
Abstract
Background
Oculocutaneous albinism (OCA) is a clinically and genetically heterogenic group of pigmentation abnormalities characterized by variable hair, skin, and ocular hypopigmentation. Six known genes and a locus on human chromosome 4q24 have been implicated in the etiology of isolated OCA forms (OCA 1–7).
Methods
The most frequent OCA types among Caucasians are OCA1, OCA2, and OCA4. We aimed to investigate genes responsible for the development of these OCA forms in Hungarian OCA patients (n = 13). Mutation screening and polymorphism analysis were performed by direct sequencing on TYR, OCA2, SLC45A2 genes.
Results
Although the clinical features of the investigated Hungarian OCA patients were identical, the molecular genetic data suggested OCA1 subtype in eight cases and OCA4 subtype in two cases. The molecular diagnosis was not clearly identifiable in three cases. In four patients, two different heterozygous known pathogenic or predicted to be pathogenic mutations were present. Seven patients had only one pathogenic mutation, which was associated with non-pathogenic variants in six cases. In two patients no pathogenic mutation was identified.
Conclusions
Our results suggest that the concomitant screening of the non-pathogenic variants—which alone do not cause the development of OCA, but might have clinical significance in association with a pathogenic variant—is important. Our results also show significant variation in the disease spectrum compared to other populations. These data also confirm that the concomitant analysis of OCA genes is critical, providing new insights to the phenotypic diversity of OCA and expanding the mutation spectrum of OCA genes in Hungarian patients.
Similar content being viewed by others

Background
Oculocutaneous albinism (OCA) is a clinically and genetically heterogenic group of rare monogenic diseases characterized by diffuse reduced melanin production in the skin, hair, and/or eyes [1]. Eye symptoms including photophobia, nystagmus, strabismus, foveal hypoplasia, reduced iris, and retinal pigmentation and reduction in visual acuity are present in all types of albinism. To date, six genes and a locus on 4q24 human chromosomal region have been implicated in the development of the isolated OCA forms (OCA 1–7) [2]. Tyrosinase gene (TYR; OMIM 606933) is responsible for the development of OCA type 1 (OCA1) [3, 4]. Mutations of the oculocutaneous albinism two gene (OCA2; OMIM 611409) are associated with OCA type 2 (OCA2) [5]. Pathogenic variants of the tyrosinase-related protein gene (TYRP; OMIM 115501) are linked with OCA type 3 (OCA3) [6]. Mutations in a membrane-associated transporter gene (SLC45A2; OMIM 606202) are implicated in OCA type 4 (OCA4) [7]. OCA5 phenotype is linked to an unknown gene on human chromosome 4q24 [8]. Mutations of the sodium/calcium/potassium exchanger 5 gene (SLC24A5; OMIM 609802) encoding a solute carrier protein are associated with a new form of OCA, named as OCA6 [9]. The mutations of chromosome 10 open reading frame 11 gene (C10ORF11; OMIM 614537) are responsible for OCA7 type of albinism [10]. Furthermore, an additional 10 genes have been associated with syndromic OCA variants such as Hermansky–Pudlak syndrome (HPS) and Chediak–Higashi syndrome (CHS) [2]. Noted, there is a form of albinism, ocular albinism (OA1), affecting the eyes, but does not affect the hair and skin, which is caused by the mutation in G protein-coupled receptor 143 gene (GPR143; OMIM 300808) [11].
Oculocutaneous albinism affects approximately one in 20,000 individuals worldwide; however, the prevalence of its subtypes varies among different populations [12]. Although OCA1, OCA2, and OCA4 are present in Caucasian populations, the most common form is OCA1 [6]. OCA3 is present in mainly Africans and is rarely seen in other populations [6], OCA5 has been found in one Pakistani family to date [8], OCA6 is recently discovered in one Chinese family [9] and OCA7 has been explored in several Faroese families (Denmark) [10].
