem Res. 2006;31:533–9.
Newmark HL, Newmark J. Vitamin D and Parkinson’s disease--a hypothesis. Mov Disord. 2007;22:461–8.
Fullard ME, Xie SX, Marek K, Stern M, Jennings D, Siderowf A, et al. Vitamin D in the Parkinson associated risk syndrome (PARS) study. Mov Disord. 2017;32:1636–40.
Dawson-Hughes B, Mithal A, Bonjour JP, Boonen S, Burckhardt P, Fuleihan GE, et al. IOF position statement: vitamin D recommendations for older adults. Osteoporos Int. 2010;21:1151–4.
Ding H, Dhima K, Lockhart KC, Locascio JJ, Hoesing AN, Duong K, et al. Unrecognized vitamin D3 deficiency is common in Parkinson disease: Harvard Biomarker Study. Neurology. 2013;81:1531–7.
Hatem AK, Lateef HF. The state of Vitamin D in Iraqi patients With Parkinson disease. Al Kindy Coll Med J. 2017;13:137–41.
van den Bos F, Speelman AD, van Nimwegen M, van der Schouw YT, Backx FJ, Bloem BR, et al. Bone mineral density and vitamin D status in Parkinson's disease patients. J Neurol. 2013;260:754–60.
Evatt ML, Delong MR, Khazai N, Rosen A, Triche S, Tangpricha V. Prevalence of vitamin d insufficiency in patients with Parkinson disease and Alzheimer disease. Arch Neurol. 2008;65:1348–52.
Suzuki M, Yoshioka M, Hashimoto M, Murakami M, Kawasaki K, Noya M, et al. 25-hydroxyvitamin D, vitamin D receptor gene polymorphisms, and severity of Parkinson’s disease. Mov Disord. 2012;27:264–71.
Peterson AL. A review of vitamin D and Parkinson's disease. Maturitas. 2014;78:40–4.
Zhou Z, Zhou R, Zhang Z, Li K. The association between vitamin D status, vitamin D supplementation, sunlight exposure, and Parkinson's disease: a systematic review and meta-analysis. Med Sci Monit. 2019;25:666–74.
Liu Y, Zhang BS. Serum 25-hydroxyvitamin D predicts severity in Parkinson’s disease patients. Neurol Sci. 2014;35:67–71.
Luo X, Ou R, Dutta R, Tian Y, Xiong H, Shang H. Association between serum vitamin D levels and Parkinson’s disease: a systematic review and meta-analysis. Front Neurol. 2018;9:909.
Mrabet S, Ben Ali N, Achouri A, Dabbeche R, Najjar T, Haouet S, et al. Gastrointestinal dysfunction and neuropathologic correlations in Parkinson disease. J Clin Gastroenterol. 2016;50:e85–90.
Wang L, Evatt ML, Maldonado LG, Perry WR, Ritchie JC, Beecham GW, et al. Vitamin D from different sources is inversely associated with Parkinson disease. Mov Disord. 2015;30:560–6.
Evatt ML, DeLong MR, Kumari M, Auinger P, McDermott MP, Tangpricha V, et al. High prevalence of hypovitaminosis D status in patients with early Parkinson disease. Arch Neurol. 2011;68:314–9.
Petersen MS, Bech S, Christiansen DH, Schmedes AV, Halling J. The role of vitamin D levels and vitamin D receptor polymorphism on Parkinson’s disease in the Faroe Islands. Neurosci Lett. 2014;561:74–9.
Knekt P, Kilkkinen A, Rissanen H, Marniemi J, Saaksjarvi K, Heliovaara M. Serum vitamin D and the risk of Parkinson disease. Arch Neurol. 2010;67:808–11.
Wang J, Yang D, Yu Y, Shao G, Wang Q. Vitamin D and sunlight exposure in newly-diagnosed Parkinson’s disease. Nutrients. 2016;8:142.
Zhu D, Liu GY, Lv Z, Wen SR, Bi S, Wang WZ. Inverse associations of outdoor activity and vitamin D intake with the risk of Parkinson’s disease. J Zhejiang Univ Sci B. 2014;15:923–7.