Since the most frequent forms of OCA in Caucasian population are OCA1, OCA2, and OCA4, we performed mutation screening of the TYR, OCA2, and SLC45A2 genes to promote the understanding of disease heterogeneity, to assess the independent and cumulative contributions of these three genes to the disease development, and to compare relative and cumulative frequencies of disease variants in a representative Hungarian OCA population.
Patients and methods
Examined individuals
The individuals (n = 13) participating in this study were recruited at the Mór Kaposi Teaching Hospital of the Somogy County (Kaposvár, Hungary), at the Hospital of Zala County (Zalaegerszeg, Hungary) and at the Department of Dermatology and Allergology, University of Szeged (Szeged, Hungary). In the enrolled patients, the diagnosis of OCA was established in the presence of skin and hair hypopigmentation and distinctive ocular changes such as nystagmus, reduced iris pigmentation, reduced retinal pigmentation, and foveal hypoplasia (Table 1). All investigated individuals were Hungarians.
The investigation was approved by the Internal Ethical Review Board of the University of Szeged. Written informed consent was obtained from the patients and the healthy controls, and the study was conducted according to the Principles of the Declaration of Helsinki.
Genetic investigation
Blood was drawn from the enrolled individuals, and genomic DNA was isolated using a BioRobot EZ1 DSP Workstation (QIAGEN; Godollo, Hungary). The entire coding regions and the flanking introns of the TYR, OCA2, and SLC45A2 genes were amplified (primer sequences used were taken from the UCSC Genome Browser). Since a pseudogene of TYR, tyrosinase-like gene (TYRL; OMIM 191270) is known, which shows 98.55% identity with the 3′region of TYR (exon 4 and 5), specific primers were used for amplification of these regions [13]. Direct sequencing of PCR products was performed on an ABI 3100 sequencer and compared with the wild-type gene sequences using the Ensemble Genome Browser.
Pathogenicity predictions
As in previous study [14], in silico tools were applied to identify the functional impact of the newly detected missense mutations. Here we used SIFT (Sorting Intolerant from Tolerant, PolyPhen 2.0 (Polymorphism Phenotyping), Mutation Taster, PROVEAN (Protein Variation Effect Analyzer) and PANTHER (Protein ANalysis THrough Evolutionary Relationships) tools. SIFT is based on the evolutionary conservation and predicts whether an amino acid substitution affects protein function. SIFT prediction score ranges from 0 to 1, and the amino acid substitution is predicted damaging if the score is less than an equal to 0.05, and tolerated if the score is greater than 0.05 [15]. PolyPhen 2.0 is based on structural and comparative evolutionary considerations and predicts the possible impact of an amino acid substitution on the stability and function of a protein. PolyPhen 2.0 uses the same range than SIFT (0–1) and the substitution is predicted to be possibly/probably damaging at greater than an equal to 0.5 value [16]. Mutation Taster is a prediction software based on the physicochemical properties of amino acids and scores substitutions according to the degree of difference between the original and the new amino acid (0–215) [17]. PROVEAN prediction is based on the sequence homology. If the PROVEAN score is equal to or below the default score threshold (−2.5), the protein variant is predicted to be deleterious and if the score is above the threshold, the variant is predicted to be neutral [18]. PANTHER program is a library of protein family and subfamily, which predicts the occurrence frequency of an amino acid in evolutionary conserved protein sequences. If the score is −3 or less, the variant is predicted has deleterious effect [19].
Results
During the investigation of TYR, OCA2, and SLC45A2 genes, we have identified pathogenic mutations in 84% (n = 11) of the examined individuals (n = 13), as shown in Table 2. In 4 cases, two heterozygous mutations have been found, suggesting a compound heterozygous state. Seven patients carried only one disease-causing mutation. Furthermore, in 11 cases out of 13, we have detected one or more common polymorphisms.
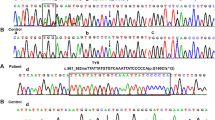
Direct sequencing of the TYR gene revealed pathogenic mutations in 69% (n = 9) of the investigated patients. Only one patient carried two heterozygous mutations, a Thymin-base duplication (c.74dupT, p.Ser26Leufs*2) and a nonsense (c.346C>T, p.Arg116*) mutation in the first exon of TYR gene, suggesting a compound heterozygous state. Three patients carried the c.1037−7T>A splice site mutation in homozygous form. Out of these three patients, two (Patient 2 and 3) are related to each other and one (Patient 4) is not aware of any relationship with the other two mutation carriers. We have detected the c.1204C>T p.Arg402* nonsense mutation heterozygously in one case and the c.650G>A p.Arg217Gln missense mutation heterozygously in three patients. The heterozygous c.1366+4A>G splice site mutation has been detected in one patient. This patient was additionally heterozygous for the SLC45A2 c.1099G>A p.Val367Ile mutation. Considering the common polymorphisms of TYR gene, the c.575C>A p.Ser192Tyr, and c.1205G>A p.Arg402Gln were detected in seven patients. The p.Ser192Tyr variant affects a copper-binding domain of the protein; all the other exonic pathogenic and non-pathogenic variants are located outside of the known functional domains of the enzyme (Fig. 1a).

a Distribution of the detected TYR variants on the tyrosinase protein, b OCA2 variant on the transporter protein, and c SLC45A2 variants on the transporter protein (SP signal peptide, EGF-like domain epidermal-growth-factor-like domain, CuA copper-binding domain, CuB copper-binding domain, TM transmembrane domain)
No pathogenic OCA2 mutation was identified in the investigated OCA individuals. However, one patient with the pathogenic c.1037−7T>A TYR mutation carried the common c.913C>T p.Arg305Trp polymorphism of the OCA2 gene in heterozygous form. This variant does not affect any known functional domains of the OCA2 protein (Fig. 1b).
Based on our results, 3 of 13 patients carried SLC45A2 pathogenic mutation. Two patients carried the combination of two novel mutation previously described by our workgroup: the c.1226G>A p.Gly409Asp missense mutation and the c.1459C>T p.Gln487* nonsense mutation. These mutations are not present in any SNP database (ExAC, 1000 Genome Project, ClinVar). Prediction analyses were performed to identify the functional role of the missense mutation. All prediction software suggested that the p.Gly409Asp mutation is deleterious (SIFT score: 0.002, damaging; PolyPhen 2.0 score: 0.996, probably damaging; Mutation Taster score: 94, disease causing; PROVEAN score: −3.25, deleterious; PANTHER score: −4.26, deleterious). The nonsense mutation was deemed to be pathogenic. One patient carried the c.1099G>A p.Val367Ile missense mutation beside the c.1366+4A>G splice site mutation on the TYR gene. In eight patients, only the non-pathogenic c.1122G>C p.Leu374Phe missense polymorphism was detected. All the detected pathogenic and non-pathogenic variants are located within transmembrane domains of the encoded protein (Fig. 1c).
Discussion
This study reports the concomitant investigation of three genes (TYR, OCA2, and SLC45A2) in 13 Hungarian OCA patients, which have been implicated in the development of isolated OCA forms. The TYR gene encodes the tyrosinase enzyme, which catalyzes the first and second steps in melanin synthesis: the hydroxylation of tyrosine to l-DOPA and the oxidation of l-DOPA to DOPA-quinone [3]. The OCA2 and SLC45A2 genes encode transporter proteins, which are implicated in the trafficking of tyrosinase to melanosomes [20, 21].
Pathogenic TYR mutations were present in 69% (n = 9) of the patients. However, the sample size of this study is small, our results correlate well with previous findings that OCA1 is the most common isolated OCA subtype and TYR mutations account for approximately 25–50% of the isolated OCA cases worldwide [6, 22].