Kenborg L, Lassen CF, Ritz B, Schernhammer ES, Hansen J, Gatto NM, et al. Outdoor work and risk for Parkinson’s disease: a population-based case-control study. Occup Environ Med. 2011;68:273–8.
Kravietz A, Kab S, Wald L, Dugravot A, Singh-Manoux A, Moisan F, et al. Association of UV radiation with Parkinson disease incidence: a nationwide French ecologic study. Environ Res. 2017;154:50–6.
Shrestha S, Lutsey PL, Alonso A, Huang X, Mosley TH Jr, Chen H. Serum 25-hydroxyvitamin D concentrations in mid-adulthood and Parkinson's disease risk. Mov Disord. 2016;31:972–8.
Larsson SC, Singleton AB, Nalls MA, Richards JB, International Parkinson’s Disease Genomics C. No clear support for a role for vitamin D in Parkinson’s disease: A Mendelian randomization study. Mov Disord. 2017;32:1249–52.
Scherzer CR, Eklund AC, Morse LJ, Liao Z, Locascio JJ, Fefer D, et al. Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc Natl Acad Sci U S A. 2007;104:955–60.
Uitterlinden AG, Fang Y, Van Meurs JB, Pols HA, Van Leeuwen JP. Genetics and biology of vitamin D receptor polymorphisms. Gene. 2004;338:143–56.
Uitterlinden AG, Fang Y, van Meurs JB, van Leeuwen H, Pols HA. Vitamin D receptor gene polymorphisms in relation to Vitamin D related disease states. J Steroid Biochem Mol Biol. 2004;89–90:187–93.
Jurutka PW, Whitfield GK, Hsieh JC, Thompson PD, Haussler CA, Haussler MR. Molecular nature of the vitamin D receptor and its role in regulation of gene expression. Rev Endocr Metab Disord. 2001;2:203–16.
Kim JS, Kim YI, Song C, Yoon I, Park JW, Choi YB, et al. Association of vitamin D receptor gene polymorphism and Parkinson’s disease in Koreans. J Korean Med Sci. 2005;20:495–8.
Torok R, Torok N, Szalardy L, Plangar I, Szolnoki Z, Somogyvari F, et al. Association of vitamin D receptor gene polymorphisms and Parkinson's disease in Hungarians. Neurosci Lett. 2013;551:70–4.
Niu MY, Wang L, Xie AM. ApaI, BsmI, FokI, and TaqI polymorphisms in the vitamin D receptor gene and Parkinson’s disease. Chin Med J (Engl). 2015;128:1809–14.
Han X, Xue L, Li Y, Chen B, Xie A. Vitamin D receptor gene polymorphism and its association with Parkinson’s disease in Chinese Han population. Neurosci Lett. 2012;525:29–33.
Tanaka K, Miyake Y, Fukushima W, Kiyohara C, Sasaki S, Tsuboi Y, et al. Vitamin D receptor gene polymorphisms, smoking, and risk of sporadic Parkinson’s disease in Japan. Neurosci Lett. 2017;643:97–102.
Arai H, Miyamoto K, Taketani Y, Yamamoto H, Iemori Y, Morita K, et al. A vitamin D receptor gene polymorphism in the translation initiation codon: effect on protein activity and relation to bone mineral density in Japanese women. J Bone Miner Res. 1997;12:915–21.
Gatto NM, Paul KC, Sinsheimer JS, Bronstein JM, Bordelon Y, Rausch R, et al. Vitamin D receptor gene polymorphisms and cognitive decline in Parkinson's disease. J Neurol Sci. 2016;370:100–6.
Gatto NM, Sinsheimer JS, Cockburn M, Escobedo LA, Bordelon Y, Ritz B. Vitamin D receptor gene polymorphisms and Parkinson’s disease in a population with high ultraviolet radiation exposure. J Neurol Sci. 2015;352:88–93.
Kang SY, Park S, Oh E, Park J, Youn J, Kim JS, et al. Vitamin D receptor polymorphisms and Parkinson’s disease in a Korean population: revisited. Neurosci Lett. 2016;628:230–5.
Invernizzi M, Carda S, Viscontini GS, Cisari C. Osteoporosis in Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:339–46.