In our study, three missense variants were detected for the TYR gene. One of these variants is considered pathogenic (p.Arg217Gln) and two (p.Ser192Tyr, p.Arg402Gln) are considered common polymorphisms. The p.Arg217Gln mutation is located in a non-conservative region of tyrosinase protein, at this amino acid position two other known missense mutations are described (p.Arg217Gly, p.Arg217Trp) [23]. The polymorphisms of TYR gene were not directly related to pigmentation phenotypes in normal Caucasians, but their impact should be taken into account as an important modifier of human skin, hair, and eye color [24]. Functional studies reported that 192Tyr allele reduced tyrosinase activity, significant reduction in heterozygous and consistent decrease in homozygous form were observed, and the presence of 402Gln allele resulted significantly less TYR protein, displayed altered trafficking and glycosylation, with reduced DOPA oxidase [24]. The p.Arg402Gln polymorphism exhibits reduced tyrosinase activity at physiological temperature and is considered a temperature-sensitive variant [25,26,27]. This variant alone is unable to cause OCA; however, its increased frequency in OCA patients with one heterozygous pathogenic TYR mutation suggests that it can contribute to the development of OCA in combination with a pathogenic mutation [28]. Two Hungarian patients carried the p.Arg402Gln polymorphism in combination with the p.Arg217Gln pathogenic variant. Previous report suggests this combination might contribute to the development of the OCA symptoms of the patient [26].
In one case, compound heterozygosity for two mutations was found. A heterozygous T-base duplication (c.74dupT p.Ser26Leufs*2) and a nonsense mutation (c.346C>T, p.Arg116*) were identified on TYR gene. Both mutations lead to the development of a premature termination codon. Due to these changes, the translated mutant TYR protein is highly truncated and we assume that these enormous truncations of the mutant TYR protein may lead to its dysfunction.
Two of the nine Hungarian OCA patients with TYR mutations carry a combination of two pathogenic mutations. In three of nine, a splice site mutation was identified in homozygous form. In four of nine, only one heterozygous pathogenic mutation was identified. These results correlate well with the recently reported investigation of an Iranian OCA population: pathogenic TYR variants were identified in 19 of 30 patients, and in this study, six patients carried only one pathogenic TYR mutation, and any pathogenic mutation were not identified in five patients [4].
The pathogenic TYR mutations detected in the Hungarian OCA patients have been previously identified in OCA patients of different ethnicity. The p.Arg217Gln mutation was detected in Caucasians from USA, Canada, and Northern-Europe; the p.Arg402* in Caucasians from Lebanon; the c.1037−7T>A in Caucasians and Japanese; the c.1366+4A>G in Caucasians; and the c.346C>T in Caucasians, Japanese, and Germans [22, 29]. The frequency of pathogenic mutations differs in different populations, and, therefore, these populations might vary in their genetic susceptibility to certain diseases. Based on our results and the results of previous studies, the identified pathogenic TYR mutations are not specific to the Hungarian population, as they have been detected worldwide in OCA patients [22, 29].
No pathogenic OCA2 mutation was identified in the examined Hungarian individuals, although one patient carried the common p.Arg305Trp polymorphism in heterozygous form. This variant has been associated with human eye color and might be an inherited biomarker of cutaneous cancer risk [28, 30].
Three mutation and a common polymorphisms were detected on the SLC45A2 gene. Two patients carried two heterozygous variants of the SLC45A2 gene previously described by our workgroup: the p.Gly409Asp missense and the p.Gln487* nonsense mutations [31]. Both mutations are situated in transmembrane domains of the MATP protein (Uniprot: Q9UMX9). The locations of the mutations suggest that they impair the transport function of the MATP protein. MATP dysfunction might cause an acidic melanosomal lumen, leading to incorrect incorporation of copper into tyrosinase. The reduced tyrosinase activity could lead to the development of the OCA phenotype [32]. Besides, the p.Leu374Phe polymorphism was detected in nine Hungarian OCA patients. This variant has a striking population distribution, exists almost exclusively in Europeans, and has also been implicated in the development of different shades of hair, skin, and eye color [33].
Our results emphasize the importance of the parallel analysis of multiple genes for studying disease phenotypes. The OCA cases presented in this study and many other cases reported in the literature call our attention to the fact that clinical symptoms—which may overlap in many cases—are not sufficient for a diagnosis of different OCA forms: the molecular genetic investigation of all OCA genes is required to determine the subtype of the disease. The Hungarian OCA patients in this study exhibited identical clinical features (Table 1); however, molecular genetic investigation identified the OCA1 subtype in eight cases, the OCA4 subtype in two cases, and the molecular diagnosis was not clearly defined in three patients (Table 2). Even with these data, the genetic basis of the disease in seven patients carrying only one pathogenic TYR or SLC45A2 mutation is still not completely explained. We wish to emphasize the screening of the non-pathogenic variants, which alone could not lead to the development of OCA, should be carried out in association with pathogenic variants that might have clinical significance. Further targeted sequencing of the genes involved in the syndromic OCA variants, including HPS and CHS, as well as genes involved in human pigmentation, is hoped elucidate the underlying disease-causing variant(s) [22].
Our results and previously reported studies suggest that, among the investigated genes, the majority of the mutations are located within the TYR gene [3, 4]. This result correlates well with the results obtained in other populations, as TYR mutations are the most common for OCA worldwide [1]. Screening of the TYR gene is, therefore, of primary importance for diagnostics. Mutations in the investigated Hungarian and in other previously reported OCA patients were found most frequently in exons 1 and 4 of the TYR gene [4]. In light of the fact that the majority of the identified TYR mutations are located within exon 1 and 4, we recommend screening these exons first.
According to our current knowledge, 10–25% of the isolated and syndromic OCA cases are not explained by paired, trans-oriented mutations in known genes [34, 35]. Based on our results and the results of previous studies [22], we suggest that screening non-Mendelian OCA-associated genes might elucidate the causative genetic variant for these cases.
Conclusions
The genetic heterogeneity of OCA is extremely complex: both rare mutations of Mendelian genes and common variants of non-Mendelian genes can contribute to the development of the disease. Our multi-gene study provides novel data for the genetic diversity of OCA in Hungarians and indicates that approaches that take this complexity into account, including large-scale studies, are needed to complete our understanding of the genetic heterogeneity of this disease.
Abbreviations
- OCA:
-
oculocutaneous albinism
- OCA1:
-
oculocutaneous albinism type 1
- OCA2:
-
oculocutaneous albinism type 2
- OCA3:
-
oculocutaneous albinism type 3
- OCA4:
-
oculocutaneous albinism type 4
- OCA5:
-
oculocutaneous albinism type 5
- OCA6:
-
oculocutaneous albinism type 6
- OCA7:
-
oculocutaneous albinism type 7
- TYR :
-
tyrosinase gene
- OCA2 :
-
oculocutaneous albinism two gene
- TYRP :
-
tyrosinase-related protein gene
- SLC45A2 :
-
solute carrier family 45, member 2 gene
- SLC24A5 :
-
sodium/calcium/potassium exchanger 5 gene
- C10ORF11 :
-
chromosome 10 open reading frame 11 gene
- HPS:
-
Hermansky–Pudlak syndrome
- CHS:
-
Chediak–Higashi syndrome
- OA1:
-
ocular albinism
- GPR143 :
-
G protein-coupled receptor 143 gene
- TYRL :
-
tyrosinase-like gene
- MATP:
-
membrane-associated transport protein
References
Mártinez-García M, Montoliu L. Albinism in Europe. J Dermatol. 2013;40:319–24.
Montoliu L, Grønskov K, Wei AH, Martínez-García M, Fernández A, Arveiler B, et al. Increasing the complexity: new genes and new types of albinism. Pigment Cell Melanoma Res. 2014;27:11–8.
King RA, Pietsch J, Fryer JP, Savage S, Brott MJ, Russell-Eggitt I, et al. Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA1): definition of the phenotype. Hum Genet. 2003;113:502–13.