Lee CK, Choi SK, Shin DA, Yi S, Kim KN, Kim I, et al. Parkinson’s disease and the risk of osteoporotic vertebral compression fracture: a nationwide population-based study. Osteoporos Int. 2018;29:1117–24.
Vaserman N. Parkinson’s disease and osteoporosis. Joint Bone Spine. 2005;72:484–8.
Bezza A, Ouzzif Z, Naji H, Achemlal L, Mounach A, Nouijai M, et al. Prevalence and risk factors of osteoporosis in patients with Parkinson’s disease. Rheumatol Int. 2008;28:1205–9.
Park SB, Chung CK, Lee JY, Lee JY, Kim J. Risk factors for vertebral, hip, and femoral fractures among patients with Parkinson’s disease: a 5-year follow-up in Korea. J Am Med Dir Assoc. 2019;20:617–23.
Metta V, Sanchez TC, Padmakumar C. Osteoporosis: a hidden nonmotor face of Parkinson’s disease. Int Rev Neurobiol. 2017;134:877–90.
Sato Y, Kaji M, Tsuru T, Oizumi K. Risk factors for hip fracture among elderly patients with Parkinson’s disease. J Neurol Sci. 2001;182:89–93.
van den Bos F, Speelman AD, Samson M, Munneke M, Bloem BR, Verhaar HJ. Parkinson’s disease and osteoporosis. Age Ageing. 2013;42:156–62.
Sato Y, Kikuyama M, Oizumi K. High prevalence of vitamin D deficiency and reduced bone mass in Parkinson’s disease. Neurology. 1997;49:1273–8.
Lyell V, Henderson E, Devine M, Gregson C. Assessment and management of fracture risk in patients with Parkinson’s disease. Age Ageing. 2015;44:34–41.
Binks S, Dobson R. Risk factors, epidemiology and treatment strategies for metabolic bone disease in patients with neurological disease. Curr Osteoporos Rep. 2016;14:199–210.
Ozturk EA, Gundogdu I, Tonuk B, Kocer BG, Tombak Y, Comoglu S, et al. Bone mass and vitamin D levels in Parkinson’s disease- is there any difference between genders. J Phys Ther Sci. 2016;28:2204–9.
Malochet-Guinamand S, Durif F, Thomas T. Parkinson’s disease: a risk factor for osteoporosis. Joint Bone Spine. 2015;82:406–10.
Bischoff-Ferrari HA, Dawson-Hughes B, Staehelin HB, Orav JE, Stuck AE, Theiler R, et al. Fall prevention with supplemental and active forms of vitamin D: a meta-analysis of randomised controlled trials. BMJ. 2009;339:b3692.
Chitsaz A, Maracy M, Basiri K, Izadi Boroujeni M, Tanhaei AP, Rahimi M, et al. 25-hydroxyvitamin d and severity of Parkinson’s disease. Int J Endocrinol. 2013;2013:689149.
Sleeman I, Aspray T, Lawson R, Coleman S, Duncan G, Khoo TK, et al. The role of vitamin D in disease progression in early Parkinson’s disease. J Parkinsons Dis. 2017;7:669–75.
Peterson AL, Mancini M, Horak FB. The relationship between balance control and vitamin D in Parkinson’s disease-a pilot study. Mov Disord. 2013;28:1133–7.
Feart C, Helmer C, Merle B, Herrmann FR, Annweiler C, Dartigues JF, et al. Associations of lower vitamin D concentrations with cognitive decline and long-term risk of dementia and Alzheimer’s disease in older adults. Alzheimers Dement. 2017;13:1207–16.
Peterson AL, Murchison C, Zabetian C, Leverenz JB, Watson GS, Montine T, et al. Memory, mood, and vitamin D in persons with Parkinson’s disease. J Parkinsons Dis. 2013;3:547–55.
Hooshmand B, Lokk J, Solomon A, Mangialasche F, Miralbell J, Spulber G, et al. Vitamin D in relation to cognitive impairment, cerebrospinal fluid biomarkers, and brain volumes. J Gerontol A Biol Sci Med Sci. 2014;69:1132–8.
Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. Striatal amyloid deposition in Parkinson disease with dementia. J Neuropathol Exp Neurol. 2008;67:155–61.