Ghodsinejad Kalahroudi V, Kamalidehghan B, Arasteh Kani A, Aryani O, Tondar M, Ahmadipour F, et al. Two novel tyrosinase (TYR) gene mutations with pathogenic impact on oculocutaneous albinism type 1 (OCA1). PLoS ONE. 2014;12(9):e106656.
Durham-Pierre D, Gardner JM, Nakatsu Y, King RA, Francke U, Ching A, et al. African origin of an intragenic deletion of the human P gene in tyrosinase positive oculocutaneous albinism. Nat Genet. 1994;7:176–9.
Rooryck C, Morice-Picard F, Elcioglu NH, Lacombe D, Taieb A, Arveiler B. Molecular diagnosis of oculocutaneous albinism: new mutations in the OCA1-4 genes and practical aspects. Pigment Cell Melanoma Res. 2008;21:583–7.
Inagaki K, Suzuki T, Shimizu H, Ishii N, Umezawa Y, Tada J, et al. Oculocutaneous albinism type 4 is one of themost common types of albinism in Japan. Am J Hum Genet. 2004;74:466–71.
Kausar T, Bhatti MA, Ali M, Shaikh RS, Ahmed ZM. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin Genet. 2013;84:91–3.
Wei AH, Zang DJ, Zhang Z, Liu XZ, He X, Yang L, et al. Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism. J Invest Dermatol. 2013;133:1834–40.
Grønskov K, Dooley CM, Østergaard E, Kelsh RN, Hansen L, Levesque MP, et al. Mutations in c10orf11, a melanocyte-differentiation gene, cause autosomal-recessive albinism. Am J Hum Genet. 2013;92:415–21.
Schiaffino MV, Tacchetti C. The ocular albinism type 1 (OA1) protein and the evidence for an intracellular signal transduction system involved in melanosome biogenesis. Pigment Cell Res. 2005;18:227–33.
Gargiulo A, Testa F, Rossi S, Iorio V, Fecarotta S, de Berardinis T, et al. Molecular and clinical characterization of albinism in a large cohort of Italian patients. Invest Ophthalmol Vis Sci. 2011;52:1281–9.
Chaki M, Mukhopadhyay A, Ray K. Determination of variants in the 3′-region of the tyrosinase gene requires locus specific amplification. Hum Mutat. 2005;26:53–8.
Kamaraj B, Purohit R. Mutational analysis on membrane associated transporter protein (MATP) and their structural consequences in oculocutaneous albinism type 4 (OCA4)—a molecular dynamics approach. J Cell Biochem. 2016;117:2608–19.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7:e46688.
Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–41.
Newton JM, Cohen-Barak O, Hagiwara N, Gardner JM, Davisson MT, King RA, et al. Mutations in the human orthologue of the mouse under white gene (uw) underlie a new form of oculocutaneous albinism, OCA4. Am J Hum Genet. 2001;69:981–8.
Costin GE, Valencia JC, Vieira WD, Lamoreux ML, Hearing VJ. Tyrosinase processing and intracellular trafficking is disrupted in mouse primary melanocytes carrying the under white (uw) mutation. A model for oculocutaneous albinism (OCA) type 4. J Cell Sci. 2003;116:3203–12.
Simeonov DR, Wang X, Wang C, Sergeev Y, Dolinska M, Bower M, et al. DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Hum Mutat. 2013;34:827–35.
Spritz RA, Oh J, Fukai K, Holmes SA, Ho L, Chitayat D, et al. Novel mutations of the tyrosinase (TYR) gene in type I oculocutaneous albinism (OCA1). Hum Mutat. 1997;10:171–4.
Jagirdar K, Smit DJ, Ainger SA, Lee KJ, Brown DL, Chapman B, et al. Molecular analysis of common polymorphisms within the human tyrosinase locus and genetic association with pigmentation traits. Pigment Cell Melanoma Res. 2014;27:552–64.
Berson JF, Frank DW, Calvo PA, Bieler BM, Marks MS. A common temperature-sensitive allelic form of human tyrosinase is retained in the endoplasmic reticulum at the nonpermissive temperature. J Biol Chem. 2000;275:12281–9.
Halaban R, Svedine S, Cheng E, Smicun Y, Aron R, Hebert DN. Endoplasmic reticulum retention is a common defect associated with tyrosinase-negative albinism. Proc Natl Acad Sci USA. 2000;97:5889–94.
Tripathi RK, Giebel LB, Strunk KM, Spritz RA. A polymorphism of the human tyrosinase gene is associated with temperature-sensitive enzymatic activity. Gene Expr. 1991;1:103–10.
Rebbeck TR, Kanetsky PA, Walker AH, Holmes R, Halpern AC, Schuchter LM, et al. P gene as an inherited biomarker of human eye color. Cancer Epidemiol Biomarkers Prev. 2002;11:782–4.
Hutton SM, Spritz RA. A comprehensive genetic study of autosomal recessive ocular albinism in caucasian patients. Invest Ophthalmol Vis Sci. 2008;49:868–72.
Jannot AS, Meziani R, Bertrand G, Gérard B, Descamps V, Archimbaud A, et al. Allele variations in the OCA2 gene (pink-eyed-dilution locus) are associated with genetic susceptibility to melanoma. Eur J Hum Genet. 2005;13:913–20.
Tóth L, Fábos B, Farkas K, Sulák A, Tripolszki K, Széll M, Nagy N. Identification of two novel mutations in the SLC45A2 gene in a Hungarian pedigree affected by unusual OCA type 4. BMC Med Genet. 2017;18:27.
Bin BH, Bhin J, Yang SH, Shin M, Nam YJ, Choi DH, et al. Membrane-associated transporter protein (MATP) regulates melanosomal pH and influences tyrosinase activity. PLoS ONE. 2015;10:e0129273.
Graf J, Hodgson R, van Daal A. Single nucleotide polymorphisms in the MATP gene are associated with normal human pigmentation variation. Hum Mutat. 2005;25:278–84.
Chaki M, Sengupta M, Mukhopadhyay A, Subba Rao I, Majumder PP, Das M, et al. OCA1 in different ethnic groups of india is primarily due to founder mutations in the tyrosinase gene. Ann Hum Genet. 2006;70:623–30.
Wei A, Wang Y, Long Y, Wang Y, Guo X, Zhou Z, et al. A comprehensive analysis reveals mutational spectra and common alleles in chinese patients with culocutaneous albinism. J Invest Dermatol. 2010;130:716–24.
Authors’ contributions
BF and KF performed the mutation analysis and wrote the manuscript. LT, AS, and KT participated in the mutation analysis. BF, MT, RN, KV, and ZC cared for patients. LK and MS were mentors who guided the research study. NN designed the study and helped in drafting the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank all the participants for the blood donation from the Mór Kaposi Teaching Hospital of the Somogy County, Kaposvár, the Hospital of Zala County, Zalaegerszeg and the Department of Dermatology and Allergology, University of Szeged, Szeged, Hungary.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All available data are included within the article.
Consent for publication
All the reported patients gave their informed consent to the publication.
Ethics approval and consent to participate
The investigation was approved by the Internal Review Board of the University of Szeged. Written informed consent was obtained from the patients and unrelated healthy individuals. The study was conducted according to the Principles of the Declaration of Helsinki.
Funding
This study was supported by the Hungarian TÁMOP-4.2.2.A-11/1/KONV-2012-0035 Grant, TÁMOP-4.2.4.A/2-11-1-2012-0001 Grant, TÁMOP-4.2.2.A3 Grant, and GINOP-2.3.2-15-2016-00039 Grant.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fábos, B., Farkas, K., Tóth, L. et al. Delineating the genetic heterogeneity of OCA in Hungarian patients. Eur J Med Res 22, 20 (2017). https://doi.org/10.1186/s40001-017-0262-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40001-017-0262-0