Grimm MOW, Thiel A, Lauer AA, Winkler J, Lehmann J, Regner L, et al. Vitamin D and its analogues decrease amyloid-beta (Abeta) formation and increase Abeta-degradation. Int J Mol Sci. 2017;18:1-21.
Jia J, Hu J, Huo X, Miao R, Zhang Y, Ma F. Effects of vitamin D supplementation on cognitive function and blood Abeta-related biomarkers in older adults with Alzheimer’s disease: a randomised, double-blind, placebo-controlled trial. J Neurol Neurosurg Psychiatry. 2019;90:1347–52.
Irwin DJ, Lee VM, Trojanowski JQ. Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci. 2013;14:626–36.
Phillipson OT. Alpha-synuclein, epigenetics, mitochondria, metabolism, calcium traffic, & circadian dysfunction in Parkinson’s disease. An integrated strategy for management. Ageing Res Rev. 2017;40:149–67.
Latimer CS, Brewer LD, Searcy JL, Chen KC, Popovic J, Kraner SD, et al. Vitamin D prevents cognitive decline and enhances hippocampal synaptic function in aging rats. Proc Natl Acad Sci U S A. 2014;111:E4359–66.
Lee EY, Eslinger PJ, Du G, Kong L, Lewis MM, Huang X. Olfactory-related cortical atrophy is associated with olfactory dysfunction in Parkinson’s disease. Mov Disord. 2014;29:1205–8.
Baba T, Kikuchi A, Hirayama K, Nishio Y, Hosokai Y, Kanno S, et al. Severe olfactory dysfunction is a prodromal symptom of dementia associated with Parkinson’s disease: a 3 year longitudinal study. Brain. 2012;135:161–9.
Takeda A, Baba T, Kikuchi A, Hasegawa T, Sugeno N, Konno M, et al. Olfactory dysfunction and dementia in Parkinson’s disease. J Parkinsons Dis. 2014;4:181–7.
Kim JE, Oh E, Park J, Youn J, Kim JS, Jang W. Serum 25-hydroxyvitamin D3 level may be associated with olfactory dysfunction in de novo Parkinson’s disease. J Clin Neurosci. 2018;57:131–5.
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211.
Kwon KY, Jo KD, Lee MK, Oh M, Kim EN, Park J, et al. Low serum vitamin D levels may contribute to gastric dysmotility in de novo Parkinson’s disease. Neurodegener Dis. 2016;16:199–205.
Jang W, Park J, Kim JS, Youn J, Oh E, Kwon KY, et al. Vitamin D deficiency in Parkinson’s disease patients with orthostatic hypotension. Acta Neurol Scand. 2015;132:242–50.
Pertile RAN, Cui X, Hammond L, Eyles DW. Vitamin D regulation of GDNF/ret signaling in dopaminergic neurons. FASEB J. 2018;32:819–28.
Cui X, Pelekanos M, Burne TH, McGrath JJ, Eyles DW. Maternal vitamin D deficiency alters the expression of genes involved in dopamine specification in the developing rat mesencephalon. Neurosci Lett. 2010;486:220–3.
Vinh Quoc Luong K, Thi Hoang Nguyen L. Vitamin D and Parkinson’s disease. J Neurosci Res. 2012;90:2227–36.
Claro da Silva T, Hiller C, Gai Z, Kullak-Ublick GA. Vitamin D3 transactivates the zinc and manganese transporter SLC30A10 via the vitamin D receptor. J Steroid Biochem Mol Biol. 2016;163:77–87.
Jiang P, Zhang LH, Cai HL, Li HD, Liu YP, Tang MM, et al. Neurochemical effects of chronic administration of calcitriol in rats. Nutrients. 2014;6:6048–59.
Ebert R, Schutze N, Adamski J, Jakob F. Vitamin D signaling is modulated on multiple levels in health and disease. Mol Cell Endocrinol. 2006;248:149–59.
Airavaara M, Voutilainen MH, Wang Y, Hoffer B. Neurorestoration. Park Relat Disord. 2012;18:S143–6.
Weissmiller AM, Wu C. Current advances in using neurotrophic factors to treat neurodegenerative disorders. Transl Neurodegener. 2012;1:14.
Eyles D, Brown J, Mackay-Sim A, McGrath J, Feron F. Vitamin d3 and brain development. Neuroscience. 2003;118:641–53.
McGrath JJ, Feron FP, Burne TH, Mackay-Sim A, Eyles DW. Vitamin D3-implications for brain development. J Steroid Biochem Mol Biol. 2004;89–90:557–60.
Sariola H, Saarma M. Novel functions and signalling pathways for GDNF. J Cell Sci. 2003;116:3855–62.
García-Martínez JM, Pérez-Navarro E, Gavaldà N, Alberch J. Glial cell line-derived neurotrophic factor promotes the arborization of cultured striatal neurons through the p42/p44 mitogen-activated protein kinase pathway. J Neurosci Res. 2006;83:68–79.
Chao CC, Lee EH. Neuroprotective mechanism of glial cell line-derived neurotrophic factor on dopamine neurons: role of antioxidation. Neuropharmacology. 1999;38:913–6.
Garcion E, Sindji L, Leblondel G, Brachet P, Darcy F. 1,25-Dihydroxyvitamin D3 regulates the synthesis of γ-glutamyl transpeptidase and glutathione levels in rat primary astrocytes. J Neurochem. 1999;73:859–66.
Berridge MJ. Vitamin D cell signalling in health and disease. Biochem Biophys Res Commun. 2015;460:53–71.
Wiseman H. Vitamin D is a membrane antioxidant. Ability to inhibit iron-dependent lipid peroxidation in liposomes compared to cholesterol, ergosterol and tamoxifen and relevance to anticancer action. FEBS Lett. 1993;326:285–8.
Lin AM, Chen KB, Chao PL. Antioxidative effect of vitamin D3 on zinc-induced oxidative stress in CNS. Ann N Y Acad Sci. 2005;1053:319–29.
Calvello R, Cianciulli A, Nicolardi G, De Nuccio F, Giannotti L, Salvatore R, et al. Vitamin D treatment attenuates neuroinflammation and dopaminergic neurodegeneration in an animal model of Parkinson’s disease, shifting M1 to M2 microglia responses. J NeuroImmune Pharmacol. 2017;12:327–39.
Lima LAR, Lopes MJP, Costa RO, Lima FAV, Neves KRT, Calou IBF, et al. Vitamin D protects dopaminergic neurons against neuroinflammation and oxidative stress in hemiparkinsonian rats. J Neuroinflammation. 2018;15:249.
Sanchez B, Relova JL, Gallego R, Ben-Batalla I, Perez-Fernandez R. 1,25-Dihydroxyvitamin D3 administration to 6-hydroxydopamine-lesioned rats increases glial cell line-derived neurotrophic factor and partially restores tyrosine hydroxylase expression in substantia nigra and striatum. J Neurosci Res. 2009;87:723–32.
Rcom-H’cheo-Gauthier AN, Meedeniya AC, Pountney DL. Calcipotriol inhibits alpha-synuclein aggregation in SH-SY5Y neuroblastoma cells by a Calbindin-D28k-dependent mechanism. J Neurochem. 2017;141:263–74.
McLeary FA, Rcom-H’cheo-Gauthier AN, Goulding M, Radford RAW, Okita Y, Faller P, et al. Switching on endogenous metal binding proteins in Parkinson's disease. Cells. 2019;8:179.
Rcom-H'cheo-Gauthier AN, Osborne SL, Meedeniya AC, Pountney DL. Calcium: alpha-synuclein interactions in alpha-synucleinopathies. Front Neurosci. 2016;10:570.
Santner A, Uversky VN. Metalloproteomics and metal toxicology of alpha-synuclein. Metallomics. 2010;2:378–92.
Pertile RA, Cui X, Eyles DW. Vitamin D signaling and the differentiation of developing dopamine systems. Neuroscience. 2016;333:193–203.
Li H, Jang W, Kim HJ, Jo KD, Lee MK, Song SH, et al. Biochemical protective effect of 1,25-dihydroxyvitamin D3 through autophagy induction in the MPTP mouse model of Parkinson’s disease. Neuroreport. 2015;26:669–74.
Cui X, Pertile R, Liu P, Eyles DW. Vitamin D regulates tyrosine hydroxylase expression: N-cadherin a possible mediator. Neuroscience. 2015;304:90–100.
Sequeira VB, Rybchyn MS, Tongkao-On W, Gordon-Thomson C, Malloy PJ, Nemere I, et al. The role of the vitamin D receptor and ERp57 in photoprotection by 1alpha,25-dihydroxyvitamin D3. Mol Endocrinol. 2012;26:574–82.
Landel V, Stephan D, Cui X, Eyles D, Feron F. Differential expression of vitamin D-associated enzymes and receptors in brain cell subtypes. J Steroid Biochem Mol Biol. 2018;177:129–34.
Aureli C, Cassano T, Masci A, Francioso A, Martire S, Cocciolo A, et al. 5-S-cysteinyldopamine neurotoxicity: influence on the expression of alpha-synuclein and ERp57 in cellular and animal models of Parkinson's disease. J Neurosci Res. 2014;92:347–58.
Kuang XL, Liu F, Chen H, Li Y, Liu Y, Xiao J, et al. Reductions of the components of the calreticulin/calnexin quality-control system by proteasome inhibitors and their relevance in a rodent model of Parkinson’s disease. J Neurosci Res. 2014;92:1319–29.
Kim-Han JS, O’Malley KL. Cell stress induced by the parkinsonian mimetic, 6-hydroxydopamine, is concurrent with oxidation of the chaperone, ERp57, and aggresome formation. Antioxid Redox Signal. 2007;9:2255–64.
Grillo C, D’Ambrosio C, Scaloni A, Maceroni M, Merluzzi S, Turano C, et al. Cooperative activity of Ref-1/APE and ERp57 in reductive activation of transcription factors. Free Radic Biol Med. 2006;41:1113–23.
Hiller AL, Murchison CF, Lobb BM, O’Connor S, O'Connor M, Quinn JF. A randomized, controlled pilot study of the effects of vitamin D supplementation on balance in Parkinson’s disease: does age matter? PLoS One. 2018;13:e0203637.
Muir SW, Montero-Odasso M. Effect of vitamin D supplementation on muscle strength, gait and balance in older adults: a systematic review and meta-analysis. J Am Geriatr Soc. 2011;59:2291–300.
Asaka T, Yahata K, Mani H, Wang Y. Modulations of muscle modes in automatic postural responses induced by external surface translations. J Mot Behav. 2011;43:165–72.
Torres-Oviedo G, Ting LH. Muscle synergies characterizing human postural responses. J Neurophysiol. 2007;98:2144–56.
Lawton M, Baig F, Toulson G, Morovat A, Evetts SG, Ben-Shlomo Y, et al. Blood biomarkers with Parkinson’s disease clusters and prognosis: the oxford discovery cohort. Mov Disord. 2020;35:279–87.
Mark KA, Dumas KJ, Bhaumik D, Schilling B, Davis S, Oron TR, et al. Vitamin D promotes protein homeostasis and longevity via the stress response pathway genes skn-1, ire-1, and xbp-1. Cell Rep. 2016;17:1227–37.
Vieth R. Vitamin D supplementation, 25-hydroxyvitamin D concentrations, and safety. Am J Clin Nutr. 1999;69:842–56.
Dudenkov DV, Yawn BP, Oberhelman SS, Fischer PR, Singh RJ, Cha SS, et al. Changing incidence of serum 25-hydroxyvitamin D values above 50 ng/mL: a 10-year population-based study. Mayo Clin Proc. 2015;90:577–86.
Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53–8.
Acknowledgements
We are grateful for the support from the National Natural Science Foundation of China (81430025 and U1801681) and the Key Field Research Development Program of Guangdong Province (2018B030337001).
Funding
The authors of this review were supported by the National Natural Science Foundation of China (81971201) and the National Science Foundation of Hunan Province (2019JJ40450).
Author information
Authors and Affiliations
Contributions
LLL reviewed the literature, drafted and revised the manuscript; WCY and TJQ gave suggestions for the article. All other authors critically revised the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lv, L., Tan, X., Peng, X. et al. The relationships of vitamin D, vitamin D receptor gene polymorphisms, and vitamin D supplementation with Parkinson’s disease. Transl Neurodegener 9, 34 (2020). https://doi.org/10.1186/s40035-020-00213-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-020-00213-2